
Abstract
Adenosquamous carcinoma of the pancreas is one of the most aggressive forms of pancreatic cancer. Molecular characterizations of this rare tumor subtype are sparse. Understanding the common molecular and pathologic features of pancreatic adenosquamous carcinomas could provide critical information for identifying therapeutic targets. Herein, we analyzed the pathologic and molecular features of our series of eight pancreatic adenosquamous carcinomas. We found KRAS2 gene mutations at codon 12 in all eight cases. All the cases showed loss of p16 protein. In three of these cases the loss was attributed to an exon 2 homozygous deletion in the p16/CDKN2a gene. The majority of the cases had loss of Dpc4 protein and strong nuclear p53 positivity, similar to the molecular signature found in pancreatic ductal adenocarcinoma. We found that E-cadherin was either lost or reduced in all cases and that epidermal growth factor receptor was overexpressed in all cases. The squamous component was positive for p63 staining and thus p63 labeling was helpful in identifying squamous differentiation in adenosquamous carcinomas with an acantholytic growth pattern. In summary, although pancreatic adenosquamous carcinoma and ductal adenocarcinoma have overlapping pathologic and molecular characteristics, there are distinct differences that may be helpful in diagnostic and therapeutic strategies.
Similar content being viewed by others

Main
Pancreatic ductal adenocarcinomas is one of the most lethal malignancies as reflected by an incidence rate that approximates its mortality rate.1 The poor prognosis associated with pancreatic adenocarcinoma is primarily due to the advanced stage of disease at the time of clinical presentation and the refractory nature of pancreatic adenocarcinoma to conventional chemotherapy and radiotherapy regimens. A number of histologic variants of pancreatic cancer have been described.2 One of the variants, adenosquamous carcinoma, is characterized by an aggressive course, with higher potential for metastasis and an even worse prognosis than conventional ductal adenocarcinoma.3 Only one large series has described the clinical, pathologic and selective molecular markers of this type of pancreatic cancer.4 Thus, a better understanding of this aggressive form of pancreatic cancer may aid in finding new molecular markers and targets for this disease.
In the current study, we performed KRAS2 mutational and p16/CDKN2a exonic deletion analyses on microdissected tissue from separately microdissected glandular and squamous components of a well-characterized series of adenosquamous carcinomas of the pancreas, to define the function of these genetic alterations in the pathogenesis of this distinctive neoplasm. We also performed immunohistochemical labeling for products of two tumor suppressor genes frequently mutated in pancreatic adenocarcinoma, TP53 and DPC4/MAD4, to evaluate their status in adenosquamous carcinomas. Expression levels of E-cadherin, a cell adhesion molecule often lost or reduced in undifferentiated pancreatic ductal adenocarcinoma,5 and epidermal growth factor receptor (EGFR), a target of a recently approved small molecule inhibitor for treatment of pancreatic adenocarcinoma,6 were both evaluated. We also further evaluated pathologic features of this rare, aggressive tumor.
Materials and methods
Case Selection
Adenosquamous carcinomas were obtained from the pathology files at the Thomas Jefferson University. The study was approved by the institutional review board. Initial diagnostic hematoxylin and eosin slides were reviewed for all cases. At least 30% of the carcinoma had to show squamous differentiation for inclusion in the study. Presence of deeply eosinophilic cytoplasm, well-defined cell borders, intercellular bridges, keratinization and focal or extensive cytoplasmic clearing were considered features of squamous differentiation. In addition to tumor grade and pathologic stage the following histologic parameters were evaluated for each case: type of margin (pushing or infiltrating), percentage of squamous differentiation, presence of necrosis (macroscopic or microscopic), cystic degeneration and squamous metaplasia in nonneoplastic pancreatic ducts. Patient follow-up and outcome information were obtained by chart review and from the tumor registry.
