
Abstract
Major consensus conferences held over a decade ago laid the foundations for the current (2004) WHO classification of renal carcinoma. Clear cell, papillary and chromophobe carcinomas account for 85–90% carcinomas seen in routine practice. The remaining 10–15% of carcinomas consist of rare sporadic and hereditary tumors, some of which had been long recognized, but many of which only emerged as distinct entities in the decade leading up to the WHO publication. Collecting-duct carcinoma is a rare, often lethal form of carcinoma. Medullary carcinoma associated with sickle cell trait, has emerged as a distinctive tumor showing some overlapping features with upper tract urothelial carcinoma. Mucinous tubular and spindle-cell carcinoma and tubulocystic carcinoma were earlier considered as patterns of low-grade collecting-duct carcinoma, but are now recognized as separate tumor entities. Carcinomas associated with somatic translocations of TFE3 and TFEB comprise a significant proportion of pediatric renal carcinomas. Oncocytoid renal carcinomas in neuroblastoma survivors was recognized as a unique tumor category in the WHO classification. Renal carcinoma associated with end-stage renal disease is now recognized as having distinct morphological patterns and behavior. In addition there is a group of rare recently described carcinomas, including clear cell papillary carcinoma, oncocytic papillary renal cell carcinoma, follicular renal carcinoma and leiomyomatous renal cell carcinoma. It behooves the surgical pathologist to not only be capable of diagnosing the common forms of renal cancer, but also to be aware of the rare types of renal carcinoma, many of which have emerged in recent years.
Similar content being viewed by others
Main
The taxonomy of renal epithelial neoplasms has evolved greatly over the past two decades.1 Landmark consensus conferences held in Heidelberg (1996) and Rochester (1997) laid the foundations for our modern classification system.2, 3 Detailed morphological studies incorporating contemporary immunohistochemical and molecular techniques have resulted in the current classification of renal epithelial neoplasms as outlined in the 2004 World Health Organization (WHO) monograph.4 The common renal cell carcinomas of clear cell, papillary and chromophobe types, account for 85–90% of the renal tubular malignancies encountered in routine practice. The remaining 10–15% includes a variety of uncommon sporadic and familial carcinomas, some of which have been recently described, along with a group of unclassified carcinomas. Selected uncommon, recently described and emerging forms of renal cell carcinoma will be discussed in this paper (Table 1). Although rare familial renal cell carcinomas can be seen, emphasis will be placed on the uncommon sporadic carcinomas. In particular, the following topics will be reviewed—collecting-duct carcinoma, renal medullary carcinoma, mucinous tubular spindle-cell carcinoma, tubulocystic carcinoma, translocation carcinoma, carcinoma in neuroblastoma survivors, dialysis associated carcinoma and an interesting group of very recently described and incompletely characterized renal epithelial tumors.
Collecting-duct carcinoma
Collecting-duct carcinoma was first described in 19495 and was recognized as a separate entity of renal cell carcinoma in 1986.6 Since then several case series have been reported, with these tumors now being recognized as an aggressive form of renal neoplasia.7, 8, 9, 10, 11, 12
Collecting-duct carcinomas comprise approximately 1% of renal epithelial malignancies and often present with advanced stage disease.13 They have been described as occurring in a wide patient age range, but generally affect patients in the 4th to 7th decades (mean age 55 years), with a male predominance of approximately 2:1. These tumors are frequently symptomatic, with typical presenting features being one or more of the classic renal tumor triad of hematuria, abdominal mass and intermittent flank/back pain, and also fatigue and weight loss. The frequency of symptomatic presentation of these tumors reflects their rapid growth and early metastatic spread, with approximately one-third of patients being shown to have metastases at the time of diagnosis.
Evidence of a collecting-duct origin for these tumors is that when small, the primary tumor was usually confined to the renal medulla. Despite this these tumors are usually of large size when diagnosed and involve both the renal cortex and medulla. Typically collecting-duct carcinomas are white to gray and have a firm consistency on sectioning (Figure 1). Tumor necrosis is typically present although hemorrhage is not usually seen macroscopically. These tumors may extend into the renal pelvis and on imaging studies may mimic pelvic urothelial carcinoma.7

Gross photograph of collecting-duct carcinoma showing infiltrating gray–white tumor centered on renal medulla.
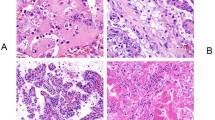
The microscopic features of collecting-duct carcinoma may be somewhat variable, however, the morphologic criteria for diagnosis are the presence of an infiltrative tubular or tubulopapillary pattern, associated with a desmoplastic stromal reaction (Figure 2a and b). The tumor cells typically exhibit a high grade of nuclear pleomorphism and nuclear atypia is seen in the epithelium of adjacent renal tubules (Figure 3).14 Mitotic figures are frequently present and histochemically both acid and neutral mucin may be seen.15 In addition to the tubulopapillary architecture, these tumors may also contain compact papillary structures, solid sheet-like areas of tumor cells and microcysts. Occasionally foci of spindle cells are present, however, if this is more than a rare occurrence, the tumor should be considered to be a sarcomatoid carcinoma arising within a collecting-duct carcinoma. There is usually an associated chronic active inflammatory cell infiltrate in and adjacent to the tumor, and in some cases a neutrophilic infiltrate can be quite pronounced. Tumor architecture may be recapitulated in extra-renal metastases (Figure 4).

Photomicrographs of collecting-duct carcinoma. (a) Note infiltrating irregular tubules embedded in fibro-inflammatory stroma. (b) Tubules are lined by cells showing high-grade nuclear atypia.

Photomicrograph showing dysplasia in medullary tubules adjacent to collecting-duct carcinoma.

Low-power photomicrograph showing metastatic collecting-duct carcinoma in lung. Note respiratory bronchial on right side. The metastatic tumor displays a similar pattern to the primary neoplasm.
The immunohistochemical expression of collecting-duct carcinoma reflects the origins of the tumor from the collecting duct of the distal nephron.7, 8, 16, 17 Tumors usually show positive reactions to lectins such as Ulex europeaus agglutinin-1 and peanut lectin, also e-cadherin, c-KIT, and both high and low molecular weight cytokeratins. Vimentin staining of tumor cytoplasm may also be present. There is a variable expression of Leu M1 and EMA, whereas markers of proximal renal tubules (CD10, RCC antigen and AMACR) are almost always negative.
There are limited studies on the genetics of collecting-duct carcinoma and the results to date are inconclusive, with no consistent genetic abnormality being noted. The karyotype of one tumor was reported as showing trisomy for chromosomes 4, 7, 8, 17 and 20 and loss of chromosomes 14, 18 and 22.12 This compares with monosomy of chromosomes 1, 6, 14, 15 and 22 noted in three further cases.10 Loss of chromosomes 4 and 18 was also found in two of these tumors, although in this latter series the diagnosis of some of the cases has been questioned.14 In further studies loss of heterozygosity of 8p and 13q has been reported.18
The differential diagnosis of collecting-duct carcinoma includes papillary renal cell carcinoma, renal medullary carcinoma, metastatic carcinoma and urothelial carcinoma with glandular differentiation.
Papillary renal cell carcinoma usually only poses a problem if it is of high grade, but usually lacks the desmoplasia and infiltrative pattern typical of collecting-duct carcinoma. Immunohistochemical staining can be useful in differentiating between these two tumor types with papillary renal cell carcinoma frequently showing positivity for CD10, AMACR and RCC antigen. Medullary carcinoma may show a morphologic overlap with collecting-duct carcinoma, but usually exhibits reticular and solid patterns of growth. The constant association with sickle cell trait and young patient age at diagnosis are further indicators in favor of a diagnosis of medullary carcinoma. Metastatic adenocarcinoma should always be considered in the differential diagnosis of these tumors, as there is usually a marked desmoplastic response to tumor associated with a brisk inflammatory cell infiltrate. A previous history of malignancy may be of diagnostic assistance and appropriate clinical and immunohistochemical investigations should be undertaken to further characterize tumors as metastatic rather than primary.
Upper tract urothelial carcinoma may cause some difficulty in differentiation from collecting-duct carcinoma, especially when glandular elements are identified within the tumor. In such cases, the presence of in situ urothelial carcinoma in adjacent calyces or within the renal pelvis argues against the diagnosis of collecting-duct carcinoma. Immunohistochemical staining may also be useful in differentiating between these two tumors, with positive CK20 and low molecular weight cytokeratin indicative of urothelial carcinoma. Results of staining for markers of the distal nephron should be interpreted with caution as expression of cytokeratin AE1/AE3 and Ulex Europeaus agglutinin-1 has been reported in high-grade urothelial carcinoma arising from the renal papilla and exhibiting adjacent carcinoma in situ.19 Recently immunoexpression of transmembrane enzyme complex carbonic anhydrase IX has been shown to differentiate urothelial carcinoma (diffuse to multifocal strong membrane staining in 90% of cases) from collecting-duct carcinoma (scanty, focal and weak membrane staining in 60% of cases).20
Survival data from case reports and small case series indicates that these tumors have a poor prognosis.14 From these reports up to 40% of patients have metastatic spread of tumor at presentation and most patients die within 1–3 years of diagnosis. The largest case series to consider outcome to date was reported from Japan and consisted of 81 patients.21 Regional lymph node metastases were present in 44% of the patients in this series at diagnosis, whereas 32% had distant metastases. The 1-, 3- and 10-year disease-specific survivals in this series were 69, 45 and 14%, respectively.
Renal medullary carcinoma
Renal medullary carcinoma was recognized as a novel form of renal malignancy in a series published by Colonel Davis of the Armed Forces Institute of Pathology in 1995.22 This study was based upon retrospective analysis of tumors coded as renal pelvic carcinoma from patients aged less than 40 years. Where race had been recorded, all patients were black and all had sickled erthrocytes on histological examination. On this basis the tumor, which was termed medullary carcinoma on morphologic grounds, was designated the seventh sickle cell nephropathy (the others being gross hematuria, papillary necrosis, nephrotic syndrome, renal infarction, inability to concentrate urine and pyelonephritis);23 since the original study over 160 cases have been reported.24, 25, 26, 27, 28, 29, 30, 31, 32, 33
Patient age at diagnosis ranges from 5 to 69 years with a mean age of 19 years. There is a male predominance with a male/female ratio of 2:1, although for patients <10 years the male/female ratio is 5:1.26 Where ethnicity was reported the overwhelming majority of patients were African American, whereas 15 patients were Hispanic/Brazilian. Medullary carcinoma has only rarely (<10 cases) been reported in Caucasians. Virtually all patients have sickle cell trait/disease although the tumor has been reported from a patient with a normal blood profile.26
Virtually all patients are symptomatic at diagnosis, with pain and hematuria predominating, whereas other presenting symptoms are abdominal mass, dysuria and weight loss.
Medullary carcinoma more commonly occurs in the right kidney (>75%) and is generally a solitary, poorly circumscribed mass. On sectioning the tumor is tan or gray–white, with macroscopic evidence of necrosis and hemorrhage. Microscopic examination shows an infiltrative poorly differentiated carcinoma consisting of solid sheets with poorly formed vacuoles (Figure 5). The tumor may also form cords, and occasionally vacuolated nests (Figure 6), microcysts, tubules and areas resembling yolk sac tumors (Figure 7) or sarcomatoid carcinoma may be seen. There is usually a pronounced desmoplastic reaction with an associated chronic active inflammatory cell infiltrate and areas of necrosis are often present (Figure 6).

