
Abstract
It remains obscure in myxofibrosarcoma about the basis of tumorigenesis, progression, and metastasis. Chromosome 7 gains are common in some sarcomas, including myxofibrosarcoma, whereas the specific oncogenes are yet to be characterized. We performed an integrative study of MET gene at 7q31.2 to elucidate its implication in myxofibrosarcoma. Focused on candidate oncogenes on chromosome 7, 385K array comparative genomic hybridization was used to profile DNA copy number alterations of 12 samples. MET transcript was successfully quantified by real-time RT–PCR for 16 laser-microdissected tumors and two myxofibrosarcoma cell lines (NMFH-1, OH931). MET immunoexpression was assessable in 86 primary localized tumors with follow-up. To analyze endogenous MET expression and activation, NMFH-1 and OH931 cells, both with wild-type MET gene, were subjected to Western blotting and hepatocyte growth factor-treated NMFH-1 cells were evaluated for the kinetics of MET tyrosine phosphorylation. Non-random large-scale gains on 7q were detected in five cases, delineating three recurrent amplicons, 7q21.11–7q21.3, 7q22.1–22.3, and 7q31.1–7q32.3, in which the locus of MET displayed increased copy number, among others. MET mRNA was upregulated in OH931, NMFH-1, and nine tumors (56%), whereas neither gene dosage nor mRNA expression of MET was associated with clinicopathological factors. In contrast, MET protein overexpression, present in 67% of cases, was highly related to deep location (P=0.004), higher grades (P=0.001), and more advanced stages (P<0.001). Importantly, MET overexpression independently portended inferior metastasis-free survival (P=0.004) and overall survival (P=0.0221). Expressing activating phospho-MET at Tyr1234/Tyr1235, OH931 cells had more abundant total MET than NMFH-1 cells, whereas the latter became promptly phosphorylated on stimulation of hepatocyte growth factor. In primary myxofibrosarcomas, MET overexpression, as a frequent event, is likely driven by 7q gains with mRNA upregulation, associated with important prognosticators, and independently predictive of worse outcomes, highlighting its possible causative function in tumor aggressiveness and potentiality as a therapeutic target.
Similar content being viewed by others

Main
Myxofibrosarcoma is a common sarcoma characterized histologically by multinodular growth of spindle to polygonal sarcoma cells within variably myxoid stroma containing long curvilinear vessels.1 Clinically, increased tumor grading and staging are frequently seen in myxofibrosarcomas after relentless local recurrences, which may eventually lead to metastatic diseases.1, 2, 3 Especially for those at the low-grade end, it seems challenging to predict acquisition of invasive and metastatic properties for individual cases by histological evaluation.1, 2, 3 Furthermore, metastatic myxofibrosarcomas are often refractory to current treatment strategies and constitute the primary cause of sarcoma-related death.1, 3, 4 To develop novel therapeutic interventions, it is highly desirable to elucidate the molecular determinants correlated with tumor invasion and metastatic spread in myxofibrosarcoma progression.
Somatic alterations in cellular DNA underlie most human cancers. Gains of gene copy number drive expression of oncogenes, whereas decreased gene dosage by hemizygous and/or homozygous deletion results in inactivation of tumor suppressor genes.5 The prospect of genomic profiling with derived targeted therapies is now prompting increasing efforts to characterize cancer genomes.5 There is mounting evidence that regional gains and/or high-level amplifications on chromosomal arm 7q are recurrently found in various types of bone and soft tissue sarcomas,6, 7, 8, 9, 10, 11, 12 including myxofibrosarcomas.8 However, as for myxofibrosarcomas, the pathogenetically relevant target oncogenes on 7q and their prognostic implications remain largely unknown.
Compared with conventional comparative genomic hybridization (CGH), array CGH (aCGH) is promising to narrow down imbalanced chromosomal regions.5, 8, 13, 14 In this series, we applied ultrahigh-resolution tiling-path aCGH to pinpoint breakpoints of DNA copy number alterations (CNAs) in chromosome 7 for 12 myxofibrosarcomas, so as to identify candidate oncogenes implicating its tumorigenesis. Special attention was given to chromosomal segments harboring the established oncogenes, such as hepatocyte growth factor (HGF) at 7q21 and MET at 7q31. Acting as a multi-functional cytokine on cells, HGF is typically secreted by mesenchymal cells as a single-chain propeptide that requires proteolytic cleavage to generate an active α/β heterodimer.15, 16, 17 MET encodes the 170-kd precursor of a transmembrane receptor tyrosine kinase, which constitutes the only known high-affinity receptor of HGF after cleavage into the 50-kd α subunit and 140-kd β subunit and linkage by disulfide bonds.15, 16, 17
In human cancers, aberrant c-Met signaling has been shown to result from diverse HGF ligand-dependent and -independent mechanisms, including activating mutation, autocrine/paracrine HGF stimulation, and overexpression with or without gene amplification.15, 16, 17, 18, 19 A subset of common carcinomas is known to harbor amplified MET gene (eg gastric cancers, 10–20%; lung cancers, 7%; metastatic colorectal cancers, 9–18%),15, 17, 20 which is therapeutically relevant in prediction of response to selective MET tyrosine kinase inhibitors.20 Despite more frequent MET overexpression in higher-grade lesions, MET gene amplification was not found by Southern blots in 35 various soft tissue tumors tested,21 unlike that observed in 7,12-dimethylbenz(a)anthracene-induced rat sarcomas.21, 22 Given its crucial function in tumor biological function, MET has emerged as a promising target for cancer therapy.15, 17, 23 In this context, we conducted by far the first systematic study to elucidate the underlying molecular aberrations and mRNA abundance of MET in selected fresh tumor samples and its protein expression and prognostic significance in 86 archival primary localized myxofibrosarcomas. Furthermore, the endogenous expression level and activation status of MET were examined by Western blotting for two myxofibrosarcoma cell lines (OH931 and NMFH-1), with the lower MET-expressing NMFH-1 cells also evaluated for the kinetic alteration of MET phosphorylating activation in response to exogenous HGF treatment.
