
Abstract
Rosai–Dorfman disease and Langerhans cell histiocytosis are both disorders of accessory immune cells. Two cases have been previously reported of concurrent Langerhans cell histiocytosis and Rosai–Dorfman disease. In this report, we characterize the findings and selected molecular studies in nine additional cases. Histology was reviewed. Immunohistochemical stains were performed on all cases in which slides or blocks were available. A combination of CD1a, S-100, CD3, CD20, langerin, CD68, CD163, CD21, CD35 and CD123 immunohistochemical stains were performed. High-resolution array comparative genomic hybridization was performed on six samples from five cases. In these cases, seven were female and two male, with an average age of 25 years (15 months−59 years). A majority of the cases were identified in lymph node. Areas of Langerhans cell histiocytosis had a typical appearance with the existence of bland ‘coffee-bean’ nuclei, clear cytoplasm and associated eosinophils. The immunophenotype was typical, including expression of CD1a, S100, CD68 and langerin. In areas of Rosai–Dorfman disease, there was emperipolesis seen in all cases. Cells were intermediate-large in size with large round nuclei and ample clear or pale cytoplasm. The lesional cells were positive for S100, CD68, CD163, without expression of langerin or CD1a. Array comparative genomic hybridization showed gains and/or losses in four of the six samples. One case showed no gains or losses and one additional case showed gains and losses in the Langerhans cell histiocytosis, while no abnormalities were discovered in the Rosai–Dorfman disease component. These findings are comparable to those seen in previous studies of Langerhans cell histiocytosis. We report the clinical and pathologic findings of the combination of Langerhans cell histiocytosis and Rosai–Dorfman disease. Furthermore, we suggest on the basis of evidence from our cases that, when simultaneous, the two entities may be pathophysiologically related.
Similar content being viewed by others

Main
Rosai–Dorfman disease and Langerhans cell histiocytosis are both disorders of accessory immune cells. Langerhans cells are specialized dendritic cells that typically reside in the skin, which migrate to lymph nodes on activation to serve as specialized antigen-presenting cells. Langerhans cell histiocytosis is a clonal proliferation of abnormal, S100 and CD1a-positive Langerhans-like cells and may present as unifocal or multisystem disease affecting a variety of sites including bone, skin, neurohypophysis, oral cavity, anogenital region, lungs, liver, spleen and lymph node. The most common sites of nodal involvement are cervical, inguinal, supraclavicular and axillary.1 Limited disease usually has an excellent clinical outcome while systemic disease is typically associated with significant morbidity and mortality.2 Several findings support the notion that Langerhans cell histiocytosis is a clonal proliferation of phenotypically immature Langerhans cells including occurrence in homozygous twins in very rare cases. The broad histologic differential diagnosis for LCH includes Kimura disease, dermatopathic lymphadenitis, classical Hodgkin lymphoma and allergic granulomatosis.1
In contrast, Rosai–Dorfman disease is characterized by a non-neoplastic proliferation of S100-positive, CD1a-negative histiocytes with unusual features including emperipolesis of lymphocytes and other hematopoietic cells. Nodal involvement is most common, with 80% of cases showing cervical adenopathy and 43% of patients showing involvement in extranodal sites. Rosai–Dorfman disease is most often a self-limited disease, but recurrences over the course of years are not infrequent, and fatality can be seen in 5–11% of cases.3 Rosai–Dorfman disease has a broad histologic differential diagnosis including sarcoidosis, Hodgkin and non-Hodgkin lymphomas, Erdheim–Chester disease, foreign body giant cell reactions, aspiration pneumonia (in lung), Epstein–Barr virus-associated lymphoproliferative disorders, infectious mononucleosis, histiocytic sarcoma, storage diseases, metastatic melanoma, metastatic carcinoma, and fungal and mycobacterial infections.
In the English-language literature, two cases have previously been reported of concurrent Langerhans cell histiocytosis and Rosai–Dorfman disease.4, 5 The co-occurrence of these two rare histiocytic disorders sharing somewhat similar phenotypes raises questions of whether these two disorders are related and possibly represent a spectrum of disease. In this report, we characterize the pathologic and clinical findings, as well as selected molecular studies, in nine cases that show combinations of these two histiocytic disorders.