Medullary carcinoma showing small sheets of poorly differentiated tumor infiltrated by neutrophils.

Medullary carcinoma displaying vacuolated nests, irregular infiltration and necrosis.

Medullary carcinoma showing a yolk sac-like morphology. Note prominent microvesicular pattern.
Tumor cells have an eosinophilic/granular cytoplasm and there is usually a marked degree of nuclear pleomorphism with prominent nucleoli. Mitotic figures are common and cells resembling those seen in rhabdoid dedifferentiation in renal cell carcinoma may also be identified.32 Sickle-cell erythrocytes are a frequent finding within the tumor and adjacent renal tissue (Figure 8).

Renal medulla adjacent to medullary carcinoma. Note prominent sickling of the erythrocytes in dilated capillaries.
Ultrastructural studies show variable findings and no consistent features were reported from one study.24 Recently it has been noted that tumor cells contain large intracytoplasmic vesicles lined by long slender microvilli. Condensed fibrillary electron-dense deposits were also present. Prominent desmosomes were identified, whereas glycogen and lipids were universally absent.33
The immunohistochemical expression of medullary carcinoma has been reported from several studies.24, 25, 31 All tumors show positive and diffuse expression for cytokeratin AE1/AE3, low molecular weight cytokeratin, EMA, vimentin, HIF and VEGF. Cytokeratin 7 and carcinoembryonic antigen was focally expressed in all cases, whereas there was a variable expression of high molecular weight cytokeratin, Ulex europeaus agglutinin-1 and TP53. There was no expression of Her-2neu.
Genetic studies on medullary carcinoma are limited. On CGH no gains or losses were found in eight of nine tumors studied, whereas one case showed loss of chromosome 22. Gene expression profiling showed 487 genes to be expressed differently from that of other types of renal tumor, with an expression most closely resembling urothelial carcinoma.25 In three cases FISH analysis showed amplification of the ABL gene, with increased expression of ABL protein in two cases. In this series no evidence of BCR-ABD translocation was detected, in contradiction to the report of Stahlschmidt et al34 based on a single case.
The differential diagnosis for medullary carcinoma is high-grade invasive urothelial carcinoma and collecting-duct carcinoma. As noted above urothelial carcinoma appears closely related to medullary carcinoma on the basis of genetic profile. Clinical evidence of sickle cell trait and young patient age are seen with medullary carcinoma, whereas cytokeratin 20 positivity and the presence of adjacent urothelial carcinoma in situ are often seen in urothelial carcinoma. The infiltrative elements of medullary carcinoma may show tubules that resemble collecting-duct carcinoma and this has been considered to be evidence that these two tumors are related. Sickle cell trait is not associated with collecting-duct carcinoma, which is usually positive for high molecular weight cytokeratin and Ulex europeaus agglutinin-1, and may be negative for EMA.
Medullary carcinoma has a poor prognosis22, 24, 25, 26, 31, 33 Metastatic disease is seen at presentation in 95% of patients with metastases encountered, in descending frequency, in lymph nodes (retroperitoneal and mediastinal), lungs, liver and adrenal glands. Metastases to breast, bone and contralateral kidney at the time of diagnosis have also been reported. In a published series post-presentation survival for patients ranged from 1 day to 68 weeks with a mean survival of approximately 18 weeks. Three patients with organ-confined disease were alive 9 months, 2 years and 8 years post-nephrectomy.26, 31 One patient with pleural, mediastinal and retroperitoneal metastases was alive 85 weeks post-diagnosis, following nephrectomy and post-operative cisplatin, gemcitabene and paclitaxel chemotherapy, with tandem autologous bone marrow transplantation.33
Mucinous tubular and spindle-cell carcinoma
Mucinous tubular and spindle-cell carcinoma is an uncommon and recently described variant of renal cell carcinoma, which is recognized as a distinct entity in the 2004 World Health Organization tumor classification.35 The current WHO terminology is descriptive reflecting the three salient histology elements—tubules, spindle-cell areas and mucinous stroma. Previously, this tumor had been referred to under a variety of terms including low-grade collecting-duct carcinoma,36 unusual renal cell carcinoma with prominent spindle-cell change possibly related to loop of Henle,37, 38 low-grade myxoid renal epithelial neoplasm with distal nephron differentiation,39 low-grade tubular mucinous renal neoplasm40 and spindle and cuboidal renal cell carcinoma.41 Other large series have also been reported.42, 43
Mucinous tubular and spindle-cell carcinoma has a wide age range and female predominance (F:M=4:1). An association with nephrolithiasis has been noted.41 The tumor may be found in a cortical or central location and generally displays a uniform homogenous tan, gray or pale yellow cut surface (Figure 9). There is a wide size range from less than 1 cm diameter to greater than 18 cm, with most tumors being 2–4 cm in width. Areas of necrosis may be seen, but these are unusual.

Mucinous tubular and spindle-cell carcinoma. Note tumor circumscription and uniform light tan coloration.
Under the microscope the neoplasm shows elongated tubules, which may be curvilinear and stretched, separated by variable amounts of mucinous stroma (Figure 10). Parallel and collapsed tubules impart a spindle-cell appearance and when this pattern predominates, the tumor may mimic a mesenchymal neoplasm such as leiomyoma (Figure 11). The nuclei are uniform and display low nuclear grade characteristics. Foam cells, lymphocytic infiltrates and small psammoma bodies may also be present (Figure 12a and b). Occasionally necrosis, solid tubular growth and areas of high nuclear grade change may be identified.

Photomicrograph of mucinous tubular and spindle-cell carcinoma showing classical pattern with elongated tubules, spindle-cell areas and stromal mucin.

Mucinous tubular and spindle-cell carcinoma showing prominent spindle-cell pattern simulating a smooth muscle neoplasm.

Mucinous tubular and spindle-cell carcinoma. Note collections of foam cells and lymphocytes in tumor (a) and focal microcalcification (b).
A recent publication has highlighted non-classic patterns of mucinous tubular and spindle-cell carcinoma including mucin-poor tumors (Figure 13) and those showing focal papillary change.44 Mucinous tubular and spindle-cell carcinoma with focal neuroendocrine differentiation have also been described.45 More recently examples of mucinous tubular and spindle-cell carcinoma with sarcomatoid change have been reported.46, 47, 48