Materials and methods
Cell Culture
The OH931 and NMFH-1 myxofibrosarcoma cell lines were established and kindly provided by Dr Bridge24 and Dr Ogose,25 respectively. Both sarcoma cell lines were grown in RPMI 1640 medium supplemented with fetal bovine serum (20% for OH931; 10% for NMFH-1), 1% penicillin/streptomycin, and 1% L-glutamine. In addition, 1% HEPES was also added to the RPMI1640 medium to culture OH931 cells. The CCD966SK dermal fibroblast cell line was purchased from the repository of Taiwan BCRC and cultured in minimum essential medium (Eagle) in Earle's BSS containing 10% of heat-inactivated fetal bovine serum, 1 mM sodium pyruvate, and 1% of non-essential amino acids.
Sequencing of MET Gene
Both OH931 and NMFH-1 myxofibrosarcoma cells were extracted for genomic DNA to perform extensive sequencing of MET gene, covering the extracellular semaphorin domain, IPT repeats, juxtamembrane domain, and tyrosine kinase domain, based on the earlier reported mutation frequency and methodologies with minor modifications.16 The sequences of primer pairs will be provided on request.
Patients and Tumor Materials
The criteria of histopathological diagnosis and assessment for various parameters were elaborated in our earlier publications.3, 4, 26 Histological grading and staging were classified according to updated French Federation of Cancer Centers (FNCLCC) scheme27 and the 6th edition of American Joint Committee on Cancer (AJCC) system, respectively. Retrospective clinical data collection and tissue procurement were in accordance with the local guidelines of research ethics (97-1110A3). For genomic profiling, aCGH was used to evaluate somatic CNAs in 12 fresh tumor specimens (six primary, six recurrent). To evaluate the reliability of CNAs obtained from aCGH profiling, 17 selected fresh specimens of myxofibrosarcomas were subjected to laser capture microdissection (LCM) for quantification of MET mRNA expression by real-time reverse-transcription PCR (RT–PCR). To perform MET immunostain, we retrieved representative paraffin-embedded blocks of primary localized myxofibrosarcomas of 86 patients from the archives of three tertiary medical centers. In the absence of preoperative neoadjuvant radiation or chemotherapy, these 86 patients, all with available clinical follow-up, received surgical excision with curative intent between 1986 and 2005. Postoperative adjuvant radiotherapy (n=21) and/or chemotherapy (n=7) were randomly given in a minor subset of high-grade or recurrent cases without consistent guidelines. Details on clinicopathological characteristics of cohorts used in aCGH analysis and immunoexpression were summarized in Tables 1 and 2.
DNA Preparation, Hybridization, and Data Analysis of aCGH
On the basis of H&E sections, regions suitable for DNA extraction were dissected from 12 fresh specimens after trimming necrotic and fibrous areas to assure higher than 80% of tumor content. The reference DNAs were obtained from adjacent normal tissues available in five cases and from normal lymphocytes of gender-matched donors in the remaining seven cases. For each case, 1 μg of total genomic DNA at a concentration of 500 ng/μl was extracted from fresh tissue by using ‘recoverall DNA Purification kits’ (Ambion, Austin, TX, USA) for hybridization against oligonucleotide microarrays. To ensure DNA integrity, each sample should have an A260/280 ratio of 1.7–2.0 and no sign of degradation, as evaluated by gel electrophoresis. Each tiling-path whole genomic microarray had 385K oligonucleotide probes with a median spacing of 6 kb and variable length to achieve a melting temperature of 76°C (NimbleGen Systems, Madison, WI, USA). The procedures of DNA labeling, hybridization, normalization of oligonucleotide arrays, window averaging of contained probes, and data acquisition were carried out by the facility of manufacturer as earlier reported.13 Briefly, genomic DNAs of tumor and reference samples were sonicated to a size range of 500–2000 bp before labeling by random priming with fluorescent Cy3 and Cy5, respectively. After combining the data of signal intensity with information of the genomic coordinates, the Cy3 and Cy5 signal intensities are normalized to one another using Qspline normalization, a robust non-linear normalized method for two-color experiments. The circular binary segmentation algorithm proposed by Olshen et al,28 was used for segmentation of the averaged log2 ratio data. Each segment was then assigned a log2 ratio that was the median of 10 contained probes, and the data were centered by the tallest mode in the distribution of the segmented values. To finely delineate the breakpoints of whole array probes, we defined gains and losses as log2 ratios of ≥+0.20 or ≤−0.20, respectively.