Materials and methods
Cases were obtained from several institutions and consultation files of the investigators. Case materials were obtained with attention to local guidelines for human research. Clinical information was reviewed and follow-up information obtained when possible. In all cases, histology and submitted immunohistochemical stains were reviewed by the individual contributor as well as a central review (DPO, PMB).
A panel of immunohistochemical stains was performed on all candidate cases in which slides or blocks were available. Depending on the material available, a combination of CD1a, S-100, CD3, CD20, langerin, CD68 (KP1), CD163, CD21, CD23, CD35, CD123, vimentin, in situ EBV (EBER) and pan keratin (AE1/AE3 or OSCAR) immunohistochemical stains were performed using standard methodologies.
High-resolution array comparative genomic hybridization was performed on six samples (cases 1, 2, 5, 6, and 7a and 7b). Although attempts were made to microdissect areas with Langerhans cell histiocytosis and compare with those with Rosai–Dorfman disease, these were unsuccessful. The array comparative genomic hybridization was performed at Combimatrix Molecular Diagnostics (Irvine, CA, USA) on DNA extracted from paraffin-embedded tissue samples using the HemeScan® bacterial artificial chromosome (BAC) array developed by Combimatrix Molecular Diagnostics. This array contains 1527 BAC clones corresponding to genomic loci encompassing the 22 autosomes and the sex chromosomes including loci targeted to genomic regions known to be clinically significant in hematological malignancies with an average resolution of ∼2.5 Mb. Differentially Cy3- and Cy5-labeled patient and normal sex-mismatched reference samples and dye swap duplicate reactions were co-hybridized to the microarrays, scanned with a Gen-Pix 4000B Microarray scanner and quantified using GenePix Pro microarray image analysis software (Molecular Devices, Sunnyvale, CA, USA). DNA copy number changes manifested as the simultaneous deviation of the ratio plots from the modal value of 1.0. Thresholds were set at forward/reverse reaction ratios <3.0 for mono-allelic deletions and ⩾3.0 for bi-allelic deletions.
Results
A total of 10 putative cases of combined Rosai–Dorfman disease and Langerhans cell histiocytosis were retrieved. After central review, one case was considered to represent Rosai–Dorfman disease with coincident dermatopathic lymphadenitis with increased Langerhans cells (case 10), lacking evidence of Langerhans cell histiocytosis and was excluded from further analysis. Case 7 presented initially as Langerhans cell histiocytosis, with recurrence 10 months later as Rosai–Dorfman disease, without any appreciable Langerhans cell histiocytosis present. Case 8 presented as a combination of Rosai–Dorfman disease/Langerhans cell histiocytosis, but a recurrence after 4 years showed only Rosai–Dorfman disease. Clinical findings are summarized in Table 1. Of the included patients, eight were female and two male (adult M:F 1:5; pediatric M:F 1:3), with an average age of 27.7 years (15 months–59 years). Five cases were seen in adults and five cases were in pediatric patients. Most cases (90%) were identified in lymph node. Including other cases reported in the literature, there appears to be a bimodal distribution of cases, with a mean age in pediatric patients of approximately 2 years (24 months), and in adults 41 years. No other significant differences were noted in the two age groups.4, 5
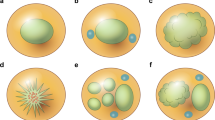
The median amount of Langerhans cell histiocytosis versus Rosai–Dorfman disease involvement was 7.5% (5–15%) versus 92.5% (80–95%), respectively. Most cases showed subtotal involvement of lymph nodes (Table 2), and necrosis was only seen in two cases. Emperipolesis was easily identified in all cases in the Rosai–Dorfman disease component (Figure 1). No significant staining was seen for CD21, CD35, pan keratin or in situ staining for Epstein–Barr virus (EBER) in any cases.

Low magnification image of case 6. Sinusoidal distribution of pale histiocytes is apparent at this magnification. These areas represent predominant Rosai–Dorfman histology, as was present in most cases. Small foci of more dense cellularity and normal residual nodal architecture are also present.