Mucinous tubular and spindle-cell carcinoma with a mucin-poor pattern. Note prominent elongated and branching tubules.
Immunohistochemical studies of mucinous tubular and spindle-cell carcinoma generally show positivity for low molecular weight cytokeratins (eg, CAM5.2) and cytokeratin 7.35, 37, 38, 39, 40, 41, 42, 43, 49, 50 High molecular weight keratins (34BE12, CK20) are usually negative. P504S (racemase) is typically positive whereas CD10 and renal cell carcinoma antigen are often negative. CD15 and RCC markers show variable expression.
At the genetic level, several studies have identified multiple chromosome losses in mucinous tubular and spindle-cell carcinoma.35, 38, 51, 52 It has been suggested that some examples of the tumor showed trisomies of chromosomes 7 and 17, but the most extensive study failed to identify this feature.52, 53
The histogenesis of mucinous tubular and spindle-cell carcinoma is unknown. Although the tumor was initially thought to arise from either a collecting duct or loop of Henle, the complex immunophenotype with cytokeratin 7 and racemase expression suggests proximal nephron origin.35, 36, 37, 38, 39, 40, 43, 50 Indeed there is significant immunohistochemical overlap between mucinous tubular and spindle-cell carcinoma and papillary renal cell carcinoma.43, 50
The differential diagnosis of mucinous tubular and spindle-cell carcinoma is relatively limited. In its classic form this tumor is a highly distinctive neoplasm and easily diagnosed. In situations where variant patterns exist, for instance when there is a paucity of mucin, diagnostic difficulties may arise.44 Recently, there has been a description of papillary renal cell carcinoma with low-grade spindle-cell foci.54 In this report of five cases, the tubular and spindle-cell areas bear some resemblance to mucinous tubular and spindle-cell carcinoma. Differences exist in that papillary carcinomas have a male predominance and they generally show areas of distinct papillation, at least focally. Papillary carcinoma and mucinous tubular and spindle-cell carcinoma show overlapping immunohistochemical features with the expression of cytokeratin 7 and P504S (racemase). However, mucinous tubular and spindle-cell carcinoma often stains negatively for CD10. In cases where genetic studies are carried out, the presence of trisomies 7 and 17 confirm the diagnosis of papillary renal cell carcinoma.52, 54
In very rare examples of mucinous tubular and spindle-cell carcinoma with spindle-cell predominance, the tumor bears resemblance to smooth muscle neoplasms such as leiomyoma and low-grade leiomyosarcoma. However, the latter have a more distinct fascicular architecture and more elongated cigar-shaped nuclei. Usually there is some evidence of tubular growth, at least focally, in mucinous tubular and spindle-cell carcinoma. Furthermore, the presence of extensive cytokeratin 7 positivity eliminates the possibility of a smooth muscle neoplasm.
Although some examples of mucinous tubular and spindle-cell carcinoma with prominent spindle-cell change have been previously interpreted as sarcomatoid renal cell carcinomas, the bland low-grade appearance of the spindle element and the relatively innocuous behavior of this tumor distinguish mucinous tubular and spindle-cell carcinoma from typical sarcomatoid renal cell carcinoma.
The great majority of mucinous tubular and spindle-cell carcinomas behave in a low-grade fashion. However, Rakozy et al40 have described one patient with multiple intra-abdominal recurrences. There are few reports demonstrating regional lymph node involvement.37, 38, 41 Recently there have been three reports describing a total of four patients with mucinous tubular and spindle-cell carcinoma showing sarcomatoid differentiation.46, 47, 48 In two patients distant metastases have been documented.46, 48 Interestingly, in one case with histologic documentation the metastatic component consisted of the high-grade sarcomatoid pattern.46 It is noteworthy that the authors have seen one example of mucinous tubular and spindle-cell carcinoma with classical morphology showing metastases to retroperitoneal lymph nodes and liver. The tumor in the liver had an appearance identical to that of the classical renal morphology.
Tubulocystic carcinoma
Tubulocystic carcinoma is a recently described entity that is not listed in the 2004 WHO classification.4 Descriptions and images of tubulocystic carcinoma are found in older literature dealing with collecting-duct carcinoma. Masson55 described ‘Bellinian epitheliome’ as a tumor of the ducts of Bellini and his superb line drawing shows many of the salient features of tubulocystic carcinoma. George Farrow56 in the Armed Forces Institute of Pathology (Series III) fascicle devoted to renal tumors described tumors identical to tubulocystic carcinoma under the rubric, low-grade collecting-duct carcinoma. In a subsequent paper from the Mayo Clinic, 13 examples of low-grade collecting ducts were described, eight of which showed typical features of tubulocystic carcinoma.36 The remaining five cases were characteristic examples of mucinous tubular and spindle-cell carcinoma.57 Recently three major series of tubulocystic carcinoma have reported 55 cases.58, 59, 60
Tubulocystic carcinoma is a tumor of adults with a wide age range, 30–94 years. Most patients are present in the fifth and sixth decade and there is a strong male predominance (male/female ratio of 7:1).58, 59, 60 Patients are often asymptomatic, although they may also present with abdominal pain, distention and hematuria. Reported tumors are more often left sided (27/44).59, 60 As the tumor has cystic elements it has a broad radiological differential diagnosis and can be classified as a Bosniak type II, III or rarely a type IV lesion.
At the macroscopic level, tubulocystic carcinoma is usually solitary, although multifocal examples may be seen.59, 60 The tumors show a variable size, ranging from 0.5 to 17 cm, with a mean of around 4 cm. The tumors are circumscribed and usually unencapsulated, and they demonstrate a white or gray spongy cut surface, often compared with ‘bubblewrap’. Co-existing renal papillomas and papillary renal cell carcinomas with their typical gross features have also been described.59
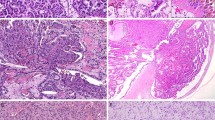
Tubulocystic carcinoma has a typical microscopic appearance. It is composed of tightly packed tubules and cysts measuring up to a few millimeters in diameter (Figure 14), separated by bland fibrous stroma (Figure 15). Solid growth is not seen. No desmoplastic reaction or cellular ovarian-like stroma is present. The lining cells are cuboidal to columnar and may have an attenuated appearance. Hobnail cells are commonly seen. The cells have abundant eosinophilic or amphophilic cytoplasm and the nuclei are large and have prominent nucleoli (Figure 16). Occasional cells with low-grade nuclear changes may be seen but they rarely predominate. Rarely minor areas with clear cell or papillary features are noted.60 Foam cells, calcospherites and hemosiderin encrustation is not found, unless there is an accompanying papillary renal cell carcinoma. Most tubulocystic carcinomas are pT1 tumors, with occasional pT2 and pT3 lesions being reported.60

Low-power photomicrograph of tubulocystic carcinoma. Note sharp circumscription of a tumor composed of variably sized tubules and small cystic spaces.

Tubulocystic carcinoma. Note variably sized tubules and cysts lined by a single layer of eosinophilic cells.

Tubulocystic carcinoma. High-power photomicrograph showing tubules lined by cells with abundant eosinophilic cytoplasm. Note high nuclear grade with prominent nucleoli. Hobnailed cells are focally present.
Immunohistochemical studies58, 59, 60 demonstrate a wide range of marker positivity with cytokeratins (CK8, CK18, CK19) and parvalbumin being consistently positive. CD10 and P504S (racemase) (Figure 17) are positive in greater than 90% of tumors. CK7 is commonly expressed, although that pattern may be weak and focal. Staining for kidney-specific cadherin and Pax2 may also be seen. High molecular weight cytokeratin (34BE12) is nearly always negative.

Tubulocystic carcinoma showing strong racemase (P504S) positivity.
Electron microscopy reveals interesting findings.60 Most cells have abundant microvilli with a brush border appearance resembling proximal convoluted tubules. Admixed cells with short, sparse microvilli and complex cytoplasmic interdigitation, reminiscent of intercalated cells of collecting duct are also seen.
Gene expression profiling studies indicate that tubulocystic carcinoma has a distinct molecular signature.59, 60 A comparative study looking at gene expression profiles of tubulocystic carcinoma and collecting-duct carcinoma showed significant differences, indicating that these two tumor types are unrelated.61 Some studies suggest a relationship between tubulocystic and papillary carcinoma.59, 62 Indeed, in one study tubulocystic carcinoma was commonly associated with papillary carcinoma, with both tumors showing gains in chromosomes 7 and 17.62
The histogenesis of tubulocystic carcinoma is unclear. The marker studies and electron microscopic observations indicate that the tumor cells express aberrant tubular differentiation.60 The tumor cells have some features of proximal nephron differentiation and others related to the distal nephron. The weight of evidence argues against an origin from cells of collecting duct or loop of Henle. The more than occasional association of tubulocystic carcinoma with papillary carcinoma is intriguing. Sometimes tubulocystic and papillary renal cell carcinoma are found separately in the kidney and at other times they are closely admixed.59, 62 The relationship between tubulocystic and papillary renal cell carcinoma, along with the overlapping immunohistochemical and molecular patterns, suggest a taxonomic relationship. We would argue that tubulocystic carcinoma is a subset of papillary renal cell carcinoma, just as multiloculated clear cell renal cell carcinoma is considered a variant of clear cell renal cell carcinoma.
The differential diagnosis is interesting and mostly includes other tumors with a multiloculated gross appearance, namely, multiloculated clear cell renal cell carcinoma, cystic nephroma, mixed epithelial and stromal tumor and cystic oncocytoma.63 Multiloculated clear cell carcinoma is lined by clear cells usually with a low nuclear grade. Furthermore, nests of clear cells are also found in the septae. The typical lining cells of tubulocystic carcinoma have eosinophilic cytoplasm and demonstrate high nuclear grade. Both tubulocystic and cystic nephroma have cysts, lined in part by hobnail cells, but in cystic nephroma the cells have a low nuclear grade. In addition, the stroma in cystic nephroma is often cellular, at least focally, whereas in tubulocystic carcinoma the stroma is paucicellular and fibrotic. Mixed epithelial and stromal tumor of kidney is usually seen in middle aged women64 and has solid areas and broad septae, in contrast to the thin fibrous septae of tubulocystic carcinoma. In addition ovarian-like cellular stroma is commonly seen in mixed epithelial and stromal tumor. A rare form of oncocytoma can have a dominant or exclusive cystic appearance, however, the constituent cells lack the nuclear atypia of tubulocystic carcinoma.65 Cystic oncocytoma usually displays, at least focally, solid nest of oncocytic cells along with the loose myxoid stroma. Although these entities are the main diagnostic considerations, tubulocystic carcinoma are not uncommonly referred to as atypical cysts or even benign cysts with unusual features. It is important to recognize that this rare tumor is a carcinoma and although appearing relatively bland, tubulocystic carcinoma can behave aggressively.
The biologic behavior of tubulocystic carcinoma has not been fully established; however, in two of the three major series published to date, a total of four patients have developed metastatic disease.59, 60 In one study a single patient developed recurrent local disease and two patients had metastases, both to bone and one to liver.60 It is very interesting that both patients with metastatic disease had focal cytoplasmic clearing in the primary tumors. In the other study, one patient with co-existing tubulocystic carcinoma and papillary renal cell carcinoma developed metastases from the latter.59 Another patient had metastases involving three pelvic lymph nodes and the morphology was typical of tubulocystic carcinoma.59 Until we have more information on the biology of tubulocystic carcinoma one should be cautious in rendering a prognosis. Although the great majority of tumors behave in an indolent fashion, one cannot at present be certain of the outcome, especially in unusual cases where there is focal clear cell or papillary change.
Translocation carcinoma
Renal translocation carcinomas are uncommon tumors generally arising in children and young adults.66, 67, 68, 69 In the pediatric literature they have sometimes been referred to as ‘juvenile carcinomas’. Most translocation carcinomas (about 90%) involve the transcription factor E3 (TFE3) located on Xp11.2.67, 69 The most common fusions are ASPL-TFE3 and PRCC-TFE3 but others have been described including PSF –TFE3, NonO-TFE3 and Clathrine-TFE3.67, 69 Xp11.2 translocation carcinoma is recognized in the 2004 WHO renal tumor classification.4 Another rare group of renal carcinomas showing a translocation [t (6; 11) (p21; q12)] involving transcription factor EB (TFEB) has been reported.67, 69, 70 In one large series TFEB carcinomas numbered only 2 of 31 cases (6.4%). Both TFE3 and TFEB belong to the microphthalmia transcription factor (MiTF) subfamily, which also includes MiTF and transcription factor EC (TFEC). Argani and Ladanyi67 have proposed to regroup these neoplasms into the category of ‘MiTF/TFE family translocation carcinomas’. Importantly, the translocations associated with TFE3 and TFEB are associated with overexpressed proteins that can be identified by immunohistochemistry. TFE3 nuclear staining is specific for the Xp11.2 translocation and nuclear TFEB staining is specific for [t (6; 12) (p21; q12)].67, 69
Translocation carcinomas are generally found in children and young adults and the average age in a recent large series was 24.7 years, with a median of 20 years and with a female to male ratio of 2.5:1.69 These tumors account for at least one-third of carcinomas seen in childhood and adolescence.67 Rare examples are found in adulthood and in one study the prevalence of renal carcinomas across all ages was estimated to be about 1%.67 Occasional examples of translocation carcinoma have been associated with a history of prior chemotherapy.67
Most patients with translocation carcinoma present symptomatically with hematuria, abdominal pain, abdominal mass or fever; however, about one-third of patients present asymptomatically. In one series, 42% of patients presented with lymph node or visceral metastases.69
The tumors are generally large (6–7 cm mean diameter) with some being up to 20 cm.67, 69 They have a macroscopic appearance similar to clear cell renal cell carcinoma. Variegated coloration and necrosis are often identified (Figure 18). There is a tendency towards extra-renal extension and regional node involvement at diagnosis.69

