
Abstract
Several oncogenes and tumor-suppressor genes have been shown to be implicated in the development, progression and response to therapy of invasive breast cancer. The phenotypic uniqueness (and thus the heterogeneity of clinical behavior) among patients’ tumors may be traceable to the underlying variation in gene copy number of these genes. To obtain a more complete view of gene copy number changes and their relation to phenotype, we analyzed 20 breast cancer-related genes in 104 invasive breast cancers with the use of multiplex ligation-dependent probe amplification (MLPA). We identified MYC gene amplification in 48% of patients, PRDM14 in 34%, topoisomerase IIα (TOP2A) in 32%, ADAM9 in 32%, HER2 in 28%, cyclin D1 (CCND1) in 26%, EMSY in 25%, IKBKB in 21%, AURKA in 17%, FGFR1 in 17%, estrogen receptor alpha (ESR1) in 16%, CCNE1 in 12% and EGFR in 9% of patients. There was a significant correlation between the number of amplified genes and the histological grade and mitotic index of the tumor. Gene amplifications of EGFR, CCNE1 and HER2 were negatively associated with estrogen receptor status whereas FGFR1, ADAM9, IKBKB and TOP2A revealed a positive association. Amplifications of ESR1, PRDM14, MYC and HER2 were associated with a high mitotic index, and PRDM14 and HER2 amplifications with high histological grade. MYC amplification was detected more frequently in ductal tumors and high-level MYC amplifications were significantly associated with large tumor size. HER2/MYC, HER2/CCNE1 and EGFR/MYC co-amplified tumors were significantly larger than tumors with either of these amplifications. Gene loss occurred most frequently in E-cadherin (CDH1) (20%) and FGFR1 (10%). In conclusion, MLPA analysis with this ‘breast cancer kit’ allowed to simultaneously assess copy numbers of 20 important breast cancer genes, providing an overview of the most frequent (co)amplifications as well as interesting phenotypic correlations, and thereby data on the potential importance of these genes in breast cancer.
Similar content being viewed by others


Main
Several genes have been shown to be involved in the development, progression and response to therapy of invasive breast cancer. Among these, HER-2/neu is likely the most important proto-oncogene. Amplification of the HER2 gene is present in approximately 15–30% of breast carcinomas and leads to protein overexpression, which correlates with a poor outcome1, 2, 3 and is associated with a good response to treatment with trastuzumab, a recombinant humanized monoclonal anti-HER2 antibody.4, 5 Furthermore, amplification of HER2 has also been shown to correlate with resistance to conventional adjuvant chemotherapy and tamoxifen.6, 7, 8, 9, 10 Topoisomerase IIα (TOP2A) gene amplification seems to be predictive of response to a class of cytostatic agents called TopoII inhibitors, which include the anthracyclines.11, 12, 13, 14, 15, 16 Recently, estrogen receptor alpha (ESR1) gene amplification has been implicated in response to tamoxifen therapy,17 but its significance was doubted by others.18 Amplification of MYC has been associated with poor prognosis and resistance to anti-estrogen therapy.19 Therapeutic or prognostic significance of other frequently amplified genes such as cyclin D1 (CCND1)20 or frequent loss of genes such as E-cadherin (CDH1) is less clear, and comparative genomic hybridization (CGH) studies have pointed to many more genes and chromosomal loci with potentially important copy number changes.21, 22, 23
Nevertheless, no single gene copy number seems to completely explain prognosis or response to therapy of individual breast cancer patients. A simultaneous analysis of copy number changes of a variety of genes involved in prognosis and therapy response may thus be very useful for molecular profiling of individual breast cancer patients. This can be achieved by array CGH but this is still a costly and labor-intensive technique that requires a relatively large amount of sample DNA and specialized personnel to deal with the complexity of the data. In this study, we used an easier and faster high-throughput PCR-based technique, called multiplex ligation-dependent probe amplification (MLPA).24 This assay determines relative gene copy numbers in a quantitative way and requires only minute quantities of small DNA fragments, which makes it very suitable for DNA isolated from paraffin-embedded material. In previous studies, we obtained promising results with HER2 MLPA in comparison with immunohistochemistry25 and in situ hybridization26 and evaluated this technique to simultaneously determine copy number changes of HER227 and TOP2A.16 The goal of this study was to apply MLPA as a technique to simultaneously detect amplifications and/or losses of a large set of breast cancer-related genes. These genes (including HER2, EGFR, TOP2A, MYC, CCND1, CCNB1, ESR1, AURKA, EMSY, CDH1, FGFR1, PRDM14, ADAM9 and IKBKB) were selected based on their prognostic and/or therapeutic implications in breast cancer, or their proven frequent copy number change by CGH. We sought to obtain a more complete view of the clinical significance of MLPA-detected gene copy number alterations and therefore analyzed their mutual interactions, as well as their associations with common prognostic factors such as age, histological type and grade, HER2 immunohistochemistry, estrogen and progesterone receptor (ER, PR) status, mitotic index28 and tumor size.