Real-Time RT–PCR to Measure MET mRNA Expression Level in Fresh Samples and Cell Lines of Myxofibrosarcoma
To circumvent the contamination of the surrounding non-neoplastic cells, we adopted LCM technology to isolate pure tumor cells. Three 7-μm sections each were serially cut from 17 snap-frozen tumor tissues, placed onto a PEN-membrane slide, and stained with HistoGene LCM Staining kit to isolate cells of interest by using the Veritas automated LCM system (Arcturus Engineering, Mountain view, CA, USA). Approximately 1500 cells were collected onto the Capsure Macro cap, extracted by Picopure RNA isolation kit at 42°C for 30 min with 50 μl of extraction buffer, and then eluted by purification column (Arcturus Bioscience, Mountain view, CA, USA). The adjacent normal fibromuscular tissues from three snap-frozen specimens were also microdissected in parallel and extracted for RNA to serve as calibrator controls. The amount and concentration of RNA obtained were measured by a nanodrop spectrophotometer (SSP-3000, infinigen, City of Industry, CA, USA). Following the manufacturer's instructions, total RNAs were extracted with RNeasy Mini kit (Qiagen, Valencia, CA, USA) from OH931, NMFH-1, and CCD966SK cells. By using ImProm-II Reverse transcription system (Promega, Madison, WC, USA), total RNA was reverse transcribed in a final volume of 40 μl with the following conditions: 2 μl of total RNA from LCM-isolated cells of each tissue sample (or 2 μg of total RNA from each cell line), 0.5 mmol/l dNTPs, 25 U of RNase inhibitor, 16 μl of RNA eluant, and 4 μl of random primers. The reactions were performed at 42°C for 60 min, followed by inactivation of the enzyme at 70°C for 15 min. Real-time PCR assays to quantify the expression levels of MET transcript were performed using the LightCycler instrument 2.0 (Roche molecular diagnostics, Mannheim, Germany). The intron-spanning primers and LON probes for cDNAs of target and housekeeping (POLR2A, a.k.a. RPII) genes were ordered from Universal Probelibrary and their corresponding nucleotide sequences were MET: sense, agtgggaattctagacacatttca, UPL probe no. 31 (tggtggaa), and antisense, cattcaagaatactgtttgacacactt; POLR2A: sense, gcatcatgaacagcgatgag, UPL probe no. 69 (ggaggaag), and antisense, tcatccatcttgtccaccac. Amplification was conducted in duplicate for each reaction for fresh specimens and in triplicate for cell lines with LightCycler TaqMan MasterMix, using 2 μl of cDNAs (or 1000-fold diluted for cell line samples), 100 nmol/l of the probes, and 200 nmol/l of the primers in a final 20 μl of reaction mixture. After 2 min incubation at 40°C to allow for uracil N-glycosylase cleavage, Taq DNA polymerase was activated by incubation for 10 min at 95°C. Each reaction of the 45 PCR cycles consisted of 10 s of denaturation at 95°C and hybridization of the probe and primers for 30 s at 60°C. After normalization to POLR2A, the relative expression fold of MET transcript was then given by 2−ΔΔCp, where ΔΔCT=ΔCT (Sarcoma cells)−ΔCT (CCD966SK) or =ΔCT (tumor specimens)−ΔCT (normal fibromuscular tissue), and ΔCT represented the CT of MET subtracted from the CT of POLR2A. Only samples with CT value <33 for POLR2A were considered to have acceptable RNA quality and included in the analyses.
Immunohistochemistry
For each of the 86 cases tested, MET immunohistochemistry was performed on one 4-μm-thick, formalin-fixed tissue section from a representative block. The tissue sections were then deparaffinized in xylene, rehydrated in graded alcohols, quenched with 3% H2O2, microwave heated for antigen retrieval in 0.01 M citrate buffer at pH 6.0 for 15 min, and incubated in 10% normal goat serum for 30 min to block non-specific immunoreactivity. Afterwards, the tissue sections were incubated at 4°C overnight with a primary antibody against total MET (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA), washed with PBS, and detected by using the streptavidin-biotin immunoperoxidase method. Diaminobenzidine was used as the chromogen and Mayer's hematoxylin was as the counterstain. One of the authors (JCL), blind to clinicopathological data, independently evaluated the slides. The labeling intensity of MET was classified as negative, weak, moderate, and strong. MET reactivity was predominantly seen in the cytoplasm, with only few cases also exhibiting distinct membranous staining. Therefore, the percentage of tumor cells with moderate or strong cytoplasmic immunoreactivity, with or without membranous staining, was recorded as expression index in 5% increment. After testing a series of cutoff values, MET protein was construed as overexpressed when the expression index was ≥30% of tumor cells.
Western Blotting Assays to Assess the Endogenous Protein Expression and Activation of MET in Myxofibrosarcoma Cell Lines
Equal amounts of total protein (25 μg) extracted from OH931, NMFH-1, and CCD966SK cell lines were separated on 4–12% gradient SDS–PAGE gel NuPAGE (Invitrogen, Carlsbad, CA, USA), transferred to PVDF membranes (Amersham Biosciences, Buckinghamshire, UK), and then blocked with 5% skimmed milk in TBST buffer at room temperature for 1 h. Afterwards, the membranes were probed with antibodies at 4°C overnight against total MET (1:200, 3D4; Zymed, South San Francisco, CA, USA), phosphorylated MET at the tyrosine residues 1234/1235 (Tyr1234/Tyr1235, 1:200, D26; Cell Signaling Technology, Beverly, MA, USA), and GAPDH (1:10000, MAB347, Chemicon) for a loading control and then incubated with the secondary antibody at room temperature for 1.5 h. Enhanced chemiluminescence reagents (Amersham Biosciences) were used to visualize the targeted proteins, which were then semi-quantitatively measured by densitometry. The endogenous expression level and activation status of MET in OH931 and NMFH-1 cells were compared with those of CCD966SK fibroblasts.
Evaluation of the Kinetic Alteration of Tyrosine Phosphorylation in Response to Exogenous HGF Stimulation in NMFH-1 Cells
To evaluate the tyrosine phosphorylation of MET, NMFH-1 cells grown in 6-cm dishes were serum starved overnight, cultured with fresh minimal essential medium containing non-essential amino acids, and treated with either 50 ng/ml recombinant human HGF (H9661, Sigma, St Louis, MO, USA) or serum-free medium alone for indicated time intervals. For Western blotting assay, total protein (40 μg each) was extracted from NMFH-1 cells harvested from various batches with or without HGF stimulation at different time intervals, with β-actin (1:3000, MAB1501, Chemicon) serving as the loading control.
Follow-Up and Statistical Analyses
Statistical analyses were performed using SPSS 14 software package. Associations and comparisons of MET mRNA expression or immunoexpression with various clinicopathological parameters were evaluated by Student's t-test, one-way ANOVA, Kruskal–Wallis, or Pearson's correlation coefficient test as appropriate. In the 86 patients tested for MET immunoexpression, the observation intervals of follow-up ranged from 2 to 229 months with the median duration of 44.8 months as of January 2008, forming the basis for prognostic analysis. The end points evaluated for survival analyses were overall survival (OS) and metastasis-free survival (MeFS). The cutoffs of various clinicopathological parameters for prognostication were described earlier.3, 4, 26 In univariate survival analysis, Kaplan–Meier curves were plotted for each variable and the difference between groups compared by the log-rank test. In Cox multivariate regression model, all significant parameters identified at the univariate level were entered to compare the independent prognostic impact. Student's t-test was used to analyze the results of real-time quantitative RT–PCR for cell line samples. For all analyses, two-sided tests of significance were used with P<0.05 considered significant.