Gross photograph of TFE3-positive translocation carcinoma. Note large size, variegated coloration and necrosis.
Histologically, papillary, nested and compact (solid) patterns of growth are seen.69 A mixture of large clear and eosinophilic cells is often present. The presence of a papillary tumor in which the papillae are lined by large clear cells (Figure 19) should raise the possibility of translocation carcinoma, especially in a young patient. A nested pattern containing cells with voluminous, clear or granular eosinophilic cytoplasm (Figure 20) should also suggest the possibility of a translocation carcinoma. Psammoma bodies and intracytoplasmic hyaline droplets are frequent findings (Figures 19, 21), although foam cells and lymphoid infiltrates are uncommon.67, 69 Necrosis is present in about one-third of cases.69 The nuclei are generally large with open chromatin and prominent nucleoli. Mitoses are regularly present.

Xp11.2 translocation carcinoma. Note papillary architecture with mixture of clear and eosinophilic cells. Focal hyaline droplets are noted.

Translocation carcinoma composed of voluminous cells in nested pattern. Note prominent eosinophilic cytoplasm and high-grade nuclear atypia.

Translocation carcinoma with papillary and compact growth patterns. Note presence of psammoma body centrally.
In the initial description of TFEB translocation carcinoma, a biphasic growth pattern consisting of nests of large polygonal eosinophilic cells and intermingled clusters of small epithelial cells centered on hyaline nodules (Figure 22), was emphasized.70 Although initially thought to be specific for the TFEB carcinomas, this biphasic pattern may also be seen in carcinomas associated with TFE3 fusion.69

TFEB-positive translocation carcinoma showing biphasic morphology. Most of the tumor consists of nests of large cells with abundant eosinophilic cytoplasm. Two areas consisting of small cells encircling hyalin droplets are noted.
By immunohistochemistry, translocation carcinomas lack or only weakly express keratins and have variable vimentin expression.67, 69 Epithelial membrane antigen and CK7 are typically negative. These tumors characteristically express CD10, renal cell carcinoma antigen, racemase (AMACR; P504S) and E-cadherin. TFEB associated tumors regularly express melanocytic markers (HMB45, Melan A), whereas occasional expression of these markers is seen in the TFE3-positive carcinomas. From a practical perspective, the typical phenotype of translocation carcinoma (CK7−, racemase+, CD10+, EMA−) helps separate them from clear cell renal carcinoma (CK7−, racemase−, CD10+, EMA+) and papillary carcinoma (CK7+, racemase+, CD10+, EMA+).69
TFE3 and TFEB immunostains are sensitive and specific markers for their respective translocation carcinomas (Figure 23), although the stains can be technically difficult to perform and are best done by laboratories where there is sufficient volume to ensure quality. The sensitivity of TFE3 staining when compared with genetic studies ranges from 82 to 97.5%.66, 67, 69 There are rare examples of genetically confirmed translocation carcinomas where weak or absent TFE3 staining was noted; however, in general a moderate to strong nuclear staining pattern should be recognizable at relatively low magnification.66, 71

Translocation carcinoma showing strong nuclear positivity for transcription factor E3 (TFE3).
Ultrastructurally, most Xp11.2 carcinomas show features, including intracytoplasmic fat and glycogen, microvilli and cell junctions, similar to clear cell renal cell carcinoma.67 Most ASPL–TFE3 carcinomas contain membrane bound cytoplasmic granules and occasionally rhomboidal crystals identical to those seen in alveolar soft part sarcoma.72 In PRCC-TFE3 carcinomas intracisternal microtubules, similar to those seen in malignant melanoma, may be noted.73
A confirmation of the diagnosis of translocation carcinoma requires immunohistochemical identification of the nuclear transcription factor (TFE3, TFEB) and/or cytogenetic or molecular genetic (FISH, PCR) identification of the translocation.67, 69 Gene expression profiling of translocation carcinomas shows them to be distinct from other well recognized subtypes of renal carcinoma.69 Furthermore, TFE3 and TFEB tumors cluster together adding support for the unifying concept of the MiTF/TFE tumor family.69
The differential diagnosis of translocation carcinoma consists of other sporadic renal cell carcinomas (clear cell, papillary and unclassified), which may occur in children and young adults. Separation among these entities requires detailed microscopy and immunohistochemistry, sometimes coupled with molecular genetic analyses. There should be a high index of suspicion for translocation carcinoma in tumors showing papillary and nested patterns, where there is a mixture of clear and eosinophilic granular cells. The presence of voluminous cells may also be a clue to the diagnosis. The presence of psammoma bodies and hyaline droplets are also helpful. The characteristic immunoprofile, with absent or weak cytokeratin expression and positivity for CD10, racemase, E-cadherin and melanoma-associated antibodies, should initiate immunohistochemical investigation for transcription factors and genetic and/or molecular studies.67, 69
From a clinical outcome perspective, TFE3 translocation carcinomas seem to have a relatively indolent course, despite their often advanced stage at presentation.67, 69, 74, 75 Late recurrences, including one over three decades after original diagnosis, have been seen.76 Although the overall follow-up interval of reported cases remains relatively short (11–81 months), only 13.6% of cases have resulted in tumor-related deaths, which is low considering the percentage of tumors that are metastatic at presentation.4 Too few TFEB translocation carcinomas have been reported in the literature to accurately comment on the biological outcome, although tumor-related death associated with this type of tumor has been reported.69
Renal cell carcinoma in neuroblastoma survivors
There have been a number of publications describing renal cell carcinomas occurring in patients surviving neuroblastoma diagnosed in early childhood. Nineteen cases were culled from the literature in a review by Eble in 200377 and since then there have been occasional cases described.78 Some tumors are clear cell carcinomas but many others have been insufficiently described or illustrated to confidently identify the histologic subtype. Although most neuroblastoma survivors with renal carcinoma had received radiation and /or chemotherapy, a few patients received no modality. A recent epidemiological study indicated that childhood neuroblastoma survivors had a 329-fold increased risk of renal carcinoma.79
In 1999 four cases of a distinctive tumor occurring post-neuroblastoma were reported by Medeiros et al80 A series of four similar tumors was described by Koyle in 2001.81 These unusual tumors did not conform to the well recognized subtypes and were thought to be sufficiently distinctive to be recognized in the 2004 WHO classification.4 Although the authors have seen an additional example in consultation practice, the tumors from the two published series will be the focus of this section.
The affected children included two boys and six girls who were diagnosed with neuroblastoma in the first 2 years of life. The interval between diagnosis of neuroblastoma and the development of renal cell carcinoma ranged from 3 to 11.5 years (mean 9 years) with the patients aged between 5 and 14 years.77 Metastases to liver and lymph nodes were seen in one patient. Two patients received no radiation or chemotherapy and the remainder received chemotherapy with or without radiation.
Limited gross findings have been reported in four cases. Four examples of multiple bilateral tumors are noted. The main tumors ranged from 3.5 to 8.0 cm in diameter. Two tumors showed involvement of renal capsule, renal vessels and peripelvic lymphatics.
Typical post-neuroblastoma renal carcinoma has papillary and solid growth patterns (Figure 24) and is composed of oncocytoid cells with abundant eosinophilic cytoplasm (Figure 25). The nuclei are irregular and nucleoli are readily seen. Mitoses are present but not plentiful. One tumor described by Medeiros80 showed high nuclear grade and displayed extensive parenchymal infiltration.

Post-neuroblastoma renal carcinoma. Note Oncocytoid cell arranged in papillary and compact nested pattern.