Materials and methods
Patient Material
Tissue samples of invasive breast cancer patients were collected between November 2004 and December 2008 at the Department of Pathology of the University Medical Center in Utrecht (UMCU), The Netherlands. This study randomly selected 104 tissue samples from this consecutive series. Anonymous use of redundant tissue for research purposes is part of the standard treatment agreement with patients in the UMCU.29 All tissue samples were analyzed with immunohistochemistry to assess HER2, ER and PR protein expression and MLPA to determine gene copy number alterations. In addition, age at diagnosis, histological type, tumor size, histological grade and mitotic activity index (MAI)30 were determined for all patients.
Immunohistochemistry
Immunohistochemistry for HER2 was performed using the Hercep test (Dako, Glostrup, Denmark) according to the manufacturers’ instructions on 4 μm thick sections from the neutral buffered formaldehyde fixed tissue blocks. Immunohistochemistry membrane staining was semiquantitatively scored as negative (0), weakly positive (1+), equivocal (2+) and strongly positive (3+) according to the DAKO FDA-approved scoring system. Interpretation of staining was conducted by two experienced breast pathologists. As control, a small tissue array containing a 0, 1+, 2+ and 3+ breast tumor samples was taken along on the same slide as the tumor to be analyzed. Immunohistochemical staining for ER (1D5, 1:80, Dako) and PR (PGR636, 1:200, Dako) was performed using a Bond-Max automated staining machine (Vision Biosystems, Newcastle, UK) with the Bond polymer refine detection kit (Vision BioSystems, cat. no DS9800). Negative controls were used throughout.
Multiplex Ligation-Dependent Probe Amplification
Invasive tumor areas as identified on serial H&E sections were harvested from one or two whole 4 μm thick paraffin sections (corresponding to approximately 1 square cm tumor tissue) with a scalpel. We have estimated the tumor percentages of all samples before MLPA and used samples with tumor percentages of at least 70%. In a previous study,26 we showed that tumor percentages >30% are already sufficient for reliable MLPA performance and that more than half of the tumors show a tumor percentage >60%. DNA was isolated from these tissue fragments by 1-h incubation in proteinase K (10 mg/ml; Roche, Almere, The Netherlands) at 56 °C followed by boiling for 10 min. This DNA solution (50–100 μl) was, after centrifugation, used in the MLPA analysis according the manufacturers’ instructions, using the P078-A1 kit (MRC Holland, Amsterdam, The Netherlands). The contents of this kit are depicted in Table 1. All tests were performed in duplicate in an ABI 9700 PCR machine (Applied Biosystems, Foster City, CA, USA). PCR products were analyzed on an ABI310 capillary sequencer (Applied Biosystems). Gene copy numbers were analyzed using Genescan (Applied Biosystems) and Coffalyser (version 7.0) software (MRC-Holland). For genes with more than one probe present in the kit, the mean of all the probe peaks of this gene in duplicate was calculated. If this mean value was below 0.7 the respective gene was defined as lost, a value between 0.7 and 1.3 was defined as normal, a value between 1.3 and 2.0 as low-level amplification and values >2.0 as high-level amplified as previously established.31, 32
Statistics
Statistics were performed using SPSS statistical software. Data were dichotomized as follows: amplified vs non-amplified, grade I vs grade II/III, age <50 vs ≥50, tumor size pT1 vs pT2/pT3, ER and PR positive vs negative, MAI <13 vs ≥13, ductal vs lobular, HER2 IHC 0/1+ vs 2+/3+. Associations were examined using χ2 test and Fisher's exact tests, if applicable. Correlations were calculated with Spearman's rho. Unsupervised hierarchical cluster analysis (Euclidean distance, average linkage analysis) was performed using the open-source R statistical software (http://www.r-project.org).