Immunohistochemical Stains
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue using the Envision Plus detection system (Dako Corporation, Carpentaria, CA, USA). The antibodies, sources, dilutions and pretreatment conditions are listed in Table 1. Two of the authors (RHH, AKW) simultaneously evaluated the immunohistochemical labeling of the cases at a multiheaded microscope and agreement between both observers was reached in all cases. Immunolabeling for p53 was interpreted as positive and predictive of TP53 mutations if >75% tumors cell show strong positive staining.7 Expression of E-cadherin was considered intact (normal) when >50% of the neoplastic cells showed membranous labeling. E-cadherin labeling was considered reduced when 10–50% of the cells labeled and, and negative when <10% of the neoplastic cells labeled.8 Loss of Dpc4 labeling was defined as previously described, as weak cytoplasmic expression and little to no expression in the nuclear compartments of all neoplastic cells.9 Nuclear expression of p63 present in >10% of the neoplastic cells was considered positive. Cases were considered positive for EGFR if complete membrane labeling was detected in >5% of the neoplastic cells.10
KRAS2 Mutational Analysis and p16/CDKN2a Amplification
Paraffin sections (7 μm) were cut using a microtome. Sections on PENfoil slides were deparaffinized in xylol for 2 min. A descending series of ethanol was used: 100% twice, 30 s; 95% twice, 30 s; 70%, 10 s; distilled water, 10 s. Slides were stained with 1% methyl green for 5 s, washed in distilled water and air-dried. Microdissection was performed using a Leica LMD system as previously described.11
Genomic DNA was isolated from tumor tissue obtained through laser capture microdissection using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA).
KRAS2 mutational analysis
PCR amplification of the KRAS2 region was carried out using specific primer sets (forward 5′-AAAGGTACTGGTGGAGTATTTG-3′ and reverse 5′-CTATTGTTGGATCATATTCG-3′). PCR reactions were carried out in 25 μl reactions using 2 μl of DNA, 0.5 units per μl of Taq polymerase (USB, Cleveland, OH, USA), 2.5 μl of 10 × PCR buffer (USB) and 0.5 μl 10 mM dNTP Mix (Invitrogen). Conditions were set for 40 cycles at (94°C for 2.5 h, 94°C for 30 s, 50°C for 30 s, 72°C for 1 min, followed by an extension of 5 min at 72°C). Sequencing reactions included PCR-purified products using DNA purification columns (Qiagen) and the above forward KRAS2 primer. Sequencing was performed by the Thomas Jefferson University, Kimmel Cancer Center DNA core facility using capillary electrophoresis.
Analysis of p16/CDKN2a
The extracted genomic DNA also underwent PCR amplification of the p16/CDKN2a region using specific primer sets (forward 5′-GAAGAAAGAGGAGGGGCTG-3′ and reverse 5′-GCGCTACCTGATTCCAATTC-3′).12 PCR reactions were performed using the same reagents and concentrations as above noted, except the additional use of 2.5 μl of 5 M betaine due to the high GC content of the gene. Conditions were set for 35 cycles at (94°C for 2.5 h, 94°C for 30 s, 56°C for 30 s, 72°C for 1 min, followed by an extension of 5 min at 72°C).
HPV DNA Detection
HPV16 detection in formalin-fixed and paraffin-embedded tissues was performed using the in situ hybridization (ISH)-catalyzed signal amplification method for biotinylated probes (GenPoint; Dako Corporation). Briefly, 5-μm tissue sections underwent deparaffinization, heat-induced target retrieval in citrate buffer and digestion using Proteinase K (Roche Diagnostics, Indianapolis, IN, USA). ISH was performed using formalin-fixed, paraffin-embedded tissue sections. Biotin-labeled HPV probe solutions (Dako Corporation) were applied to individual sections. These included a wide spectrum probe (cocktail of HPV 6, 11, 16, 18, 31, 33, 45 and 51) and separate type specific probes for HPV 16 and HPV 18. Detection of hybridized probe was performed by tyramide-catalyzed signal amplification using the Dako GenPoint kit (Dako Corporation). Chromogenic detection was performed with DAB/H2O2. Controls included tissue sections positive for HPV wide spectrum, the HeLa cell line for HPV 18 and the SiHa cell line for HPV 16. Cases with a discrete punctate reaction product specifically in tumor cell nuclei were interpreted as positive, others were considered negative.
Results
Clinical
Of the eight patients, six were women and two were men. The age at presentation ranged from 55 to 76 years (mean and median 64 years). The mean tumor size was 5.5 cm (range 3.5–11 cm). Five of the carcinomas were located in the pancreatic body and/or tail and three were in the pancreatic head. Clinicopathologic characteristics of the patients are depicted in Table 2. Associated symptoms included abdominal pain (five cases), weight loss (two cases) and obstructive jaundice (two cases). In patient one, the pancreatic mass was discovered incidentally on CT scan performed after a motor vehicle accident. Seven of eight patients died of the disease in 3–12 months after resection with the median survival of 5 months. One patient is alive with recurrent disease 11 months after surgery.