Post-neuroblastoma renal carcinoma with papillary pattern. Note cells with abundant eosinophilic cytoplasm.
There is limited immunohistochemical information. The four tumors studied by Medeiros et al80 showed positivity for cytokeratin (Cam 5.2), epithelial membrane antigen and vimentin. There was less frequent positivity for CK19 and CK20. Stains for CK7, S-100 protein and HMB45 were negative.
Although there are no molecular studies of these histologically distinctive tumors, the possibility exists that this carcinoma represents part of some unusual genetic cancer syndrome. Interestingly there is a recent report documenting a similar oncocytoid renal carcinoma in a child who received chemotherapy for cardiac leiomyosarcoma.82
Carcinoma associated with end-stage renal disease
The relationship between end-stage renal disease, and the development of renal neoplasia has been debated, although recent studies indicate an increased prevalence of carcinoma in these patients.83, 84, 85 From 852 patients with end-stage renal disease studied prospectively from 1994 to 2000,85 19 had clinical evidence of renal neoplasia. Seventeen of these patients underwent nephrectomy with 14 being subsequently diagnosed with renal cell carcinoma, giving a prevalence of 1.64%. This compares with a renal cancer incidence of 0.04% for the general population. In a multinational study of 831 patients, in 804 there was an increased risk factor for renal neoplasia of 3.6 in patients who were on maintenance dialysis.86
The spectrum of renal tumors associated with end-stage renal disease is quite varied and in single cases and small series clear cell renal cell carcinomas, papillary renal cell carcinomas, chromophobe renal carcinoma, collecting-duct carcinoma, tubulocystic carcinoma, angiomyolipoma, oncocytoma and mixed epithelial and stromal tumor have been reported.87, 88, 89, 90, 91, 92, 93, 94 In up to 70% of cases more than one tumor was present in a single kidney.95
In a detailed study of 66 tumor-bearing kidneys from patients with end-stage renal disease, a wide spectrum of renal neoplasia was noted.96 In total 261 tumors were examined with 54.5% of kidneys containing >1 tumor in each kidney ranging from 0.6 to 8.5 cm (mean 3.0 cm, median 2.6 cm). Of recognized morphotypes of renal cell carcinoma, papillary renal cell carcinomas were most frequently encountered (18%), whereas 23% were clear cell renal cell carcinoma or chromophobe renal carcinoma. Sarcomatoid dedifferentiation has been reported in association with these tumors.97
Genetic studies on carcinomas associated with end-stage kidneys are limited, however, clear cell carcinoma arising in these kidneys have shown VHL gene mutations.98, 99
Additional to recognized forms of renal neoplasia, two novel tumors associated with end-stage renal failure have been described.96 One of these designated acquired cystic disease-associated renal cell carcinoma, was present in 46% of kidneys with acquired cystic disease. These tumors are usually well circumscribed and where large, showed pseudoencapsulation—often with dystrophic calcification. Microscopically there are a variety of architectural patterns with solid, acinar, cystic and papillary patterns being present. In all cases the presence of irregular lumina give the tumor a cribriform appearance (Figure 26). In 67% of reported cases the tumor appeared to arise in a cyst.

Renal cell carcinoma in end-stage renal disease. Sheets of cells with prominent vacuolation are present.
The tumor cells contain bulky eosinophilic cytoplasm with a rounded nucleus and large nucleolus (Figure 27). Occasional cells have vacuolated cytoplasm and focally clear cells are present. Oxalate crystals are present in the majority of tumors and also calcium aggregates, rarely forming psammoma bodies, are seen. These tumors are positive for vinculin and AMACR on immunohistochemical examination, and in a proportion of cases show variable and predominantly focal staining for cytokeratin 7 and parvalbumin. These tumors have also been shown to have positive immunoexpression for cytokeratin AE1/AE3 and CD10, and variable expression for vimentin CAM 5.2 and AMACR. Staining for EMA, cytokeratin 7, high molecular weight cytokeratin is negative.99

Renal cell carcinoma in end-stage renal disease. Note prominent cytoplasmic vacuolation.
A similar tumor had earlier been described in a kidney showing acquired cystic disease and genetic analysis of the tumor showed gains of chromosomes 7 and 17.100 FISH analysis showed gains of chromosomes 1, 2 and 6 in two cases with an additional gain of chromosome 10 in one case.98 Mutations of the VHL gene have not yet been identified in these tumors.101
The second form of novel tumor associated with end-stage renal disease is papillary clear cell renal cell carcinoma (Figure 28).96 This tumor occurs in kidneys both with or without acquired cystic disease and frequently contain a prominent pseudocapsule. In 50% of cases there is a pronounced cystic component and solid, tubular and microcystic areas are also present. The tumor cells show low-grade nuclear pleomorphism with nuclei situated towards the surface of the papillary tufts. These tumors show positive staining for cytokeratin 7 and are negative for AMACR and parvalbumin.96 Although these tumors were originally described in end-stage renal disease they have also been described in apparently normal kidneys (see section on New and emerging entities).

Clear cell papillary carcinoma in acquired cystic disease. Note delicate papillae lined by cuboidal cells with clear cytoplasm. Prominent eosinophilic secretion is present on the left side.
Outcome data for acquired cystic disease-associated renal cell carcinoma are limited, with one death from metastatic disease 34 months following diagnosis, being reported. In two other cases regional lymph node metastases were seen.96 No deaths from clear cell papillary renal cell carcinoma associated with end-stage renal disease have been reported.
New and emerging entities
Follicular Renal Carcinoma
In 2004, four cases of a novel form of renal parenchymal neoplasia, with a follicular architecture resembling follicular carcinoma of the thyroid, were reported.102 A further case was described in 2006103 and this was followed in 2009 by a more detailed study in which the original series was expanded by the addition of two further cases.104
The seven tumors reported to date were from four female patients and three male patients with an age range of 29 to 83 years (median 45 years). All tumors were incidental findings and two patients had a past history of unrelated malignancies. The tumors were tan colored and ranged in size from 1.9 to 11.8 cm (median 3.0 cm). There was no evidence of extra-renal extension on macroscopic examination.
Microscopically the tumors contain a prominent pseudocapsule and are composed of cells showing low-grade pleomorphism with amphophilic to eosinophilic cytoplasm, forming micro and macrofollicles (Figure 29). Colloid-like proteinaceous fluid is present in >50% of follicles in each of the tumors. No areas of papillary architecture or clear cells have been reported. Pseudoinclusions and nuclear grooves may be present (Figure 30).

Follicular renal carcinoma. This low-power photomicrograph shows tightly packed follicles, some of which contain bright eosinophilic secretion.

Follicular renal carcinoma. Note tightly packed follicles lined by cells showing features resembling thyroid cancer. Prominent nuclear pseudoinclusions and some nuclear grooves are seen.
Of the reported cases, one showed positive immunoexpression for cytokeratin 7 and for CD10,103 whereas the remainder of cases were CD10 negative. Six of the cases were negative for RCC, WT1, vimentin, Ksp-cadherin, Pax 2, AMACR, CD56 and CD57, whereas all cases were negative for TTF1. One case showed gains of chromosome 8q24, 12 and 16, and loss of 1p36.3 and 9q21.33,103 whereas gene expression profiling showed widespread underexpression or overexpression, particularly involving chromosomes 1, 2, 3, 5, 6, 10, 11, 16 and 17.104 Follow-up data were available for all seven of the cases and all patients remained tumor free after 6–84 months.
The main differential diagnosis for these tumors is metastases from either a primary thyroid follicular carcinoma or thyroid carcinoma arising in a teratoma. In view of this, evaluation of TTF1 expression should always be undertaken in tumors showing this morphology to exclude metastatic disease.
Clear Cell Papillary and Cystic Renal cell carcinoma
Although clear cell papillary renal cell carcinoma is one of the forms of renal cell carcinoma associated with end-stage renal disease,96 this morphotype has also been described in otherwise normal kidneys.105
There were three male patients and one female patient in this small reported series, with ages of patients ranging from 55 to 64 years. The tumors were 1.6–5 cm in maximum diameter with a thin pseudocapsule. Histologically clear cell papillary structures were associated with cysts lined by epithelial cells with clear cytoplasm. The papillae had prominent fibro-vascular cores and were covered with a single layer of clear cells with predominantly apical nuclei, exhibiting low-grade nuclear pleomorphism (Figure 31). Tumor cells showed immunoexpression of CA-IX and cytokeratin 7. In one case focal expression of CD10 was seen, while AMACR and TFE3 were negative. FISH analysis showed gains of chromosome 17 in two tumors.

Clear cell papillary and cystic carcinoma. Note delicate papillary structures lined by cells with mostly clear cytoplasm.
In all cases the tumors were confined to the kidney at diagnosis and no recurrence or metastases were reported in follow-up, which ranged from 1 to 48 months.
The principal differential diagnosis for these tumors in TFE3 translocation carcinomas with a prominent papillary pattern. Translocation carcinomas often require confirmation of TFE3 immunoexpression for diagnosis. Translocation carcinoma may also be differentiated by the absence of epithelial-lined cystic spaces surrounding papillary structures and the frequent presence of psammoma-like calcification.
In view of the constant occurrence of the papillary structures within cysts lined by clear cells in these tumors, the term clear cell papillary and cystic renal cell carcinoma is preferred to that of clear cell papillary renal cell carcinoma.
Oncocytic Papillary Renal Cell Carcinoma
Although the great majority of papillary renal cell carcinomas are classified as type 1 or type 2, principally on the absence or presence of nuclear pseudostratification and cytoplasmic eosinophilia,106, 107 a third variant, designated oncocytic papillary renal cell carcinoma was first reported in 2006. These tumors are characterized by the presence of a papillary architecture, with tumor cells exhibiting a voluminous and intensely eosinophilic cytoplasm.108
In series published to date,108, 109, 110, 111 there is a strong gender association with 87% of cases occurring in male patients. Age at diagnosis is similar to that for other forms of adult renal cell carcinoma with patient ages ranging from 40 to 80 years (mean 65 years). Reflecting the current trend for renal neoplasms to be diagnosed at an early stage through imaging studies, oncocytic papillary renal cell carcinomas reported to date ranged in size from 0.8 to 27 cm at diagnosis (mean 4.9, median 3 cm). Macroscopically the tumors are well demarcated with a cut surface of varying shades of brown. Intratumoral hemorrhage is a relatively frequent finding, even in small tumors.
Histologically the tumors are composed of papillae and trabeculae, with fibro-vascular cores covered by tumor cells with eosinophilic cytoplasm exhibiting a low nuclear/cytoplasmic ratio (Figure 32). In the majority of cases the nuclei are round although in some tumors nuclei show varying degrees of pleomorphism. The distribution of nuclear grades from published series to date are grade 1; 17 cases, grade 2; 18 cases and grade 3; 11 cases,108, 109, 110, 111 with grading apparently being based upon the degree of nucleolar prominence. Ultrastructure studies show the tumor cytoplasm to be packed with mitochondria with lamella cristae.109, 111 The position of the nuclei within the cytoplasm of the neoplastic cells appears to be somewhat variable in these tumors. In the majority of reported cases there was no evidence of pseudostratification, with nuclei predominantly having a luminal rather than basal distribution.108, 110 In other reported cases pseudostratification of nuclei was occasionally observed.111, 112

Oncocytic papillary renal carcinoma. Note papillae lined by eosinophilic cells resembling oncocytes.
In some cases aggregates and individual foamy macrophages were frequently seen, whereas tumor necrosis appears to be a common feature.108, 111 Occasionally psammoma bodies have also been identified.110, 111
Although these tumors commonly have a papillary architecture, solid variants have been reported (Figure 33).112 In these cases the identification of the tumors as papillary renal cell carcinoma was based upon the presence of foamy tumor cells (Figure 34), abortive papillary structures and necrosis, and immunoexpression of neoplastic cells.