Results
Amplifications and Losses
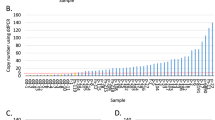
Frequencies of gains and losses for the 20 analyzed genes in 104 invasive breast cancers are depicted in Figure 1 and Table 2. All analyzed regions were involved in amplification with varying frequencies. A majority of the amplifications (low and high level) were found on chromosome 8 (particularly MYC, PRDM14 and ADAM9 in 48%, 34% and 32% of the patients, respectively) and on chromosome 17 (particularly TRAF4, CDC6, TOP2A and HER2 in 36, 35, 32 and 28% of the patients, respectively). Although MYC showed amplification in almost half the patients, only 16% of these amplifications were high level (ratio > 2.0). For HER2, in contrast, most amplifications (72%) were high level. In a previous study, we already established a good correlation between HER2 gene amplification by MLPA and HER2 gene amplification by in situ hybridization.26 Of 56 out of 104 (54%) patients, we had previously determined HER2 chromogenic in situ hybridization data: of 11 out of 21 MLPA amplified patients there were CISH data available and all 11 patients showed CISH amplification. Of 41 out of 75 MLPA HER2 normal patients, CISH data were available and all 41 tumors were normal by CISH. Of four out of eight HER2 MLPA low-level amplified patients, CISH data were available: three-fourths were normal and one-fourth was amplified by CISH. CCND1 amplification was found in 26% of the patients, and 56% of these amplifications were high level. ESR1 amplification was found in 16% of the patients although most were low level and only rarely high level (2% of all patients).

Amplifications (green) and losses (red) for 20 oncogenes and tumor-suppressor genes as found by analysis of 104 invasive breast cancer patients with the P078-A1 breast cancer dedicated MLPA kit. The chromosome numbers of the genes are shown on the horizontal axis.
Several regions showed loss by MLPA. The two regions with the most frequent loss were CDH1 on chromosome 16 (20% of patients: 13 out of 21 of ductal and 6 out of 21 of lobular type) and FGFR1 on chromosome 8 (10% of the patients).
On average, this study found five amplifications per patient (range 0–17 of the 20 analyzed genes) of which two were high-level amplifications (range 0–10). Only five patients (5%) did not show any amplification or loss for the analyzed regions. Five other patients showed no amplifications but did show loss of one or more genes. Of these five patients, there were three patients with only loss of CDH1 (two-thirds were ductal carcinomas). One other patient with a lobular carcinoma showed a loss of CDH1 accompanied by IKBKB, CCND1 and LASP1 loss, and the fifth patient presented with a PRDM14 and FGFR1 loss. Ten patients showed amplifications of just one gene: PRDM14 (3 out of 10), MYC (3 out of 10), EMSY (2 out of 10) and a high-level amplification of AURKA and EGFR in one patient each.
Co-amplified Regions, Loss of Regions and Their Association
A majority of the genes were never found amplified or lost alone. Nine of the 104 patients (9%) showed amplifications for all five analyzed chromosome 8 genes, possibly pointing to polysomy 8. In 13 patients (13%), both genes on chromosome 11q were amplified. Two patients (2%) were amplified for all eight chromosome 17q genes analyzed, possibly pointing to gain of 17q. Of these two patients, one was also amplified for all chromosome 8 and 11 genes, and the other patient was amplified for all chromosome 8 genes. None of the patients showed loss for all analyzed chromosome 8, 11 and/or 17 genes.
In all, 15% of all patients showed a co-amplification of HER2 and MYC, 13% of HER2 and TOP2A, 9% of HER2 and CCND1 and 7% of HER2 and CCNE1. Eighteen percent of all patients showed a co-amplification of MYC and TOP2A and 16% of MYC and CCND1. Figure 2 shows the percentage of co-amplifications of HER2, MYC, CCND1, CCNE1 and TOP2A amplified breast tumors. Of the 27 HER2 amplified breast cancers, 52 and 45% were MYC and TOP2A co-amplified, respectively. Of the 12 CCNE1 amplified patients, 10 were also MYC amplified (83%). When only high-level amplifications (MLPA ratio >2.0) were considered relevant, 5 out of 21 HER2 high-level amplified patients were also CCND1 (high level) amplified, 4 out of 21 TOP2A co-amplified, 1 out of 21 MYC co-amplified and 1 out of 21 CCNE1 co-amplified. Of the 15 CCND1 high-level amplified patients, 5 out of 15 were HER2 co-amplified, 2 out of 15 TOP2A co-amplified and 1 out of 15 was MYC amplified. Patients with an amplification of EGFR had an increased likelihood to also have CCNE1 amplifications (P<0.001) and tumors with a TOP2A amplification had an increased probability of EMSY amplification (P=0.004). Furthermore, patients with high-level HER2 amplifications had an increased probability to have high-level TOP2A amplifications (P=0.017) and patients with high-level EGFR amplification a higher risk of having high-level MYC amplifications (P=0.023).

Co-amplifications of HER2, TOP2A, MYC, CCND1 and CCNE1 amplified breast tumors in a series of 104 invasive breast cancers analyzed by MLPA.