Mutation Analysis
KRAS2 mutational analysis
KRAS2 sequencing of codons 12 and 13 was performed on the genomic DNA extracted from the squamous and adenocarcinomatous components of the eight adenosquamous pancreatic carcinomas. All mutations were heterozygous in nature. Sequences too close to call were confirmed by re-amplification and sequencing, from either the same template or microdissection from the same sample. We detected KRAS2 gene mutations in all eight cases, all at codon 12. KRAS2 gene mutations were consistently detected in both the squamous and adenocarcinomatous portions. One wild-type KRAS2 gene sequence was found in the squamous component. The mutations changed the amino-acid sequence from a glycine to a valine in two cases, a glycine to an asparagine in two cases, a glycine to an aspartic acid in two cases and a glycine to a cysteine in one case (Table 3). The amino-acid changes in the KRAS2 gene were consistent between the two components except for two cases (of the informative cases) (Table 1; Figure 1). Interestingly, we found two unique activating mutations (G>R and G>D) in the same case from different components (Figure 1). In two cases, the small size of the adenocarcinomatous component precluded its microdissection.

KRAS2 gene sequencing at codon 12. (a) Identification of two different KRAS2 activating mutations in the squamous and adeno components in case 7. (b) An example of two identical KRAS2 activating mutations as seen in the majority of cases.
p16/CDKN2a homozygous deletion screen of exon 2
We screened for p16/CDKN2a homozygous deletions based on a previously described assay.12 All genomic DNA templates were validated for integrity by amplification of another gene product (PRSS1 or KRAS2). Each template was amplified for 35–40 cycles for at least three separate PCR reactions and when a homozygous deletion was found up to five separate reactions were performed to validate the deletion. We found loss of exon 2 in both components of one case and in the squamous components of two cases (where no DNA was available from the adenocarcinomatous component). These deletions were confirmed by immunohistochemistry analysis (Tables 3 and 4).
Pathologic Features
Six carcinomas consisted of distinct squamous and glandular components and two tumors had intermingled squamous and mucinous cells. Areas of squamous differentiation ranged between 30 and 90% of the tumor volume. Carcinomas composed predominantly of a squamous component had more circumscribed borders than carcinomas with predominantly glandular differentiation. The squamous component was well differentiated in two cases, moderately differentiated in three cases and poorly differentiated in three cases. Foci of tumor cell necrosis were present in all cases. In five cases, necrosis was extensive and seen on gross examination, whereas in three cases small foci of necrosis were only detected microscopically. Cystic change, corresponding to dilated pancreatic ducts, often lined by malignant squamous epithelium was present in five cases. Extensive clear cell change was noted in two cases and all carcinomas showed perineural invasion.
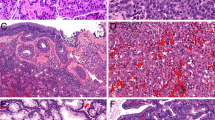
Adjacent pancreatic ducts showed squamous metaplasia in six of eight cases (Figure 2a). In addition, in five cases there was a pagetoid spread of malignant squamous epithelium to adjacent ducts (Figure 2b). Pancreatic intraepithelial neoplasia 1–3 were present in seven of eight cases and in a few cases squamous metaplasia and PanIN coexisted in one duct (Figure 2c).

(a) Normal pancreatic ducts showing squamous metaplasia. (b) Pagetoid spread of malignant squamous epithelium to benign ducts; arrowhead indicates residual normal ductal epithelial cells. (c) Squamous metaplasia and PanIN involving one pancreatic duct; black arrowhead points PanIN and white arrowhead indicates squamous metaplastic cells (original magnification × 200).
Immunohistochemistry
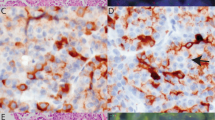
The results of immunohistochemical labeling are summarized in Table 4. The malignant squamous epithelium showed strong, diffuse nuclear p63, labeling in all cases. Normal pancreatic ducts and PanIN lesions did not label for p63 whereas nonneoplastic squamous metaplasia showed nuclear labeling. Positive p63 labeling was helpful in confirming squamous differentiation in adenosquamous carcinomas with acantholytic growth pattern (Figure 3). Nuclear p53 labeling was present in five of eight cases in both glandular and squamous components (Figure 4a). As expected, the benign squamous metaplasia did not label for p53, but p53 labeling did highlight neoplastic squamous epithelium involving pancreatic ducts by pagetoid extension (Figure 4b). Dpc4 expression was lost in the glandular and squamous components in five of the cases with intact labeling present in the desmoplastic stroma and normal pancreas (Figure 5a). All cases showed loss of p16 labeling with intact internal control nuclear p16 staining present in islet cells, nerves and normal pancreatic acini (Figure 5b). Strong, membranous E-cadherin labeling was present in glandular and solid squamous areas in one case, was reduced in four cases and absent in three cases (Figure 5c). In general, single infiltrating neoplastic cells and poorly differentiated areas showed reduced or absent E-cadherin labeling. EGFR immunolabeling was present in 40–100% of the neoplastic cells in the squamous component of all cases, and was present in the neoplastic glands in six of eight cases (Figure 5d).