Oncocytic papillary renal carcinoma. Note solid growth pattern of cells with oncocyte-like cytoplasm.

Oncocytic papillary renal carcinoma. Note foam cells in papillary cores. Tumor cells are large with abundant granular cytoplasm-resembling oncocytes.
Immunohistochemical expression of oncocytic papillary renal cell carcinoma varied somewhat between series,108, 109, 110, 111 with CD10 and AMACR showing strong diffuse cytoplasmic staining in virtually all cases. Expression of cytokeratin 7, cytokeratin 19, e-cadherin, RCC antigen and vimentin was variable, with positive cytoplasmic staining observed in 50, 65, 50, 50 and 60% of cases respectively. EMA was either negative or only weakly positive.
The genetics of these tumors appears to be similar to those of typical papillary renal cell carcinoma with trisomy 7, (13 of 19 cases), trisomy 17 (14 of 19 cases) and loss of Y (5 of 14 cases) being shown on FISH.110, 111 Karyotyping to date is limited to four cases and three of these showed loss of chromosomes 1, 14 and Y, whereas in one case trisomy 3 was present.108 In a single case CGH showed partial loss of the long arm of chromosome 11.108
The main differential diagnosis for these tumors is oncocytoma and chromophobe renal carcinoma. Although papillary architecture is impermissible in oncocytoma, oncocytic papillary renal cell carcinoma with solid architecture can resemble oncocytoma. The presence of foamy macrophages, foamy tumor cells and extensive microscopic necrosis favors oncocytic papillary renal cell carcinoma, as does positive immunoexpression for AMACR, CD10, RCC, cytokeratin 7 and vimentin. Eosinophilic chromophobe renal carcinoma usually contains foci of classic chromophobe cells, albeit focally. Immunohistochemical staining may provide further diagnostic evidence as AMACR and vimentin are usually negative, whereas EMA is frequently positive in chromophobe renal carcinoma.
There may be some overlap between oncocytic papillary renal cell carcinoma and type 2 papillary renal cell carcinoma. In those cases where there is no evidence of pseudostratification then type 2 papillary renal cell carcionoma can be excluded. In those cases where this is present, diagnosis relies on assessment of the cytoplasmic volume of tumor cells, although in reality it may be that these two tumor types are closely related morphotypes of papillary renal cell carcinoma.
Assessment of outcome for oncocytic papillary renal cell carcinoma is, to date, limited. Virtually all tumors in published series were organ confined at diagnosis108, 110, 111 (pT1a, 16 cases; pT1b, 8 cases; pT2, 3 cases; pT3-2). One of the pT3 cases was staged on the basis of spread of a co-existing clear cell renal cell carcinoma. Outcome data were available for 29 cases with follow-up ranging from 3.5 to 144 months. One pT2 tumor recurred 2 years post-nephrectomy and the patient died 2 years later. For all other cases the patient was either alive without evidence of recurrence or had died of unrelated causes.
Leiomyomatous Renal cell carcinoma
In 2006, five cases of renal cell carcinoma described as having angioleiomyoma-like stroma were described.113 Four similar cases had been previously reported in which angioleiomyoma-like stroma was admixed with clear cells that were interpreted as being either renal cell carcinoma or benign.114, 115, 116, 117 Since the publication of the original series two further reports have detailed the features of a further six cases.118, 119
Clinical details of these tumors were available for 12 cases. There were eight female and four male patients with ages ranging from 18 to 93 years (mean 54 years). In nine of the cases there was no significant past medical history. Two of the patients had co-existing cancer (breast carcinoma and papillary renal cell carcinoma) and one patient had tuberous sclerosis. In the majority of cases the tumors were incidental findings, although three patients presented with hematuria.
Grossly the tumors measured 1.8–14 cm (mean 4.6 cm) and were variously described as tan, brown, yellow or white with the frequent presence of a thick investing capsule. Microscopically the tumors are composed of nests, cords and sheets of epithelial cells (Figure 35) frequently forming solid areas, tubules or papillary structures. There is minimal nuclear pleomorphism with abundant clear cytoplasm. The stroma has the appearance of mature smooth muscle (Figure 36) sometimes with scattered, often dilated vascular spaces. The stromal component of the tumor is often pronounced at the periphery and in some cases appears to extend into adjacent renal tissue or into perirenal fibrofat.

Leiomyomatous renal cell carcinoma. Nests, cords and tubules lined by clear cells are embedded in background spindle cells.

Leiomyomatous renal carcinoma. Note nests and tubules lined by clear cells embedded in smooth muscle.
The epithelial component of the tumor showed positive immunoexpression of cytokeratins AE1/AE3, 7 and CAM 5.2, CD-10, S-100 protein (focal), EMA and vimentin. There was variable expression of 34βE12, whereas smooth muscle actin and HMB45 was negative. The stroma component was positive for α smooth muscle actin (Figure 37), caldesmon, desmin, vimentin and negative for HMB45, CD117, cytokeratins, EMA, ER and PR. Ultrastructural studies are limited to two cases and these showed the epithelial component to have features similar to clear cell renal cell carcinoma, whereas the stroma was typical of smooth muscle cells.