Results
Genomic Profiling of Chromosome 7 by Ultrahigh-Resolution aCGH
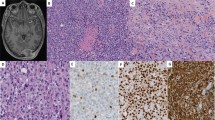
Chromosomal imbalances of varying size were detected in all 12 samples subjected to aCGH profiling. Examples of aCGH profiles are shown in Figure 1. Although large-scale chromosomal gains were only seen in two (MFS64, MFS96b) cases on 7p and five cases on 7q (MFS19b, MFS64, MFS84b, MFS96b, MFS99b), non-contiguous segmental copy number gains interspersed by small interstitial regions of no alteration were frequently detected on at least one of both arms. The later finding might be attributed to the higher resolution of current aCGH platform to localize exact breakpoints. Besides, there was no apparent correlation of CNAs of chromosome 7 with the tumor grading, staging, or primary vs recurrent status of myxofibrosarcoma. To unravel causal genes showing copy number-driven deregulated expression, we filtered the imbalanced chromosomal regions with non-random alterations at the identical DNA segments in at least 40% of samples. Accordingly, recurrent regions of gain and/or amplification were mapped to 7p14.3 and 7p14.1 on the p arm and 7q21.11–7q21.3, 7q22.1–22.3, and 7q31.1–7q32.3 on the q arm. Among the three amplicons on the q arm, we found that nine, three, and six loci of DNA gains in the chromosomal regions 7q21.11–7q21.3, 7q22.1–22.3, and 7q31.1–7q32.3, respectively, are known to harbor reported putative or established oncogenes implicating tumorigenesis. These included RPIP9 (7q21.12, a.k.a, RUNDC3B),29 PFTK1 (7q21.13),14 CDK6 (7q21.2),30 SRPK2 (7q22.3),31 PBEF1 (7q22.3),32 NRCAM (7q31.1),33 MET (7q31.2),15, 20, 23 PTPRZ1 (7q31.32),34 POT1 (7q31.33),35 and SND1 (7q32.1).36 However, complete genomic DNA gains at the probes spanning HGF were only observed in three cases (Figure 1). On the other hand, DNA losses tended to be small focal changes and recurrently mapped to 7p22, 7p12.3, and 7q11.23 in which three earlier reported candidate tumor suppressor genes, that is MAD1L1 (7p22.3),37 TNS3 (7p12.3),38 and CLDN4 (7q11.23),39 were located.

Array comparative genomic hybridization (aCGH) profiling of myxofibrosarcoma showed a colorimetric gene copy number matrix of chromosome 7. Intensities of red and green coloration generated by Java TreeView indicate an increased or decreased signal ratio for each averaged window of 10 probes, respectively. Each row corresponds to an individual average window and each column represents the aCGH profile of chromosome 7 in a tumor sample. Cytoband pattern of chromosome 7 is shown to the left. Recurrent regions of alterations are denoted by vertical lines in the middle, with gains and deletions of candidate oncogenes and tumor suppressor genes indicated in green and red brackets, respectively. The close-up views of loci harboring genes of most interest are shown to the right. (b) Horizontal karyograms of chromosomal region mapped to candidate oncogenes, HGF, CDK6, and MET, in three representative myxofibrosarcoma samples. MFS86 (upper panel) showed no apparent CNAs across these three genes. High-level DNA gains were seen in MFS96b (middle panel) at the loci harboring CDK6 and MET, but not so for HGF. In addition to CDK6 and MET genes, MFS99b (lower panel) showed slight DNA gains at the locus of HGF.
MET Transcript Was Frequently Upregulated in Myxofibrosarcoma Specimens but not Associated with Clinicopathological Factors
Next, we addressed whether the mRNA expression of MET gene was upregulated in at least a subset of myxofibrosarcoma specimens as inferred from the aCGH findings and whether there was association between the expression level of MET transcript and clinicopathological factors. Real-time RT–PCR quantification could be successfully determined with sufficient RNA yields in LCM-isolated pure tumor cells (Figure 2a) of 16 fresh myxofibrosarcoma samples, including four grade 1, seven grade 2, and five grade 3 cases. As compared with the normal adjacent fibromuscular tissues, we found that 9 out of 16 cases tested showed more than threefold higher expression of MET transcripts, suggesting their frequent upregulation in human myxofibrosarcomas. However, there was no significant difference among myxofibrosarcoma specimens regarding various clinicopathological characters, including grading (Figure 2b), staging, and primary vs recurrent status, which was generally in keeping with the aCGH findings.

Pure myxofibrosarcoma cells were precisely isolated by LCM technology (a) without contamination of surrounding vasculature or inflammatory cells for downstream quantitative real-time RT–PCR assay (b) that showed frequent overexpression of MET mRNA, but no apparent difference among cases of various grades (P=0.115 by Kruskal–Wallis test).
Expression and Activation of Endogenous MET Kinase in Myxofibrosarcoma Cell Lines
Given activating MET mutations identified in few cancer types,15, 16, 17, 23 we extensively sequenced the genomic DNAs of OH931 and NMFH-1 myxofibrosarcoma cell lines and found wild-type MET gene in both (data not shown). Real-time RT–PCR quantification in triplicate measurements (Figure 3a) showed that the expression levels of MET mRNA were significantly higher in two myxofibrosarcoma cell lines than in the non-transformed CCD966SK fibroblasts (P<0.001). In addition, OH931 myxofibrosarcoma cells also had significantly more abundant MET mRNA when compared with NMFH-1 (P<0.001). It has become explicit that the Tyr1234/Tyr1235 residues of MET kinase must be first activated by phosphorylation to relay MET-mediated signaling, a prerequisite critical for tumor growth and progression.15, 19, 23, 40 We, therefore, further compared the endogenous expression and activation status of MET protein in myxofibrosarcoma cells vs CCD966SK fibroblasts. By Western blotting, OH931 myxofibrosarcoma cells showed high endogenous expression of total MET, whereas it was comparatively lower in abundance in NMFH-1 cells and barely detectable in CCD966SK fibroblasts (Figure 3b, row 2). However, endogenous phosphorylation of MET at Tyr1234/Tyr1235 was only detected in OH931 myxofibrosarcoma cells (Figure 3b, row 1).