Immunohistochemical stain for p63 in adenosquamous carcinoma. (a) Clear cell adenosquamous carcinoma with acantholytic growth pattern. (b) p63 labeling confirms squamous differentiation (original magnification × 200).

Immunohistochemical stain for p53 in adenosquamous carcinoma. (a) Nuclear p53 labeling of adenosquamous carcinoma. (b) Pagetoid extension of p53-positive neoplastic cells to a benign duct; arrowhead points to residual nonneoplastic duct lining (original magnification × 200).

Dpc4, p16, E-cadherin and EGFR immunohistochemical stains. (a) Dpc4 stain lost in adenosquamous carcinoma with intact staining present in desmoplastic tumor stroma. (b) Neoplastic cells do not label with antibodies to p16, with intact nuclear labeling present in normal pancreas (arrowheads). (c) Loss of E-cadherin labeling in the majority of neoplastic cells (arrowheads), with small cluster of neoplastic cells showing intact expression. (d) Membranous EGFR immunolabeling (original magnification × 200).
HPV DNA Detection
All cases lacked detectable HPV DNA by ISH.
Discussion
We report the morphologic features of eight cases of adenosquamous carcinoma seen in one institution and provide immunohistochemical and molecular characterization of this rare variant of ductal adenocarcinoma. This is the most extensive molecular study of these tumors, and the first to utilize microdissection techniques on this tumor type. In the past two decades, our understanding of molecular events that occur in pancreatic tumorigenesis has markedly evolved.1, 13 Genes frequently mutated in pancreatic ductal adenocarcinoma including KRAS2, p16/CDKN2a, TP53 and DPC4/MAD4 have been identified.1
Previously Kardon et al4 performed a limited molecular analysis of 25 cases of pancreatic adenosquamous carcinomas. These investigators screened for KRAS2 gene mutations in 12 cases of ‘amplifiable genomic DNA’ and found 50% to have heterozygous, activating mutations in codon 12. These mutations changed the KRAS2 codon 12 from a glycine to a valine or an aspartic acid. Except for KRAS2 gene sequencing, no other molecular marker related to pancreatic tumorigenesis was analyzed. Further, these investigators did not perform these studies on microdissected tissue samples.4
Some of the most common molecular features of pancreatic ductal adenocarcinoma include loss of the DPC4/MAD4 gene (55%), p16/CDKN2a loss either through epigenetic or genetic alteration silencing (>90%)14 and KRAS2 activating mutations (>90%).13 In this study, we found a similar frequency in our cohort. We found loss of p16 protein expression in all cases. Homozygous deletions were detected in only three of the eight cases (Tables 1 and 2). Thus, we deduced that p16 protein expression may be lost in the other cases due to other genetic alterations such as an intragenic mutation coupled with loss of heterozygosity or other modes of gene silencing such as DNA methylation.12, 15, 16
In the majority of our cases and the previously reported cases,4 presence of the same KRAS2 gene mutations in both components suggests a common precursor cell of origin for both components. It is interesting to note that in one case we found two different activating KRAS2 gene mutations, in the adenocarcinoma and squamous components (Figure 1). However, these data taken together with the fact that all cases harbored a KRAS2 mutation support the notion that KRAS2 activation is critical for some aspect of the development of this rare tumor.
We observed loss of Dpc4 protein expression in five of eight cases. Wilentz et al9 showed that immunohistochemical analysis for the Dpc4 protein performed on pancreatic resection specimens is an extremely sensitive and specific technique to classify DPC4 gene status. The DPC4 gene (also known as MAD4) is a tumor suppressor that is inactivated in approximately 55% of pancreatic adenocarcinomas, yet its expression has not been investigated until now in adenosquamous carcinomas of the pancreas.17 The high rate of Dpc4 loss in our cases is comparable with the rate described in advanced unresectable pancreatic ductal adenocarcinoma, and possibly reflects a more aggressive nature of these neoplasms. Importantly, inactivation of DPC4 is rare in extrapancreatic malignancies, therefore loss of immunolabeling for this protein may be an informative diagnostic marker in evaluating whether this carcinoma originated from the pancreas.