Leiomyomatous renal carcinoma. The smooth muscle cells are highlighted by the smooth muscle actin stain.
Genetic studies on these tumors are contradictory.118, 119 In three cases FISH showed loss of VHL and FHIT, with loss of chromosome 3 in one case and 3p in another. In a separate study there was no evidence of 3p deletion in the three cases examined.
The differential diagnosis for these tumors is clear cell renal cell carcinoma, angiomyolipoma with co-existing clear cell renal cell carcinoma and sarcomatoid renal cell carcinoma, although the presence of a pronounced smooth muscle stroma should present few diagnostic difficulties. Unlike clear cell renal cell carcinoma, the epithelial components of these tumors are positive for cytokeratin 7 and 34βE12. The stromal component of leiomyomatous renal cell carcinoma has bland cytological features unlike that of sarcomatoid renal cell carcinoma and further there is no merging of the epithelial and stromal components.
Outcome studies for leiomyomatous renal cell carcinoma are limited. Of cases for which details are available, four were pT1a and one pT1b at diagnosis. No evidence of recurrence or metastases was seen in these and two other cases followed from 6 months to 5 years (mean 3 years).
References
Delahunt B, Eble JN . History of the development of the classification of renal cell neoplasia. Clin Lab Med 2005;25:231–246.
Kovacs G, Akhtar M, Beckwith JB, et al. The Heidelberg classification of renal cell tumours. J Pathol 1997;183:131–133.
Störkel S, Eble JN, Adlakha K, et al. Classification of renal carcinoma. Cancer 1997;80:987–989.
Eble JN, Sauter G, Epstein JI, et al. (eds). World Heatlh Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press: Lyon, 2004.
Foot NC, Papanicolaou GN . Early renal carcinoma in situ detected by means of smears of fixed urinary sediment. JAMA 1949;139:356–358.
Fleming S, Lewi HJE . Collecting duct carcinoma of the kidney. Histopathology 1986;10:1131–1141.
Kennedy SM, Meriino MJ, Linehan WM, et al. Collecting duct carcinoma of the kidney. Hum Pathol 1990;21:449–456.
Rumpelt HJ, Störkel S, Moll R, et al. Bellini duct carcinoma: further evidence for this rare variant of renal cell carcinoma. Histopathology 1991;18:115–122.
Carter MD, Tha S, McLoughlin MG, et al. Collecting duct carcinoma of the kidney: a case report and review of the literature. J Urol 1992;147:1096–1198.
Füzesi L, Cober M, Mittermayer C . Collecting duct carcinoma: cytogenetic characterization. Histopathology 1992;21:155–160.
Bielsa O, Arango O, Corominas JM, et al. Collecting duct carcinoma of the kidney. Br J Urol 1994;74:127–128.
12 Cavazzana AO, Prayer-Galetti T, Tirabosco R, et al. Bellini duct carcinoma, a clinical and in vitro study. Eur Urol 1996;30:340–344.
Delahunt B, Eble JN . Renal cell neoplasia. Pathology 2002;34:13–20.
Srigley JR, Eble JN . Collecting Duct Carcinoma of Kidney. Semin Diagn Pathol 1998;15:54–67.
Halenda G, Sees Jr JN, Belis JA, et al. Atypical renal adenocarcinoma with features suggesting collecting duct origin and mimicking a mucinous adenocarcinoma. Urology 1998;41:165–168.
Dimopoulos MA, Logothetis CJ, Markowitz A, et al. Collecting duct carcinoma of the kidney. Br J Urol 1993;71:388–391.
Kobayashi N, Matsuzaki O, Shirai S, et al. Colelcting duct carcinoma of the kidneys: an immunohistochemical evaluation of the use of antibodies for differential diagnosis. Hum Pathol 2008;39:1350–1359.
Schoenberg M, Cairns P, Brooks JD, et al. Frequent loss of chromosome arms 8p and 13q in collecting duct carcinoma (CDC) of the kidney. Genes Chromosomes Cancer 1995;12:76–80.
Orsola A, Trias I, Raventós CX, et al. Renal collecting (Bellini) duct carcinoma displays similar characteristics to upper tract urothelial cell carcinoma. J Urol 2005;65:49–54.
Gupta R, Balzar B, Picken M, et al. Diagnostic implications of transcription factor Pax 2 protein and transmembrane enzyme complex carbonic anhydrase IX immunoreactivity in adult renal epithelial neoplasms. Am J Surg Pathol 2009;33:241–247.
Tokuda N, Naito S, Matsuzaki O, et al. Collecting duct (Bellini duct) renal cell carcinoma in Japan: a nationwide survey in Japan. J Urol 2006;176:40–43.
Davis CJ, Mostofi FK, Sesterhenn IA . Renal medullary carcinoma: the seventh sickle cell nephropathy. Am J Surg Pathol 1995;19:1–11.
Berman LB . Sickle cell nephropathy. JAMA 1974;228:1279.
Swartz MA, Karth J, Schneider DT, et al. Renal medullary carcinoma: clinical, pathologic, immunohistochemical and genetic analysis with pathogenetic implications. Urology 2002;60:1083–1089.
Yang XJ, Sugimura J, Tretiakova MS, et al. Gene expression profiling of renal medullary carcinoma: potential clinical relevance. Cancer 2004;100:976–985.
Simpson L, He X, Pins M, et al. Renal medullary carcinoma and ABL gene amplification. J Urol 2005;173:1883–1888.
Sathyamoorthy K, Teo A, Atallah M . Renal medullary carcinoma in a patient with sickle-cell disease. Nature Clin Pract Urol 2006;3:279–283.
Leitao VA, da Silva W, Ferreira U, et al. Renal medullary carcinoma. Case report and review of the literature. Urol Int 2006;77:184–186.
Baig MA, Lin Y-S, Rasheed J, et al. Renal medullary carcinoma. J Natl Med Assoc 2006;98:1171–1174.
Hakimi AA, Koi PT, Milhoua PM, et al. Renal medullary carcinoma: the Bronx experience. Urology 2007;70:878–882.
Watanabe IC, Billis A, Guimaraes MS, et al. Renal medullary carcinoma: report of seven cases from Brazil. Mod Pathol 2007;20:914–920.
Cheng JX, Tretiakova M, Gong C, et al. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol 2008;21:647–652.
Wartchow EP, Trost BA, Tucker JA, Albano EA, Mierau GW . Renal medullary carcinoma: ultrastructural studies may benefit diagnosis. Ultrastruct Pathol 2008;32:252–256.
Stahlschmidt J, Cullinane C, Roberts P, Picton SV . Renal medullary carcinoma: prolonged remission with chemotherapy, immunohistochemical chararcterisation and evidence of bcr/abl rearrangement. Med Pediatr Oncol 1999;33:551–557.
Weber A, Srigley J, Moch H . Muzinöses, tubuläres und spindelzelliges nierenkarzinom. Pathologie 2003;24:453–459.
MacLennan GT, Farrow GM, Bostwick DG . Low-grade collecting duct carcinoma of the kidney: report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology 1997;50:679–684.
Srigley JR, Eble JN, Grignon DJ, Hartwick RWJ . Unusual renal cell carcinoma (RCC) with prominent spindle cell change possibly related to the loop of Henle. Mod Pathol 1999;12:107A.
Srigley JR, Kapusta L, Reuter V, et al. Phenotypic, molecular and ultrastructural studies of a novel low grade renal epithelial neoplasm possibly related to the loop of Henle. Mod Pathol 2002;15:182A.
Parwani AV, Husain AN, Epstein JI, Beckwith JB, Argani P . Low-grade myxoid renal epithelial neoplasms with distal nephron differentiation. Hum Pathol 2001;32:506–512.
Rakozy C, Schmahl GE, Bogner S, Störkel S . Low-grade tubular-mucinous renal neoplasms: morphologic, immunohistochemical and genetic features. Mod Pathol 2002;15:1162–1171.
Hes O, Hora M, Perez-Montiel DM, et al. Spindle and cuboidal renal cell carcinoma, a tumour having frequent association with nephrolithiasis:report of 11 cases including a case with hybrid conventional renal cell carcinoma/spindle and cuboidal renal cell carcinoma components. Histopathology 2002;41:549–555.
Ferlicot S, Allory Y, Compérat E, et al. Mucinous tubular and spindle cell carcinoma: a report of 15 cases and a review of the literature. Virchows Arch 2005;447:978–983.
Shen SS, Ro JY, Tamboli P, et al. Mucinous tubular and spindle cell carcinoma of kidney is probably a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol 2007;11:13–21.
Fine SW, Argani P, DeMarzo AM, et al. Expanding the histologic spectrum of mucinous tubular and spindle cell carcinoma of the kidney. Am J Surg Pathol 2006;30:1554–1560.
Kuroda N, Nakamura S, Miyazaki E, et al. Low-grade tubular-mucinous renal neoplasm with neuroendocrine differentiation: A histological, immunohistochemical and ultrastructural study. Pathol Int 2004;54:201–207.
Dhillon J, Amin MB, Selbs E, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid change. Am J Surg Pathol 2009;33:44–49.
Pillay N, Ramdial PK, Cooper K, Batuule D . Mucinous tubular and spindle cell carcinoma with aggressive histomorphology-a sarcomatoid variant. Hum Pathol 2008;39:966–969.
Simon RA, di Sant′agnese PA, Palapattu GS, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid differentiation. Int J Clin Exp Pathol 2008;1:180–1844.
Kuehn A, Paner GP, Skinnider BF, et al. Expression analysis of kidney-specific cadherin in a wide spectrum of traditional and newly recognized renal epithelial neoplasms: diagnostic and histogenetic implications. Am J Surg Pathol 2007;31:1528–1533.
Paner GP, Srigley JR, Radhakrishnan A, et al. Immunohistochemical analysis of mucinous tubular and spindle cell carcinoma and papillary renal cell carcinoma of the kidney. Am J Surg Pathol 2006;30:13–19.
Brandal P, Lie AK, Bassarova A, et al. Genomic aberrations in mucinous tubular and spindle cell renal cell carcinomas. Mod Pathol 2005;19:186–194.
Cossu-Rocca P, Eble JN, Delahunt B, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006;19:488–493.
Srigley JR . Mucinous tubular and spindle cell carcinoma WHO classification of tumors In: Eble JN, Sauter G, Epstein JI, Sesterhenn IA (eds). Tumors of the Genito-urinary and Male Genital Organs. IARC Press: IARC, Washington DC, 2004.
Argani P, Netto GJ, Parwani AV . Papillary renal cell carcinoma with low-grade spindle cell foci. A mimic of mucinus tubular and spindle cell carcinoma. Am J Surg Pathol 2008;32:1353–1359.
Masson P . Tumeurs Humaines 1955. Human Tumors, Histology, Diagnosis and Technique. Wayne State University Press: Detroit, 1970.
Murphy W, Beckwith JB, Farrow GM . Tumors of the Kidney and Bladder. Armed Forces Institute of Pathology: Washington, DC, 1994.
MacLennan GT, Bostwick DG . Tubulocystic carcinoma, mucinous tubular and spindle cell carcinoma, and other recently described rare renal tumors. Clin Lab Med 2005;25:393–416.
Azoulay S, Vieillefond A, Paraf F, et al. Tubulocystic carcinoma of the kidney: a new entity among renal tumors. Virchows Arch 2007;451:905–909.
Yang XJ, Zhou M, Ondrej H, et al. Tubulocystic carcinoma of the kidney. Clinicopathologic and molecular characterization. Am J Surg Pathol 2008;32:177–187.
Amin MB, MacLennan GT, Gupta R, et al. Tubulocystic carcinoma of the kidney clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am J Surg Pathol 2009;33:384–392.
Osunkoya AO, Young AN, Wang W, Netto GJ, Epstein JI . Comparison of gene expression profiles in tubulocystic carcinoma and collecting duct carcinoma of the kidney. Am J Surg Pathol 2009, e-pub ahead of print.
Zhou M, Yang Ximing, Lopez JI, et al. Renal tubulocystic carcinoma is closely related to papillary renal cell carcinoma: implications for pathologic classification. Am J Surg Pathol 2009, e-pub ahead of print.