Morphology and Immunohistochemistry: Langerhans Cell Histiocytosis
As with typical cases, the usual lesional cells of Langerhans cell histiocytosis had bland, ovoid ‘coffee-bean’ nuclei, with notable nuclear grooves or folds. These cells had pink-granular cytoplasm and variable numbers of associated eosinophils. In contrast to areas of Rosai–Dorfman disease, the areas of Langerhans cell histiocytosis were relatively well-circumscribed and appeared more densely cellular (Figure 2). Similarly, while there were many infiltrating mononuclear cells in the areas of Rosai–Dorfman disease, in the Langerhans cell histiocytosis areas there were mostly Langerhans cells and eosinophils, without other cells types. The degree and type of tissue involvement is summarized in Table 2. The immunophenotype was as expected including expression of CD1a, S100, CD68 and langerin (Table 3). Case 7a showed some minimal staining for CD123 in Langerhans cell histiocytosis cells. Careful morphologic examination revealed that a subset of cases had rare cells (<5% of all lesional cells), which had morphologic features comparable to Rosai–Dorfman disease, with larger round nuclei and open chromatin and increased cytoplasm, but with CD1a and Langerin expression (Figure 3). These cells seemed to display transitional features between a lesional cell of Langerhans cell histiocytosis and Rosai–Dorfman disease. No convincing emperipolesis was identified in CD1a-positive cells. We propose that these transitional cells would be appropriately called the eponymous ‘Banks cells’ after our co-author who initially brought them to our attention.

Intermediate magnification from case 4. Focus of Langerhans cell histiocytosis with classic histologic appearance. These cells have classic nuclear folds and grooves with ample pale pink cytoplasm. This case was not rich in eosinophils in this region.

(a, b) Immunohistochemical staining for CD1a showing cells with overlapping features between Langerhans type histiocytes and Rosai–Dorfman type histiocytes. Note the rounded appearance, relatively open nuclear chromatin, and faint, but prominent nucleoli.
Morphology and Immunohistochemistry: Rosai–Dorfman Disease
In areas of Rosai–Dorfman disease, there was emperipolesis seen in all the cases. The Rosai–Dorfman disease cells were moderate to large in size (Figure 4). The nuclei of the lesional histiocytic cells were large and round with pale, marginated chromatin. There was ample clear or pale pink cytoplasm. In all cases, there were many accompanying mononuclear cells including variable numbers of small lymphocytes, neutrophils and plasma cells. Eosinophils were infrequent. The degree and type of tissue involvement is summarized in Table 2. As is typical of Rosai–Dorfman disease, the lesional cells were positive for S100, CD68, CD163, without expression of langerin or CD1a (Table 3) (Figure 5).

Intermediate magnification from case 4. Rosai–Dorfman histiocytes with rounded nuclei, delicate chromatin, occasional prominent nucleoli and voluminous cytoplasm. Some examples of emperipolesis are seen.

H&E (a), S-100 immunohistochemical staining (b) and CD1a immunohistochemical staining (c) from case 6. Note the intimate admixture of CD1a-positive cells (Langerhans cell histiocytosis) and the much larger areas of S-100 staining (both Rosai–Dorfman disease and Langerhans cell histiocytosis).
Array Comparative Genomic Hybridization
Array comparative genomic hybridization was performed on six samples, including both cases 7a and 7b. In four of the six samples, array comparative genomic hybridization showed gains and/or losses. One case showed no gains or losses. Case 7 showed gains and losses in the Langerhans cell histiocytosis, while no abnormalities were discovered in the Rosai–Dorfman disease component. Loss of 16p11 was seen in two of the cases. Two cases had single abnormalities (case 1: 16p11.2 loss; case 5: 9p13-q12 loss). Two cases had multiple abnormalities (case 2: 1p36.21 → 36.33 loss, 13q21.1 → q21.32 gain, 19p13.11 → p13.3 loss; case 7a: 5q13.2 loss, 16p11.2 loss, 19p13.3 loss, 19p12 loss). No correlation of array comparative genomic hybridization results and clinical, morphologic or immunohistochemical results was noted. These results are summarized in Table 4.