Cluster analysis, as illustrated in Figure 3, showed one apparent cluster of HER2 (ERBB2) and PPARBP. Most other chromosome 17 genes (TOP2A, CPD, CDC6, TRAF4 and NOS2A) were located in a different cluster. ESR1, CCNE1 and all chromosome 8 genes except for FGFR1 formed another cluster.

Hierarchical cluster analysis of 104 invasive breast cancer patients (horizontal axis) analyzed by MLPA for 20 breast cancer-related genes (vertical axis).
Supplementary Table 1 shows the co-amplifications (all and high level) for all 20 genes analyzed by MLPA.
Association between Amplified Regions and Clinical Characteristics
There was a significant correlation between the number of amplifications per tumor and grade (P=0.030) and even more between the number of high-level amplifications per tumor and grade (P<0.001). There was a significant association between the number of amplifications and HER2 immunohistochemistry status. In addition, there was a significant correlation between the number of amplifications and high-level amplifications per tumor and mitotic index (P=0.015 and P=0.004, respectively) but there was no association with tumor size, with hormone receptor status nor with the patient's age. We also found significantly more high-level amplifications per tumor for tumors of the ductal subtype than for tumors of the lobular subtype (P<0.001, P=0.083) but not for all amplifications. Although not significant, we did observe more CDH1 loss in lobular tumors (36%) than in ductal tumors (17%).
Table 3 shows the association of several amplified regions with clinicopathological characteristics. There was, as expected, a significant association between HER2 immunohistochemistry and HER2 gene amplification (P<0.001), but there was also a significant association with PRDM14 gene amplification (P=0.027). ESR1 amplification was significantly associated with higher MAI (P=0.007) and showed a trend toward association with higher grade (P=0.054). EGFR amplification was significantly associated with negative ER status (P=0.005) and showed a trend toward association with negative PR status (P=0.052). FGFR1 and ADAM9 amplifications were significantly associated with positive ER status (P=0.032 and P=0.019, respectively). IKBKB was significantly associated with positive ER and PR status (P=0.026 and 0.015, respectively). PRDM14 amplification was correlated with higher grade (P=0.049) and MAI (P=0.010). MYC amplification was significantly associated with higher MAI (P=0.040) and with the ductal subtype (P=0.011). High-level MYC amplifications were significantly associated with a larger tumor size (P=0.045). HER2 amplification was associated with higher grade (P=0.040) and MAI (P=0.036) and showed a trend toward association with ER status (P=0.060, for high-level amplifications P=0.004). TOP2A amplification was significantly associated with positive ER status of the tumor (P=0.045), in contrary to CCNE1, which was significantly associated with ER negativity (P=0.004). CCND1, EMSY and AURKA amplification did not show any significant associations with clinical–pathological features. We also did not observe any association between amplified regions and age.
Tumors with HER2 and MYC co-amplification were significantly larger in size (P=0.030) than tumors with amplification of only one or neither of these genes, as were HER2–CCNE1 co-amplified tumors (P=0.017). Tumors with HER2–MYC co-amplification were also significantly associated with higher HER2 immunohistochemistry status (P<0.001). There was also a trend toward an association between tumor size and HER2–TOP2A co-amplification (P=0.061). Tumors with EGFR–MYC co-amplification were significantly associated with ER negativity (P=0.023), were significantly larger (P=0.017) and showed a trend toward higher MAI (P=0.059) than tumors with either or neither of these amplifications.
Discussion
Several chromosomal regions are frequently amplified in breast cancer. Gene amplifications are essential features of advanced cancers and have prognostic as well as therapeutic significance in clinical cancer treatment. The aim of this study was therefore to simultaneously examine the copy number status of important or promising breast cancer genes (located on different chromosomal regions) by MLPA, to study the frequency of their co-amplifications, and to couple the obtained data to clinical–pathological characteristics currently used to determine treatment and/or prognosis.