Eble JN, Bonsib SM . Extensively cystic renal neoplasms: cystic nephroma, cystic partially differential nephroblastoma, multi-locular cyst renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol 1998;15:2–20.
Adsay NV, Eble JN, Srigley JR, et al. Mixed epithelial stromal tumor of the kidney. Am J Surg Pathol 2000;24:958–970.
Leroy X, Aubert S, Lemaitre L, et al. Multilocular cystic renal oncocytoma. J Clin Pathol 2006;59:223–224.
Argani P, Ladanyi M . Recent advances in pediatric renal neoplasia. Adv Anat Pathol 2003;10:243–260.
Argani P, Ladanyi M . Translocation carcinoma of the kidney. Clin Lab Med 2005;25:363–378.
Bruder E, Passera O, Harms D, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol 2004;28:1117–1132.
Camparo P, Vasiliu V, Molinie V, et al. Renal translocation carcinomas—clinicopathologic, immunohistochemical and gene expression profiling analysis of 31 cases with a review of the literature. Am J Surg Pathol 2008;35:656–670.
Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane-production, focal HMB45 immunoreactivity, and t(6;11) (p21.1;q12) chromosome translocation. Am J Pathol 2001;158:2089–2096.
Argani P, Lal P, Hutchinson B, et al. Aberrant nuclear immunoreactivity for TFE3 in neoplasms with TFE3 gene fusions: a sensitive and specific immunohistochemical assay. Am J Surg Pathol 2003;27:750–761.
Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasm with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma. A distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol 2001;159:179–192.
Argani P, Antonescu CR, Couturier J, et al. PRCC-TFE3 renal carcinomas. Morphologic, immunohistochemical, ultrastructural and molecular analysis of an entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol 2002;26:1553–1566.
Argani P, Ladanyi M . Distinctive neoplasms characterised by specific chromosomal translocations comprise a significant proportion of pediatric renal cell carcinomas. Pathology 2003;35:492–498.
Geller JI, Argani P, Adeniran A, et al. Translocation renal cell carcinoma lack of negative impact due to lymph node spread. Cancer 2008;112:1607–1616.
Dal Cin P, Stas M, Sciot R, et al. Translocation (X:1) reveals metastasis 31 years after renal cell carcinoma. Cancer Genet Cytogenet 1998;101:58–61.
Eble JN . Mucinous tubular and spindle cell carcinoma and post-neuroblastoma carcinoma: newly recognised entities in the renal cell carcinoma family. Pathology 2003;35:499–504.
Fleitz JM, Wootton-Gorges SL, Wyatt-Ashmead J, et al. Renal cell carcinoma in long-term survivors of advanced stage neuroblastoma in early childhood. Pediatr Radiol 2003;33:540–545.
Bassal M, Mertens AC, Taylor L, et al. Risk of selected subsequent carcinomas in survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 2006;24:476–483.
Medeiros LJ, Palmedo G, Krigman HR, et al. Oncocytoid renal cell carcinoma after neuroblastoma: a report of four cases of a distinct clinicopathologic entity. Am J Surg Pathol 1999;23:772–780.
Koyle MA, Hatch DA, Furness PD, et al. Long-term urological complications in survivors younger than 15 months of advanced stage abdominal neuroblastoma. J Urol 2001;166:1455–1458.
Dhall D, Al-Ahmadie HA, Dhall G, Shen-Schwarz S, Tickoo SK . Pediatric renal cell carcinoma with oncocytoid features occurring in a child after chemotherapy for cardiac leiomyomasarcoma. Urology 2007;70:178e13–178e15.
Tosaka A, Ohya R, Vamada K, et al. Incidence and properties of renal masses and asymptomatic renal cell carcinoma detected by abdominal ultrasonography. J Urol 1990;144:1097–1099.
Gehrig JJ, Gottheiner TI, Swenson RS . Acquired cystic disease of the end-stage kidney. Am J Med 1985;79:609–620.
Farivar-Mohseni H, Perlmutter AE, Wilson S, et al. Renal cell carcinoma and end stage renal disease. J Urol 2006;175:2018–2021.
Stewart JH, Buccianti G, Agodoa L, et al. Cancers of the kidney and urinary tract in patients on dialysis for end stage renal disease: analysis of data from the United States, Europe, and Australia and New Zealand. J Am Soc Nephrol 2003;14:197–202.
Irie K, Ishizuka O, Yokoyama H, Kinebuchi Y, Satoh T, Nishizawa O . Resection of local recurrence of renal cell carcinoma in a hemodialysis patient 6 years after radical nephrectomy. Int J Urol 2004;11:550–552.
Onishi T, Igarashi T, Tobe T, Ichikawa T . A case report of chromophobe cell renal carcinoma arising from atrophic kidney during long-term haemodialysis. Acta Urol Japon 2006;52:919–921.
Ianhez LE, Lucon M, Nahas WC, et al. Renal cell carcinoma in renal transplant patients. Urology 2007;69:462–464.
Sangoi AR, Higgins JP . Bilateral mixed epithelial stroma tumor in an end-stage renal disease patient: the first case report. Hum Pathol 2008;39:142–146.
Hamamoto S, Okamura T, Mizunok, et al. Renal oncocytoma with bilateral synchronous renal cell carcinoma in a patient undergoing long term dialysis. Int J Urol 2008;15:87–89.
Nishihara K, Shomori K, Nagashima Y, et al. Chromophobe renal cell carcinoma and ‘capsulomas’ with acquired cystic disease of the kidney in a long-term hemodialysis patient. Int J Urol 2008;15:543–545.
Sakura M, Masuda H, Saitok, et al. Collecting duct carcinoma with acquired cystic disease of the kidney in a long-term hemodialysis patient. Int J Urol 2008;15:93–94.
Martin M, De Castro Olmedo C, Torrecilla Garcia-Ripoli JR, et al. Low grade collecting duct carcinoma of the kidney in a patient treated by dialysis. Acta Urol Espanol 2008;32:559–562.
Hora M, Hes O, Reischig T, et al. Tumors in end-stage kidneys. Transplant Proc 2008;40:3354–3358.
Tickoo SK, de Peralta-Venturina MN, Harik LR, et al. Spectrum of epithelial disease. An experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 2006;30:141–153.
Kuroda N, Tamura M, Taguchi T, et al. Sarcomatoid acquired cystic disesase-associated renal cell carcinoma. Histol Histopathol 2008;23:1327–1331.
Yoshida M, Yao M, Ishikawa I, et al. Somatic von Hippel-Lindau disease gene mutation in clear cell renal carcinomas associated with end-stage renal disease/acquired cystic diseases of the kidney. Genes Chromosomes Cancer 2002;35:359–364.
Inone H, Nonomura N, Kojima Y, et al. Somatic mutations of the von Hippel-Lindau disease gene in renal carcinomas occurring in patients with long-term dialysis. Nephrol Dial Transplant 2007;22:2052–2055.
Cheuk W, Lo ES, Chan AK, Chan JK . Atypical epithelial proliferations in acquired renal cystic disease harbor cytogenetic alterations. Hum Pathol 2002;33:761–765.
Cossu-Rocca P, Eble JN, Zhang S, et al. Acquired cystic disease—associated renal tumors: an immunohistochemical and fluorescence in situ hybridization study. Mod Pathol 2006;19:780–787.
Amin MB, Michal M, Radhakrishnan A, et al. Primary thyroid-like follicular carcinoma of the kidney: a histologically distinctive primary renal epithelial tumor. Mod Pathol 2004;17:137A.
Jung SJ, Chung JI, Park SH, et al. Thyroid follicular carcinoma-like tumor of kidney: a cases report with morphologic, immunohistochemical and genetic analysis. Am J Surg Pathol 2006;30:411–415.
Amin MB, Gupta R, Ondrej H, et al. Primary thyroid-like follicular carcinoma of the kidney. Report of 6 cases of a histologically distinctive adult renal epithelial neoplasm. Am J Surg Pathol 2009;33:393–400.
Gobbo S, Eble JN, Grignon DJ, et al. Clear cell papillary renal cell carcinoma. A distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008;32:1239–1245.
Delahunt B, Eble JN . Papillary renal cell carcinoma in Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs In: Eble JN, Sauter G, Epstein JI and Sesterhenn IA (eds). World Health Organization Classification of Tumours. IARC: Lyon, 2004, pp 27–29.
Delahunt B, Eble JN . Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997;10:537–544.
Lefevre M, Couturiere J, Sibony M, et al. Adult papillary renal tumor with oncocytic cells: clincopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol 2005;29:1576–1581.
Masuzawa N, Kishimoto M, Nishimura A, Shichiri Y, Yanagisawa A . Oncocytic renal cell carcinoma having papillotubular growth: rare morphological variant of papillary renal cell carcinoma. Path Int 2008;58:300–305.
Lakshmi P, Kunju, Wojno K, et al. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum Pathol 2008;39:96–101.
Hes O, Burnelli M, Michal M, et al. Oncocytic papillary renal cell carcinoma: a clinicopathologic immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann Diagn Pathol 2006;10:133–139.
Mai KT, Kohler DM, Roberston SJ, Belanger EC, Marginean EC . Oncocytic papillary renal cell carcinoma with solid architecture: mimic of renal oncocytoma. Path Int 2008;58:164–168.
Kuhn E, De Anda J, Manoni S, Netto G, Rosai J . Renal cell carcinoma associated with prominent angioleiomyoma-like proliferation. Report of 5 cases and review of the literature. Am J Surg Pathol 2006;30:1372–1381.
Honey RJ, Honey RM . Tuberous sclerosis and bilateral renal carcinoma. Br J Urol 1977;49:441–446.
Govaerts JJ, van Gooswilligen JC, Vooys GP, et al. Renal hamartoma associated with renal cell (Grawitz) tumor: another indication that Grawitz tumors are carcinosarcomas. Eur Urol 1987;13:276–280.
Canzonieri V, Volpe R, Gloghini A, et al. Mixed renal tumor with carcinomatous and fibroleiomyomatous components, associated with angiomyolipoma in the same kidney. Pathol Res Pract 1993;189:951–959.
Michal M, Hes O, Havlicek F . Benign renal angiomyoadenomatous tumor: a previously unreported renal tumor. Ann Diagn Pathol 2000;4:311–315.
Brunelli M, Menestrina F, Segala D, et al. Renal cell carcinoma associated with prominent leiomyomatous proliferation appears not to be a variant of clear cell renal cell carcinoma. Mod Pathol 2009;22:160A–161A.
Shannon BA, Cohen RG, Segal A, Baker EG, Murch AR . Clear cell renal cell carcinoma with smooth muscle stroma. Hum Pathol 2009;40:425–429.
Acknowledgements
We are grateful to Dr Glenn Taylor, Dr Mahul B Amin, Dr Hemamali Samaratunga, Dr Liang Cheng and Dr Guido Martignoni for the provision of illustrations or tissues for photographic purposes.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Srigley, J., Delahunt, B. Uncommon and recently described renal carcinomas. Mod Pathol 22 (Suppl 2), S2–S23 (2009). https://doi.org/10.1038/modpathol.2009.70
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2009.70
Keywords
This article is cited by
-
Uncommon malignant renal tumors and atypical presentation of common ones: a guide for radiologists
Abdominal Radiology (2019)
-
WHO/ISUP classification, grading and pathological staging of renal cell carcinoma: standards and controversies
World Journal of Urology (2018)
-
Distribution of Vascular Patterns in Different Subtypes of Renal Cell Carcinoma. A Morphometric Study in Two Distinct Types of Blood Vessels
Pathology & Oncology Research (2018)
-
Leiomyomatöses Nierenzellkarzinom
Der Urologe (2016)
-
Tubulocystic renal cell carcinoma: a new radiological entity
European Radiology (2016)