(a) Real-time RT–PCR to quantify the MET mRNA expression level: **P<0.001 for comparison of OH931 or NMFH-1 myxofibrosarcoma cell line with CCD966SK fibroblasts; ##P<0.001 for comparison between OH931 and NMFH-1 cells. (b) Western blotting assays: when compared with CCD966SK fibroblasts, MET protein overexpression was apparent with activating phosphorylation in OH931 myxofibrosarcoma cells as seen by probing with phospho-METTyr1234/1235, whereas there was only modest expression of total MET protein in NMFH-1 cells without phosphorylation. (c) Kinetics of tyrosine phosphorylation of MET in NMFH-1 myxofibrosarcoma cells in response to exogenous HGF treatment. Cells grown in 6-cm dishes were serum starved overnight, left unstimulated or stimulated with 50 ng/ml HGF for indicated time periods, and then probed with indicated antibodies.
Tyrosine Phosphorylation of MET in Response to Recombinant HGF Stimulation in NMFH-1 Myxofibrosarcoma Cells
As NMFH-1 cells showed relatively lower endogenous expression of MET protein without apparent phosphorylating activation, we further examined its kinetics of tyrosine phosphorylation of MET in response to exogenous HGF ligand. As shown in Figure 3c, phosphorylation of Tyr1234/Tyr1235 was observed at 5 min after HGF treatment and lasted until 10 min. Afterwards, expression of phospho-METTyr1234/Tyr1235 started to diminish. The total amount of MET protein was decreased at 60 min after HGF treatment, probably because of the degradation by proteasome after internalization.
Correlations of MET Immunoexpression with Clinicopathological Factors in Primary Myxofibrosarcomas
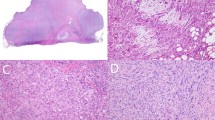
MET protein in primary myxofibrosarcomas was successfully scored for 86 cases (Table 2), including 47 males and 39 females with a median age of 64 years (range, 16–84). On the basis of FNCLCC grading scheme, 39 cases were classified as grade 1 (Figure 4a), 34 as grade 2 (Figure 4b), and 13 as grade 3 (Figure 4c). Fifteen (18%), 34 (41%), and 34 (41%) cases were AJCC stage I, stage II, and stage III, respectively. However, the tumor staging of three remote cases could not be determined due to the lack of data of tumor size. As shown in Table 2 and Figures 4d–f, MET was overexpressed in 58 cases (67%) with an expression index ≥30%, whereas MET expression in the remaining 28 cases was either absent (n=10) or low (n=18, expression index ≥5% but <30%). MET immunolabeling was significantly higher in expression index in those myxofibrosarcomas that were deep seated (P=0.004), classified as FNCLCC grade 2 or grade 3 (P=0.001), and more advanced in AJCC staging (P<0.001). In addition, MET expression index was positively related to increased tumor size (P=0.003), mitotic rate (P=0.008), and percentage of tumor necrotic area (P<0.001), but inversely associated with the proportion of myxoid component (P<0.001). However, we could not substantiate the associations of MET overexpression with age, gender, and tumor location.

MET immunohistochemical overexpression preferentially detected in myxofibrosarcomas of higher grades. Histological features of one each representative myxofibrosarcoma classified as FNCLCC grade 1 (a), grade 2 (b), and grade 3 (c). The grade 1 lesion showed no staining of MET (d), whereas overexpression of MET was readily found in grade 2 (e) and grade 3 (f) lesions.
Survival Analyses
At last follow-up, 15 patients died of the tumors, 7 died of other causes, 14 were alive with tumors, and 50 were alive without evidence of tumor. Correlations of patient survivals with immunohistochemical and various clinicopathological parameters at the univariate level are shown in Table 3 and Figure 5. The cumulative 5-year rates of OS and MeFS were 62.7 and 50.1%, respectively, in patients with primary localized myxofibrosarcomas. In agreement with our and others’ earlier reports,1, 2, 4 several clinicopathological factors significantly correlated with adverse outcomes with respect to both end points, including the deep locations (OS, P=0.0321; MeFS, P=0.0162), high mitotic count (OS, P<0.0001; MeFS, P<0.0001), higher histological grades (OS, P=0.0437; MeFS, P=0.0030), and AJCC stage III (OS, P=0.0354; MeFS, P=0.0056). However, older age (P=0.0333), larger tumor size (P=0.0124), and remarkable tumor necrosis (P=0.0010) were only significantly related to worse MeFS. More importantly, MET overexpression was found not only significantly associated with inferior OS (P=0.0032, Figure 5a), but also strongly predictive of worse MeFS (P=0.0007, Figure 5b).

Kaplan–Meier plots to predict overall survival (a) and metastasis-free survival (b) according to MET immunoexpression status.
In multivariate analysis (Table 4), MET overexpression remained as an independent adverse prognosticator for both OS (P=0.0221) and MeFS (P=0.0040), along with high mitotic count (P<0.0001 for both OS and MeFS). In addition, MET overexpression also identified patients at more than six- and ninefold higher risks of worse OS and MeFS rates, respectively. However, an older age (P=0.0003) at presentation was independently predictive of worse MeFS alone. Other significant factors at the univariate level lost their statistical significance, including histological grading and tumor staging.