It has long been known that the more advanced a cancer is, the more rearranged the genome is. We were therefore interested in verifying whether there was an association between the number of genetic alterations observed in a tumor and worse clinical–pathological tumor characteristics. On average, this study found 5 amplifications of the 20 analyzed genes per patient of which 2 were high-level amplifications. Ten patients (10%) showed single amplifications (of which 60% involved MYC or PRDM14 amplifications), and interestingly these tumors were all grade 2 or 3 and were often highly proliferative with MAI>13. Only five patients (5%) did not show any amplification or loss in the analyzed regions. In these five tumors, the MAI was smaller than five, and three of them were grade 1 while two were grade 2. Although grade 1 tumors generally showed fewer genomic events than grade 2/3 tumors, they also rarely showed more complex genomic patterns associated with more advanced tumors indicating that there is not a strict relation between genomic state and histological grade. Nevertheless, this study found a significant correlation between the number of (high level) amplifications and the histological grade and MAI. Presence of gene amplifications may not only be important because of the resulting overexpression of the oncogenes, it may also serve as a surrogate parameter for increased genetic instability of a cancer and, as such, represent an indicator of poor patient prognosis. Indeed, an association between patient survival and the number of amplifications was described by some studies.33, 34
Amplifications and Losses
Amplifications involving chromosomes 8p (FGFR1, ADAM9, IKBKB), 11q (CCND1, EMSY) and 17q (NOS2A, TRAF4, CPD, LASP1, PPARBP, HER2, CDC6, TOP2A) are among the most common high-level copy number aberrations in breast tumors, occurring, for example, in one study, in 22.8, 19.6 and 9.9% of tumors, respectively.35 Table 2 shows that the frequencies of amplification observed by MLPA for all analyzed genes in this study are in line with other studies. A majority of the MYC amplifications observed in this study were low level (84%), which is consistent with published results.36 High-level amplifications of ESR1, a gene that is possibly involved in tamoxifen response,17 were rare although we did observe 16% of patients with increased ESR1 copy numbers. In a study by Chin et al,37 low-level copy number aberrations by array CGH were not associated with reduced survival and they hypothesized that these aberrations are presumably selected during tumor development because they increase basal cell metabolism.
We found CDH1 loss in 20% of all patients (36% in lobular carcinomas and 17% in ductal carcinomas), which is less than the reported frequency of LOH on 16q (78% in lobular carcinomas and 28% in ductal carcinomas).38 FGFR1 loss, which was found in 10% of cases in this study has previously been described and has been associated with poor outcome.37
Cluster analysis of all 20 breast cancer-related genes showed one apparent cluster of HER2 (ERBB2) and PPARBP. Both genes are located near each other on chromosome 17 and have previously been shown to be often co-amplified.39 Most other chromosome 17 genes (TOP2A, CPD, CDC6, TRAF4 and NOS2A) were located in a separate cluster, indicating that these amplifications are probably independent of HER2 amplification and represent a different advantage for tumor growth or survival. Another cluster was composed of ESR1, CCNE1 and all chromosome 8 genes except for FGFR1, which was located in yet another small cluster with AURKA, CDH1 and CCND1. Co-amplification of FGFR1 on 8p12 and CCND1 on 11q13 is one of the most common co-amplifications in breast cancer.34, 40
Association of Genomic Regions with Clinical–Pathological Parameters
Amplification of 8p and 11q are most often observed in ER positive tumors whereas amplification of 17q occurs in both ER-positive and ER-negative tumors.41, 42 In our study, EGFR (7p), CCNE1 (19q) and HER2 (17q) were associated with a negative ER status, whereas FGFR1 (8p), ADAM9 (8p), IKBKB (8p) and TOP2A (17q) were associated with a positive ER status of the tumor. Contrary to the study of Holst et al,17 we did not observe a significant association between ESR1 amplification and ER protein overexpression (73% of tumors with ESR1 amplification were ER-positive compared with 69% of tumors without ESR1 amplification).
HER2 and PRDM14 amplifications were associated with positive HER2 immunohistochemistry. ESR1, PRDM14, MYC and HER2 amplifications were associated with a higher MAI and PRDM14 and HER2 amplifications were also correlated with higher grade. For ESR1, we found a strong trend toward association with higher grade, which was in contrast to a previous study,17 but this study did not examine the association with MAI. For PRDM14, one study found no correlation between its expression levels and clinicopathological characteristics, which was assumed to reflect the small number of samples analyzed.43 MYC amplification was more likely to be present in tumors of the ductal subtype compared with lobular ones, and high-level MYC amplifications were significantly associated with a larger tumor size.