Discussion
We report the clinical and pathologic findings of the apparently rare combination of Langerhans cell histiocytosis and Rosai–Dorfman disease. Our cases illustrate that while rare, these processes do indeed coexist and that it is possible that their (co-)incidence is less rare than might be assumed.
As mentioned in the publication by Sachdev and Shyama,5 previous studies have speculated on comparable cytokine-mediated mechanisms in both Langerhans cell histiocytosis and Rosai–Dorfman disease. In this same study, the authors suggested that both manifestations could be due to divergent differentiation from a common precursor, or that the histiocytes have undergone a phenotypic switch. On the basis of this study, we would suggest that both processes occur. It is possible to conceive that the initial proliferation of reactive Rosai–Dorfman histiocytes may undergo clonal transformation in rare cases. In these cases, the neoplasm develops a phenotype of Langerhans cell histiocytosis.
Diagnostic difficulties are a paramount issue in the concurrence of these two disorders. In our cases, some were so subtle as to raise the question of the true concurrence of these two disorders. In some cases, there was also coincident finding of paracortical T-cell hyperplasia or dermatopathic lymphadenitis, which is both reactive conditions with a component of benign Langerhans cells. In these circumstances, the Langerhans cells are functioning to deliver antigen from a site of immunologic challenge, usually skin, and transporting these to a regional lymph node. As there is no single immunohistochemical study that can distinguish a ‘normal’ Langerhans cell from those of Langerhans cell histiocytosis, it could be argued that some of these cases represent non-clonal, ‘normal’ Langerhans cells, rather than Langerhans cell histiocytosis. Non-clonal Langerhans cells are dendriform with cytoplasmic projections seen on immunohistochemistry and electron microscopy, features that are lacking in Langerhans cell histiocytosis cells; such dendriform morphology was absent in the Langerhans cell histiocytosis areas of our cases. In our series, the diagnosis of the areas of Rosai–Dorfman disease and Langerhans cell histiocytosis were made on populations of cells and background rather than individual cell morphology, in which there is some overlap. However, immunohistochemical evaluation showed both the individual components as well as cells with intermediate features between the two entities.
The findings in our study support those of Chikwava and Jaffe6 and Lau et al.7 In these papers, langerin staining was noted to be highly specific and sensitive for Langerhans cells with high concordance noted between CD1a and langerin staining. Our study showed comparable findings, and showed that both langerin and CD1a were useful for highlighting the presence of Langerhans cells against a background of histiocytes of Rosai–Dorfman disease.
The presence of clonal abnormalities by array comparative genomic hybridization in our cases raises important questions. Molecular genetic abnormalities have not been identified by array comparative genomic hybridization in Rosai–Dorfman disease cases to our knowledge. Arguably, it is current dogma that Rosai–Dorfman disease lacks clonal abnormalities. Although the investigators feel that this is likely true, our current results raise the possibility that a subset of Rosai–Dorfman cases could be clonal. In this case, it would change the specific interpretation of our results, but would also change some views in regards to underlying features of Rosai–Dorfman disease. It may be appropriate in future studies, to apply techniques such as array comparative genomic hybridization to large numbers of cases of Rosai–Dorfman disease, Langerhans cell histiocytosis and other histiocytic proliferations to gain more insight into their clonal versus non-clonal nature.
In contrast, array comparative genomic hybridization studies, as well as studies with comparable levels of resolution for evaluating genetic gain and loss (loss of heterozygosity testing) have shown recurrent abnormalities in several genetic loci in cases of Langerhans cell histiocytosis.8, 9 Three of four cases in this with combined Rosai–Dorfman disease/Langerhans cell histiocytosis did have measurable genetic gains and losses by array comparative genomic hybridization (Table 4). Also, in case 7, the original presentation of Langerhans cell histiocytosis showed multiple abnormalities, while a later recurrence containing only Rosai–Dorfman disease showed no abnormalities. This intriguing finding suggests that if there is a spectrum of these disorders that Langerhans cell histiocytosis may be due to an acquisition of clonal abnormalities, while these are not present in the polyclonal progenitor lesion, Rosai–Dorfman disease. Of greater importance is that in our cases with large amounts of Rosai–Dorfman disease cells present, clonal abnormalities were found and that these overlap many of those previously ascribed to Langerhans cell histiocytosis in the studies of Dacic et al9 and Murakami et al.8 Losses of 1p, 5q, 9p and 16p were described in these studies, which were also seen in the current series. Although this is not definitive evidence of a clonal relationship between Langerhans cell histiocytosis and Rosai-Dorfman disease, it does raise the possibility that this may be true in rare cases.