Discussion
The management of myxofibrosarcoma is complicated by the lack of good prognosticators to accurately determine tumor invasion and metastatic spread. However, it is perceived that increased clinical aggressiveness of myxofibrosarcoma is accompanied by phenotypic change from predominantly loose myxoid to more cellular solid histology,1, 2, 3 which provides a unique model of exploring stepwise genetic alterations in soft tissue malignancies. Presenting as both continuous large-scale and segmental interstitial gains, DNA copy number gains across both arms of chromosome 7 were detected in our series using ultrahigh-resolution aCGH. However, the frequencies and regions of recurrent CNAs appeared somewhat different from those reported by Ohguri et al8 using bacterial artificial chromosome-based aCGH,8 which comprised only 1440 target DNA clones to cover the whole genome. Despite being one of the most common CNAs (4/8, 50%) reported by Ohguri et al, gains at 7p21.1-p22.1 were present in a minor proportion of our cases, not fulfilling our threshold to define non-random aberrations in aCGH profiling. In this study, three amplicon cores were mapped to 7q21.11–7q21.3, 7q22.1–q22.3, and 7q31.1–7q32.3, respectively, which harbored established oncogenes on the q arm, including CDK6 (7q21.2) and MET (7q31.2). However, the boundaries of these recurrent CNAs did not overlap 7q33–q35, the minimal common region of DNA gains on 7q in the series of Ohguri et al.8
By using RNA extracted from LCM-isolated tumor cells for validation, 56% of 16 myxofibrosarcoma specimens showed apparently upregulated MET transcript by real-time RT–PCR. This finding suggested that MET should represent a bona fide oncogene located within regions of chromosomal gain on 7q in myxofibrosarcoma, rather than a co-amplified ‘passenger’ unrelated to tumorigenesis. Although histological grade and tumor stage of myxofibrosarcoma were identified as univariate adverse prognosticators, neither of these was associated with increased copy number gains or mRNA expression levels of MET oncogene. Intriguingly, we found that immunoexpression of MET protein was significantly higher in myxofibrosarcomas with unfavorable features, such as deeper location, higher grades, more advanced stages, and so on. Regarding the correlations with clinicopathological factors, we consider that the relatively fewer case numbers used in aCGH profiling and real-time RT–PCR quantification should not entirely account for the discrepancy in implications between MET protein expression and its gene dosage and mRNA abundance. This argument was partly in keeping with the findings of a karyotypic study of 32 myxofibrosarcomas, showing that chromosomal aberrations were not restricted to higher-grade myxofibrosarcomas, but could also be observed in low-grade tumors, albeit less frequently.41
In many tumors, MET protein is actually expressed at levels much higher than in the normal counterpart.16, 17 However, it has become more explicit that the upregulatory mechanisms of MET protein expression appear more complex than earlier thought and that most mechanisms identified thus far, such activation of other oncogenes and transcription factors, inactivation of p53 tumor suppressor, hypoxia, and so on, are known to increase MET gene transcription.15, 16, 17, 21, 23, 42, 43, 44 More recently, several small non-coding microRNAs, including miR-34b, miR-34c, miR-199a*, miRNA-1, and miRNA-206, have been proved to upregulate MET protein expression in a variety of cancers by either targeting MET mRNA for degradation or repressing its translation post-transcriptionally.43, 45, 46, 47 These microRNAs may constitute a novel and critical mechanistic basis to modulate MET expression in tumor cells. Through binding of activated HGF to its extracellular domain, MET undergoes oligomerization with subsequent phosphorylation of multiple tyrosine residues at the intracellular domains of the β subunit, thereby modulating its internalization, catalytic activity, and docking of adaptor proteins.15, 18, 19, 21, 23, 40, 44, 48 The interaction between HGF and MET has vital functions in orchestrating the ‘invasive growth program’ acquired in cells involving various physiological processes, such as embryonic organ morphogenesis and tissue repair.15, 18, 19, 23, 41, 48 Besides mitogenic and antiapoptotic activities common to many growth factor receptors, heightened MET activation can stimulate cell–cell detachment, migration, invasiveness, and angiogenesis.15, 18, 19, 23, 40, 48
However, the biological and prognostic implications of MET overexpression in human mesenchymal malignancies vary among series and may be tumor type-dependent.42, 44, 49, 50, 51 MET ovexpression is present in a high percentage of human rhabdomyosarcomas and osteosarcomas.42, 44, 49 In rhabdomyosarcomas, MET overexpression not only correlates with alveolar histology and advanced group, but also has an important function in the dissemination to bone marrow,49 whereas only expression of HGF enhances the resistance to radiochemotherapy.48 Recently, the tumorigenic property of MET oncogene in osteosarcomas was further reinforced by a study showing that MET overexpression can drive the transformation of human primary cultured osteoblasts into bone-forming sarcoma cells, both in vitro and in vivo.52 In synovial sarcomas, MET is expressed in approximately one-third of cases and co-expression of both MET and HGF, instead of MET expression alone, correlates with adverse outcomes.51 Conversely, MET overexpression was frequently found in skull base chordomas, but significantly predictive of favorable prognosis.50 In primary myxofibrosarcomas, we have identified for the first time that MET overexpression was independently predictive of shorter OS and MeFS as well as being associated with tumor size and mitotic rate. Our findings and the reported discrepancies in the implications of MET expression among various sarcoma types indicated its pleiotropic functions in tumor development, progression, and dissemination.
It has become clarified that activating mutations constitutively elicit MET signaling only in few tumor types, such as papillary renal cell carcinomas and gastric carcinomas.15, 17, 23 We performed direct sequencing for OH931 and NMFH-1 myxofibrosarcoma cells and found wild-type MET oncogene in both cell lines. Actually, MET receptor tyrosine kinase in most cancers is more frequently activated by transcriptionally upregulated or amplification-driven protein overexpression.15, 17, 23, 43 In this study, NMFH-1 myxofibrosarcoma cells, with lower endogenous MET expression, were responsive to treatment of recombinant HGF ligand by showing phosphorylation at Tyr1234/Tyr1235, two tyrosine residues essential for HGF-dependent activation of MET kinase.40 Although the activating phosphorylation induced by exogenous HFG ligand only lasted transiently, this finding suggested the possibility of an ectopic autocrine or paracrine loop in myxofibrosarcomas that mediates the HGF-MET signaling pathway. This scenario had been earlier exemplified in osteosarcomas, rhabdomyosarcomas, gliomas, and so on, when the tumors or surrounding microenvironments aberrantly express MET receptor or its HGF ligand.18, 19, 48
In conclusion, our work highlights the value of genomic profiling with ultrahigh-resolution aCGH. In myxofibrosarcomas, it shows recurrent regions of gain harboring oncogenes on 7q and the likelihood of an increased MET gene dosage in the induction of mRNA expression. Immunohistochemically, approximately two-thirds of primary localized tumors display MET protein overexpression, which correlates with adverse clinicopathological factors and independently portends worse MeFS and OS. Our findings strengthen the possible causative function of MET in conferring an aggressive phenotype, implying the potentiality of HGF/MET as an attractive target of therapeutics in myxofibrosarcoma. The latter approach seems conceivable given the availability of several HGF-neutralizing antibodies and MET-directed small molecule inhibitors undergoing clinical trials for other cancers.