Co-amplified Regions
Patients with more amplified loci had a significantly higher grade and MAI. As not only the number of amplified loci but also the function of the genes involved determine tumor characteristics, we selected pairs of frequently co-amplified genes and studied their relation to clinicopathological features. HER2–MYC co-amplification, for example, was present in 15% of the tumors, which could indicate the existence of a selective advantage associated with their co-amplification. This hypothesis is supported by our findings showing that concomitant amplification of HER2 and MYC is associated with a significant larger tumor size and higher HER2 IHC status, and by other studies that found a relationship between HER2 and MYC co-amplification and reduced survival.33, 34 From preliminary analyses from the NSABP B-31 trial, it was suggested that tumors that are HER2–MYC co-amplified have a remarkably favorable prognosis with adjuvant trastuzumab treatment.44 Although not as frequent (7%) as HER2–MYC co-amplification, this study found that HER2–CCNE1 co-amplified tumors were significantly larger than tumors with either of these amplifications. CCNE1 protein overexpression has previously been associated with positive HER2 status and poor prognosis,45 but CCNE1 amplification on itself was shown to have no prognostic role in breast cancer so far.46 Tumors with EGFR–MYC co-amplification (7%) were larger and showed a trend toward higher MAI than tumors with either or neither of these amplifications. Several other frequent co-amplifications in this study (eg, 17% MYC–TOP2A, 15% MYC–CCND1, 12% TOP2A–EMSY and 9% HER2–CCND1) did not show any association with clinicopathological characteristics. These data imply that there is no relationship between the frequency of the co-amplification and the association with current prognostic markers and that the type of genes involved in the co-amplifications determines the association with prognostic factors.
Co-amplification of 8p12 (FGFR1) has been reported in 30–40% of tumors with CCND1 (11q13) amplification. In our study, 33% (9 out of 27) of CCND1 amplifications were concomitant with FGFR1 amplifications. Co-amplification of these genes is associated with significantly reduced survival,34 but in our study this co-amplification was not associated with any clinical–pathological characteristics.
In this study 50% of EMSY amplifications were also CCND1 amplified, which is less than the 70% described by another study.47 CCND1 and EMSY amplifications have both been associated with poor overall survival,47, 48 but there is no a straightforward association between CCND1 amplification and expression, and CCND1 expression has been associated with ER and good survival.49 The mechanism for the frequent co-amplification of genes spread over different chromosomes is yet unclear.
In conclusion, this study introduces a dedicated breast cancer MLPA kit that provides data on the copy number of 20 tumor suppressor and oncogenes in a single PCR reaction on paraffin-derived DNA. MLPA is an easy and high-throughput PCR-based technique that provided potentially important information on associations with essential clinicopathological features and on the frequency of co-amplifications of different genes in breast cancer. Such detailed knowledge of potential driver oncogenes and their gene–gene interactions may help to refine patient-tailored treatment of breast cancer patients in the future.
References
Owens MA, Horten BC, Da Silva MM . HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin Breast Cancer 2004;5:63–69.
Ross JS, Fletcher JA, Bloom KJ, et al. Targeted therapy in breast cancer: the HER-2/neu gene and protein. Mol Cell Proteomics 2004;3:379–398.
Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989;244:707–712.
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783–792.
Hudis CA . Trastuzumab, mechanism of action and use in clinical practice. N Engl J Med 2007;357:39–51.
Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987;235:177–182.
Sjogren S, Inganas M, Lindgren A, et al. Prognostic and predictive value of c-erbB-2 overexpression in primary breast cancer, alone and in combination with other prognostic markers. J Clin Oncol 1998;16:462–469.
Borg A, Baldetorp B, Ferno M, et al. ERBB2 amplification is associated with tamoxifen resistance in steroid-receptor positive breast cancer. Cancer Lett 1994;81:137–144.
Carlomagno C, Perrone F, Gallo C, et al. c-erb B2 overexpression decreases the benefit of adjuvant tamoxifen in early-stage breast cancer without axillary lymph node metastases. J Clin Oncol 1996;14:2702–2708.
Tetu B, Brisson J, Plante V, et al. p53 and c-erbB-2 as markers of resistance to adjuvant chemotherapy in breast cancer. Mod Pathol 1998;11:823–830.
Tanner M, Isola J, Wiklund T, et al. Topoisomerase IIalpha gene amplification predicts favorable treatment response to tailored and dose-escalated anthracycline-based adjuvant chemotherapy in HER-2/neu-amplified breast cancer: Scandinavian Breast Group Trial 9401. J Clin Oncol 2006;24:2428–2436.
Jarvinen TA, Holli K, Kuukasjarvi T, et al. Predictive value of topoisomerase IIalpha and other prognostic factors for epirubicin chemotherapy in advanced breast cancer. Br J Cancer 1998;77:2267–2273.
Jarvinen TA, Tanner M, Rantanen V, et al. Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am J Pathol 2000;156:839–847.
O’Malley FP, Chia S, Tu D, et al. Topoisomerase II alpha and responsiveness of breast cancer to adjuvant chemotherapy. J Natl Cancer Inst 2009;101:644–650.
Coon JS, Marcus E, Gupta-Burt S, et al. Amplification and overexpression of topoisomerase IIalpha predict response to anthracycline-based therapy in locally advanced breast cancer. Clin Cancer Res 2002;8:1061–1067.