The TP53 gene on chromosome 17p is another tumor suppressor gene frequently inactivated in pancreatic adenocarcinoma, with approximately 50–75% of these carcinomas showing intragenic mutation combined with the loss of the second allele.13, 18 Strong nuclear labeling for the p53 protein was present in over 60% of our cases, again supporting the notion that adenosquamous carcinoma shows very similar molecular changes with pancreatic ductal adenocarcinoma.
Epidermal growth factor receptor is expressed in 31–58% of pancreatic ductal adenocarcinomas.19, 20 The EGFR is a member of the HER family of receptor tyrosine kinases. Stimulation of the receptor through ligand binding activates the intrinsic receptor tyrosine kinase and promotes receptor homo- or heterodimerization with HER family members. EGFR activation leads to downstream stimulation of several signaling cascades, including MAPK and PI(3)K/Akt that influence cell proliferation, angiogenesis, invasion and metastasis.21 Therefore, many strategies, including the use of small molecule tyrosine kinase inhibitors and monoclonal antibodies to target EGFR, have been developed.22 A recent randomized phase III trial of 569 patients with untreated, inoperable pancreatic cancer demonstrated a small but significant survival benefit for the combination of gemcitabine and the EGFR inhibitor, erlotinib, vs gemcitabine alone.23
EGFR/ERK signaling is influenced by E-cadherin, a cell–cell adhesion molecule.24 Absence of E-cadherin expression was recently reported in several aggressive variants of pancreatic carcinoma including anaplastic carcinoma, adenocarcinoma with osteoclast-like giant cells and signet ring carcinomas; however the expression of this protein has not been previously evaluated in pancreatic adenosquamous carcinomas.5 We demonstrated that reduced and absent E-cadherin expression was present in seven of eight adenosquamous carcinomas, possibly contributing to the aggressive nature of this tumor through its central role in the epithelial to mesenchymal transition. It has been also shown that loss of E-cadherin in pancreatic cancer cell lines is associated with resistance to EGFR inhibition. Therefore, determining E-cadherin status may have therapeutic implications in cases of adenosquamous carcinoma expressing EGFR.25 In addition, as wild-type KRAS is required for EGFR inhibitor efficacy in patients with lung adenocarcinoma and metastatic colorectal cancer, further studies might be necessary to determine efficacy of targeting EGFR in adenosquamous carcinoma of the pancreas.26, 27
Because the majority of the molecular features of adenosquamous carcinomas surveyed showed similarity to pancreatic ductal adenocarcinoma, we assessed an alternative molecular marker (HPV DNA detection) that could drive and distinguish adenosquamous carcinomas from pancreatic ductal adenocarcinoma.28 Unfortunately, we did not detect any HPV DNA by ISH. Thus, ongoing studies will be needed to uncover unique molecular features that may distinguish adenosquamous carcinomas from pancreatic ductal adenocarcinoma.
In contrast to previously reported series, we noted a high incidence of coexisting squamous metaplasia in adjacent nonneoplastic pancreatic ducts. In a few cases the PanIN lesion and squamous metaplasia coexisted in one duct, suggesting that both components might be derived from a common progenitor. We confirmed the presence of squamous differentiation in all cases using immunohistochemical labeling for p63. p63 is a member of the p53 family and a marker of squamous differentiation in benign and malignant epithelia.29 In one previous study, p63 was shown to identify areas of squamous metaplasia and the squamous component of adenosquamous carcinomas in the pancreas.30 Our study supports these results and suggests that p63 is a useful marker in identifying squamous differentiation in cancers with growth patterns not readily identified as adenosquamous carcinomas. In our series, two cases with extensive acantholytic pattern and clear cell change showed strong nuclear reactivity with p63, supporting the diagnosis of adenosquamous carcinoma rather then pancreatic ductal adenocarcinoma with clear cell change. This distinction has clinical implications because adenosquamous carcinoma has a worse prognosis than ordinary pancreatic ductal adenocarcinoma.
In summary, we report a well-characterized series of adenosquamous carcinomas of the pancreas and characterize the pattern of genetic alterations in this rare tumor type. This work, in combination with future molecular mining of genotypic differences between pancreatic ductal adenocarcinoma and adenosquamous carcinomas should provide diagnostic and prognostic value.