References
Mentzel T, Calonje E, Wadden C, et al. Myxofibrosarcoma. Clinicopathological analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol 1996;20:391–405.
Huang HY, Lal P, Qin J, et al. Low-grade myxofibrosarcoma: a clinicopathologic analysis of 49 cases treated at a single institution with simultaneous assessment of the efficacy of 3-tier and 4-tier grading systems. Hum Pathol 2004;35:612–621.
Lin CN, Chou SC, Li CF, et al. Prognostic factors of myxofibrosarcomas: implications of margin status, tumor necrosis, and mitotic rate on survival. J Surg Oncol 2006;93:294–303.
Huang HY, Huang WW, Wu JM, et al. Flow cytometric analysis of DNA ploidy and S-phase fraction in primary localized myxofibrosarcoma: correlations with clinicopathological factors, Skp2 expression, and patient survival. Ann Surg Oncol 2008;15:2239–2249.
Snijders AM, Nowak N, Segraves R, et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 2001;29:263–264.
Bridge JA, Liu J, Qualman SJ, et al. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosomes Cancer 2002;33:310–321.
Bridge Jr RS, Bridge JA, Neff JR, et al. Recurrent chromosomal imbalances and structurally abnormal breakpoints within complex karyotypes of malignant peripheral nerve sheath tumour and malignant triton tumour: a cytogenetic and molecular cytogenetic study. J Clin Pathol 2004;57:1172–1178.
Ohguri T, Hisaoka M, Kawauchi S, et al. Cytogenetic analysis of myxoid liposarcoma and myxofibrosarcoma by array-based comparative genomic hybridisation. J Clin Pathol 2006;59:978–983.
Ozaki T, Wai D, Schafer KL, et al. Comparative genomic hybridization in cartilaginous tumors. Anticancer Res 2004;24:1721–1725.
Stock C, Kager L, Fink FM, et al. Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosomes Cancer 2000;28:329–336.
Tarkkanen M, Kiuru-Kuhlefelt S, Blomqvist C, et al. Clinical correlations of genetic changes by comparative genomic hybridization in Ewing sarcoma and related tumors. Cancer Genet Cytogenet 1999;114:35–41.
Tarkkanen M, Wiklund TA, Virolainen MJ, et al. Comparative genomic hybridization of postirradiation sarcomas. Cancer 2001;92:1992–1998.
Huang HY, Li SH, Yu SC, et al. Homozygous deletion of MTAP gene as a poor prognosticator in gastrointestinal stromal tumors. Clin Cancer Res 2009;15:6963–6972.
Pang EY, Bai AH, To KF, et al. Identification of PFTAIRE protein kinase 1, a novel cell division cycle-2 related gene, in the motile phenotype of hepatocellular carcinoma cells. Hepatology 2007;46:436–445.
Corso S, Comoglio PM, Giordano S . Cancer therapy: can the challenge be MET? Trends Mol Med 2005;11:284–292.
Ma PC, Tretiakova MS, MacKinnon AC, et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 2008;47:1025–1037.
Migliore C, Giordano S . Molecular cancer therapy: can our expectation be MET? Eur J Cancer 2008;44:641–651.
Ferracini R, Di Renzo MF, Scotlandi K, et al. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995;10:739–749.
Koochekpour S, Jeffers M, Rulong S, et al. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 1997;57:5391–5398.
Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007;104:20932–20937.
Wallenius V, Hisaoka M, Helou K, et al. Overexpression of the hepatocyte growth factor (HGF) receptor (Met) and presence of a truncated and activated intracellular HGF receptor fragment in locally aggressive/malignant human musculoskeletal tumors. Am J Pathol 2000;156:821–829.
Helou K, Walentinsson A, Kost-Alimova M, et al. Hgfr/Met oncogene acts as target for gene amplification in DMBA-induced rat sarcomas: free chromatin fluorescence in situ hybridization analysis of amplicon arrays in homogeneously staining regions. Genes Chromosomes Cancer 2001;30:416–420.
Eder JP, Vande Woude GF, Boerner SA, et al. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res 2009;15:2207–2214.
Krause AK, Hinrichs SH, Orndal C, et al. Characterization of a human myxoid malignant fibrous histiocytoma cell line, OH931. Cancer Genet Cytogenet 1997;94:138–143.
Kawashima H, Ogose A, Gu W, et al. Establishment and characterization of a novel myxofibrosarcoma cell line. Cancer Genet Cytogenet 2005;161:28–35.
Huang HY, Kang HY, Li CF, et al. Skp2 overexpression is highly representative of intrinsic biological aggressiveness and independently associated with poor prognosis in primary localized myxofibrosarcomas. Clin Cancer Res 2006;12:487–498.
Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997;15:350–362.
Olshen AB, Venkatraman ES, Lucito R, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004;5:557–572.
Raguz S, De Bella MT, Slade MJ, et al. Expression of RPIP9 (Rap2 interacting protein 9) is activated in breast carcinoma and correlates with a poor prognosis. Int J Cancer 2005;117:934–941.
Nagel S, Leich E, Quentmeier H, et al. Amplification at 7q22 targets cyclin-dependent kinase 6 in T-cell lymphoma. Leukemia 2008;22:387–392.