Moelans CB, de Weger RA, van Blokland MTM, et al. Simultaneous detection of TOP2A and HER2 gene amplification by multiplex ligation dependent probe amplification in breast cancer. Mod Pathol 2009;23:62–70.
Holst F, Stahl PR, Ruiz C, et al. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet 2007;39:655–660.
Albertson DG . Conflicting evidence on the frequency of ESR1 amplification in breast cancer. Nat Genet 2008;40:821–822.
Lambros MB, Natrajan R, Reis-Filho JS . Chromogenic and fluorescent in situ hybridization in breast cancer. Hum Pathol 2007;38:1105–1122.
Elsheikh S, Green AR, Aleskandarany MA, et al. CCND1 amplification and cyclin D1 expression in breast cancer and their relation with proteomic subgroups and patient outcome. Breast Cancer Res Treat 2008;109:325–335.
Korsching E, Packeisen J, Helms MW, et al. Deciphering a subgroup of breast carcinomas with putative progression of grade during carcinogenesis revealed by comparative genomic hybridisation (CGH) and immunohistochemistry. Br J Cancer 2004;90:1422–1428.
Janssen EA, Baak JP, Guervos MA, et al. In lymph node-negative invasive breast carcinomas, specific chromosomal aberrations are strongly associated with high mitotic activity and predict outcome more accurately than grade, tumour diameter, and oestrogen receptor. J Pathol 2003;201:555–561.
Andre F, Job B, Dessen P, et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res 2009;15:441–451.
Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57.
Purnomosari D, Aryandono T, Setiaji K, et al. Comparison of multiplex ligation dependent probe amplification to immunohistochemistry for assessing HER-2/neu amplification in invasive breast cancer. Biotech Histochem 2006;81:79–85.
Moelans CB, de Weger RA, van Blokland MT, et al. HER-2/neu amplification testing in breast cancer by multiplex ligation-dependent probe amplification in comparison with immunohistochemistry and in situ hybridization. Cell Oncol 2009;31:1–10.
Moelans CB, de Weger RA, Ezendam C, et al. HER-2/neu amplification testing in breast cancer by multiplex ligation-dependent probe amplification: influence of manual- and laser microdissection. BMC Cancer 2009;9:4.
van Diest PJ, van der Wall E, Baak JP . Prognostic value of proliferation in invasive breast cancer: a review. J Clin Pathol 2004;57:675–681.
van Diest PJ . No consent should be needed for using leftover body material for scientific purposes. BMJ 2002;325:648–651.
van Diest PJ, Baak JP, Matze-Cok P, et al. Reproducibility of mitosis counting in 2469 breast cancer specimens: results from the Multicenter Morphometric Mammary Carcinoma Project. Hum Pathol 1992;23:603–607.
Bunyan DJ, Eccles DM, Sillibourne J, et al. Dosage analysis of cancer predisposition genes by multiplex ligation-dependent probe amplification. Br J Cancer 2004;91:1155–1159.
Coffa J, van de Wiel MA, Diosdado B, et al. MLPAnalyzer: data analysis tool for reliable automated normalization of MLPA fragment data. Cell Oncol 2008;30:323–335.
Al-Kuraya K, Schraml P, Torhorst J, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res 2004;64:8534–8540.
Cuny M, Kramar A, Courjal F, et al. Relating genotype and phenotype in breast cancer: an analysis of the prognostic significance of amplification at eight different genes or loci and of p53 mutations. Cancer Res 2000;60:1077–1083.
Letessier A, Sircoulomb F, Ginestier C, et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer 2006;6:245.
Blancato J, Singh B, Liu A, et al. Correlation of amplification and overexpression of the c-myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br J Cancer 2004;90:1612–1619.
Chin K, DeVries S, Fridlyand J, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006;10:529–541.
Cingoz S, Altungoz O, Canda T, et al. DNA copy number changes detected by comparative genomic hybridization and their association with clinicopathologic parameters in breast tumors. Cancer Genet Cytogenet 2003;145:108–114.
Kauraniemi P, Kallioniemi A . Activation of multiple cancer-associated genes at the ERBB2 amplicon in breast cancer. Endocr Relat Cancer 2006;13:39–49.
Kwek SS, Roy R, Zhou H, et al. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene 2009;28:1892–1903.
Loo LW, Grove DI, Williams EM, et al. Array comparative genomic hybridization analysis of genomic alterations in breast cancer subtypes. Cancer Res 2004;64:8541–8549.
Fridlyand J, Snijders AM, Ylstra B, et al. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer 2006;6:96.