The question as to the exact interrelationship of these two entities remains unanswered by this study. It is conceivable that future studies could look at more subtle genetic relatedness of the two coincident lesions. At present, we are left only to speculate. It is the impression of the investigators that these two lesions are more than coincidentally related. On the basis of the review of our cases, overlap in abnormal immunohistochemical findings in both components and the identification of cells with overlapping microscopic features (sharing those of both Rosai–Dorfman histiocytes and Langerhans cells) both suggest that the two entities are related and their co-occurrence may be under-recognized.
References
Edelweiss M, Medeiros LJ, Suster S, et al. Lymph node involvement by Langerhans cell histiocytosis: a clinicopathologic and immunohistochemical study of 20 cases. Hum Pathol 2007;38:1463–1469.
Jaffe R, Weiss LM, Facchetti F . Tumours derived from Langerhans cells In: Swerdlow SH, Campo E, Harris NL, et al (eds). WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues 4th edn. IARC: Lyon, France, 2008.
Weitzman S, Jaffe R . Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45:256–264.
Wang KH, Cheng CJ, Hu CH, et al. Coexistence of localized Langerhans cell histiocytosis and cutaneous Rosai-Dorfman disease. Br J Dermatol 2002;147:770–774.
Sachdev R, Shyama J . Co-existent Langerhans cell histiocytosis and Rosai-Dorfman disease: a diagnostic rarity. Cytopathology 2008;19:55–58.
Chikwava K, Jaffe R . Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disorders. Pediatr Dev Pathol 2004;7:607–614.
Lau SK, Chu PG, Weiss LM . Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol 2008;32:615–619.
Murakami I, Gogusev J, Fournet JC, et al. Detection of molecular cytogenetic aberrations in Langerhans cell histiocytosis of bone. Hum Pathol 2002;33:555–560.
Dacic S, Trusky C, Bakker A, et al. Genotypic analysis of pulmonary Langerhans cell histiocytosis. Hum Pathol 2003;34:1345–1349.
Acknowledgements
We thank Lawrence Weiss at City of Hope National Medical Center, Duarte, CA, USA, for performing the langerin staining. This material was presented in abstract form at the 2010 United States and Canadian Academy of Pathology Annual Meeting, Washington, DC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
O'Malley, D., Duong, A., Barry, T. et al. Co-occurrence of Langerhans cell histiocytosis and Rosai–Dorfman disease: possible relationship of two histiocytic disorders in rare cases. Mod Pathol 23, 1616–1623 (2010). https://doi.org/10.1038/modpathol.2010.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.157
Keywords
This article is cited by
-
Rosai–Dorfman Disease: A Rare Clinicopathological Presentation and Review of the Literature
Head and Neck Pathology (2021)
-
Rosai-Dorfman disease with renal involvement and associated autoimmune haemolytic anaemia in a 12-year-old girl: A case report
BMC Pediatrics (2020)
-
Systemic Histiocytosis (Langerhans Cell Histiocytosis, Erdheim–Chester Disease, Destombes–Rosai–Dorfman Disease): from Oncogenic Mutations to Inflammatory Disorders
Current Oncology Reports (2019)
-
Expression of the transcription factor ZBTB46 distinguishes human histiocytic disorders of classical dendritic cell origin
Modern Pathology (2018)
-
Bone pathologic fracture revealing an unusual association: coexistence of Langerhans cell histiocytosis with Rosai-Dorfman disease
BMC Clinical Pathology (2017)