Jang SW, Yang SJ, Ehlen A, et al. Serine/arginine protein-specific kinase 2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res 2008;68:4559–4570.
Reddy PS, Umesh S, Thota B, et al. PBEF1/NAmPRTase/Visfatin: a potential malignant astrocytoma/glioblastoma serum marker with prognostic value. Cancer Biol Ther 2008;7:663–668.
Gorka B, Skubis-Zegadlo J, Mikula M, et al. NrCAM, a neuronal system cell-adhesion molecule, is induced in papillary thyroid carcinomas. Br J Cancer 2007;97:531–538.
Muller S, Kunkel P, Lamszus K, et al. A role for receptor tyrosine phosphatase zeta in glioma cell migration. Oncogene 2003;22:6661–6668.
Kondo T, Oue N, Yoshida K, et al. Expression of POT1 is associated with tumor stage and telomere length in gastric carcinoma. Cancer Res 2004;64:523–529.
Tsuchiya N, Ochiai M, Nakashima K, et al. SND1, a component of RNA-induced silencing complex, is up-regulated in human colon cancers and implicated in early stage colon carcinogenesis. Cancer Res 2007;67:9568–9576.
Osaki M, Inoue T, Yamaguchi S, et al. MAD1 (mitotic arrest deficiency 1) is a candidate for a tumor suppressor gene in human stomach. Virchows Arch 2007;451:771–779.
Martuszewska D, Ljungberg B, Johansson M, et al. Tensin3 is a negative regulator of cell migration and all four Tensin family members are downregulated in human kidney cancer. PLoS One 2009;4:e4350.
Cao D, Polyak K, Halushka MK, et al. Serial analysis of gene expression of lobular carcinoma in situ identifies down regulation of claudin 4 and overexpression of matrix metalloproteinase 9. Breast Cancer Res 2008;10:R91.
Longati P, Bardelli A, Ponzetto C, et al. Tyrosines1234–1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994;9:49–57.
Willems SM, Debiec-Rychter M, Szuhai K, et al. Local recurrence of myxofibrosarcoma is associated with increase in tumour grade and cytogenetic aberrations, suggesting a multistep tumour progression model. Mod Pathol 2006;19:407–416.
Chen Y, Takita J, Mizuguchi M, et al. Mutation and expression analyses of the MET and CDKN2A genes in rhabdomyosarcoma with emphasis on MET overexpression. Genes Chromosomes Cancer 2007;46:348–358.
Migliore C, Petrelli A, Ghiso E, et al. MicroRNAs impair MET-mediated invasive growth. Cancer Res 2008;68:10128–10136.
Scotlandi K, Baldini N, Oliviero M, et al. Expression of Met/hepatocyte growth factor receptor gene and malignant behavior of musculoskeletal tumors. Am J Pathol 1996;149:1209–1219.
Duan Z, Choy E, Nielsen GP, et al. Differential expression of microRNA (miRNA) in chordoma reveals a role for miRNA-1 in Met expression. J Orthop Res 2010;28:746–752.
Kim S, Lee UJ, Kim MN, et al. MicroRNA miR-199a* regulates the MET proto-oncogene and the downstream extracellular signal-regulated kinase 2 (ERK2). J Biol Chem 2008;283:18158–18166.
Yan D, Dong Xda E, Chen X, et al. MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma development. J Biol Chem 2009;284:29596–29604.
Jankowski K, Kucia M, Wysoczynski M, et al. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res 2003;63:7926–7935.
Diomedi-Camassei F, McDowell HP, De Ioris MA, et al. Clinical significance of CXC chemokine receptor-4 and c-Met in childhood rhabdomyosarcoma. Clin Cancer Res 2008;14:4119–4127.
Naka T, Kuester D, Boltze C, et al. Expression of hepatocyte growth factor and c-MET in skull base chordoma. Cancer 2008;112:104–110.
Oda Y, Sakamoto A, Saito T, et al. Expression of hepatocyte growth factor (HGF)/scatter factor and its receptor c-MET correlates with poor prognosis in synovial sarcoma. Hum Pathol 2000;31:185–192.
Patane S, Avnet S, Coltella N, et al. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer Res 2006;66:4750–4757.
Acknowledgements
This work was supported in part by grants from Chang Gung Memorial Hospital (CMRPG870751, CMRPG870752) and Chi-Mei Medical Center (CMFHR 9634). The authors are deeply indebted to Dr Julia Bridge and Dr Ogose for kindly providing OH931 and NMFH-1 cell lines. We are also grateful to genomic core laboratory (CMRPG880251) and tissue bank (CMRPG870461) of Chang Gung Memorial Hospital-Kaohsiung Medical Center for critical technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work has been presented in part in the 98th annual meeting of United States and Canadian Academy of Pathology, Boston, USA, 7–13 March 2009
Rights and permissions
About this article
Cite this article
Lee, JC., Li, CF., Fang, FM. et al. Prognostic implication of MET overexpression in myxofibrosarcomas: an integrative array comparative genomic hybridization, real-time quantitative PCR, immunoblotting, and immunohistochemical analysis. Mod Pathol 23, 1379–1392 (2010). https://doi.org/10.1038/modpathol.2010.128
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.128
Keywords
This article is cited by
-
Molecular and clinicopathological analysis revealed an immuno-checkpoint inhibitor as a potential therapeutic target in a subset of high-grade myxofibrosarcoma
Virchows Archiv (2022)
-
MET-overexpressing myxofibrosarcoma frequently exhibit polysomy of chromosome 7 but not MET amplification, especially in high-grade cases: clinical and pathological review of 30 myxofibrosarcoma cases
Diagnostic Pathology (2018)
-
Integrated genetic and epigenetic analysis of myxofibrosarcoma
Nature Communications (2018)
-
The novel myxofibrosarcoma cell line MUG-Myx1 expresses a tumourigenic stem-like cell population with high aldehyde dehydrogenase 1 activity
BMC Cancer (2013)