Nishikawa N, Toyota M, Suzuki H, et al. Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res 2007;67:9649–9657.
Kim C, Bryant J, Horne Z, et al. Trastuzumab sensitivity of breast cancer with co-amplification of HER2 and cMYC suggests pro-apoptotic function of dysregulated cMYC in vivo [abstract]. Breast Cancer Res Treat 2005;94 (Suppl 1):S6. Abstract 46.
Potemski P, Kusinska R, Watala C, et al. Cyclin E expression in breast cancer correlates with negative steroid receptor status, HER2 expression, tumor grade and proliferation. J Exp Clin Cancer Res 2006;25:59–64.
Callagy G, Pharoah P, Chin SF, et al. Identification and validation of prognostic markers in breast cancer with the complementary use of array-CGH and tissue microarrays. J Pathol 2005;205:388–396.
Kirkegaard T, Nielsen KV, Jensen LB, et al. Genetic alterations of CCND1 and EMSY in breast cancers. Histopathology 2008;52:698–705.
Hughes-Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003;115:523–535.
van Diest PJ, Michalides RJ, Jannink L, et al. Cyclin D1 expression in invasive breast cancer. Correlations and prognostic value. Am J Pathol 1997;150:705–711.
Park K, Han S, Shin E, et al. EGFR gene and protein expression in breast cancers. Eur J Surg Oncol 2007;33:956–960.
Rodriguez-Pinilla SM, Jones RL, Lambros MB, et al. MYC amplification in breast cancer: a chromogenic in situ hybridisation study. J Clin Pathol 2007;60:1017–1023.
Cleton-Jansen AM . E-cadherin and loss of heterozygosity at chromosome 16 in breast carcinogenesis: different genetic pathways in ductal and lobular breast cancer? Breast Cancer Res 2002;4:5–8.
Camilleri-Broet S, Cremer I, Marmey B, et al. TRAF4 overexpression is a common characteristic of human carcinomas. Oncogene 2007;26:142–147.
Grunewald TG, Butt E . The LIM and SH3 domain protein family: structural proteins or signal transducers or both? Mol Cancer 2008;7:31.
Zhu Y, Qi C, Jain S, et al. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc Natl Acad Sci USA 1999;96:10848–10853.
Arriola E, Marchio C, Tan DS, et al. Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab Invest 2008;88:491–503.
Jensen LB, Bartlett JM, Witton CJ, et al. Frequent amplifications and deletions of G1/S-phase transition genes, CCND1 and MYC in early breast cancers: a potential role in G1/S escape. Cancer Biomark 2009;5:41–49.
Ginestier C, Cervera N, Finetti P, et al. Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res 2006;12:4533–4544.
Sen S, Zhou H, White RA . A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene 1997;14:2195–2200.
Elbauomy ES, Green AR, Lambros MB, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridisation analysis. Breast Cancer Res 2007;9:R23.
Chalmers IJ, Aubele M, Hartmann E, et al. Mapping the chromosome 16 cadherin gene cluster to a minimal deleted region in ductal breast cancer. Cancer Genet Cytogenet 2001;126:39–44.
Knoop AS, Knudsen H, Balslev E, et al. Retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil: Danish Breast Cancer Cooperative Group. J Clin Oncol 2005;23:7483–7490.
Di Leo A, Gancberg D, Larsimont D, et al. HER-2 amplification and topoisomerase IIalpha gene aberrations as predictive markers in node-positive breast cancer patients randomly treated either with an anthracycline-based therapy or with cyclophosphamide, methotrexate, and 5-fluorouracil. Clin Cancer Res 2002;8:1107–1116.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article
Cite this article
Moelans, C., de Weger, R., Monsuur, H. et al. Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol 23, 1029–1039 (2010). https://doi.org/10.1038/modpathol.2010.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.84
Keywords
This article is cited by
-
Impact of fibroblast growth factor receptor 1 (FGFR1) amplification on the prognosis of breast cancer patients
Breast Cancer Research and Treatment (2020)
-
PRDM14 promotes malignant phenotype and correlates with poor prognosis in colorectal cancer
Clinical and Translational Oncology (2020)
-
A phase Ib dose allocation study of oral administration of lucitanib given in combination with fulvestrant in patients with estrogen receptor-positive and FGFR1-amplified or non-amplified metastatic breast cancer
Cancer Chemotherapy and Pharmacology (2019)
-
The pleiotropic roles of ADAM9 in the biology of solid tumors
Cellular and Molecular Life Sciences (2018)
-
EMSY copy number variation in male breast cancers characterized for BRCA1 and BRCA2 mutations
Breast Cancer Research and Treatment (2016)