References
Maitra A, Hruban RH . Pancreatic cancer. Annu Rev Pathol 2008;3:157–188.
Hruban RH, Klimstra DS, Pitman MB . Tumors of the Pancreas. Armed Forces Institute of Pathology: Washington, DC, 2007.
Hsu JT, Yeh CN, Chen YR, et al. Adenosquamous carcinoma of the pancreas. Digestion 2005;72:104–108.
Kardon DE, Thompson LD, Przygodzki RM, et al. Adenosquamous carcinoma of the pancreas: a clinicopathologic series of 25 cases. Mod Pathol 2001;14:443–451.
Winter JM, Ting AH, Vilardell F, et al. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin Cancer Res 2008;14:412–418.
Jimeno A, Hidalgo M . Pharmacogenomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Biochim Biophys Acta 2006;1766:217–229.
Alsner J, Jensen V, Kyndi M, et al. A comparison between p53 accumulation determined by immunohistochemistry and TP53 mutations as prognostic variables in tumours from breast cancer patients. Acta Oncol 2008;47:600–607.
Rakha EA, Abd El Rehim D, Pinder SE, et al. E-cadherin expression in invasive non-lobular carcinoma of the breast and its prognostic significance. Histopathology 2005;46:685–693.
Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol 2000;156:37–43.
Abd El-Rehim DM, Pinder SE, Paish CE, et al. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br J Cancer 2004;91:1532–1542.
Feltmate CM, Mok SC . Whole-genome allelotyping using laser microdissected tissue. Methods Mol Biol 2005;293:69–77.
Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994;8:27–32.
Hruban RH, Iacobuzio-Donahue C, Wilentz RE, et al. Molecular pathology of pancreatic cancer. Cancer J 2001;7:251–258.
Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997;57:3126–3130.
Fukushima N, Sato N, Ueki T, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol 2002;160:1573–1581.
Wilentz RE, Geradts J, Maynard R, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998;58:4740–4744.
Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996;271:350–353.
DiGiuseppe JA, Hruban RH, Goodman SN, et al. Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994;101:684–688.
Dancer J, Takei H, Ro JY, et al. Coexpression of EGFR and HER-2 in pancreatic ductal adenocarcinoma: a comparative study using immunohistochemistry correlated with gene amplification by fluorescent in situ hybridization. Oncol Rep 2007;18:151–155.
Lemoine NR, Hughes CM, Barton CM, et al. The epidermal growth factor receptor in human pancreatic cancer. J Pathol 1992;166:7–12.
Citri A, Yarden Y . EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 2006;7:505–516.
Gross ME, Shazer RL, Agus DB . Targeting the HER–kinase axis in cancer. Semin Oncol 2004;31:9–20.
Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960–1966.
Fuchs BC, Fujii T, Dorfman JD, et al. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res 2008;68:2391–2399.
Buck E, Eyzaguirre A, Barr S, et al. Loss of homotypic cell adhesion by epithelial–mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther 2007;6:532–541.
Bonomi PD, Buckingham L, Coon J . Selecting patients for treatment with epidermal growth factor tyrosine kinase inhibitors. Clin Cancer Res 2007;13:s4606–s4612.
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626–1634.
Hennig EM, Kvinnsland S, Holm R, et al. Significant difference in p53 and p21 protein immunoreactivity in HPV 16 positive and HPV negative breast carcinomas. Acta Oncol (Stockholm, Sweden) 1999;38:931–938.
Wu M, Wang B, Gil J, et al. p63 and TTF-1 immunostaining. A useful marker panel for distinguishing small cell carcinoma of lung from poorly differentiated squamous cell carcinoma of lung. Am J Clin Pathol 2003;119:696–702.
Basturk O, Khanani F, Sarkar F, et al. DeltaNp63 expression in pancreas and pancreatic neoplasia. Mod Pathol 2005;18:1193–1198.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brody, J., Costantino, C., Potoczek, M. et al. Adenosquamous carcinoma of the pancreas harbors KRAS2, DPC4 and TP53 molecular alterations similar to pancreatic ductal adenocarcinoma. Mod Pathol 22, 651–659 (2009). https://doi.org/10.1038/modpathol.2009.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2009.15
Keywords
This article is cited by
-
YY1 promotes pancreatic cancer cell proliferation by enhancing mitochondrial respiration
Cancer Cell International (2022)
-
Adenosquamous carcinoma of the pancreas: two case reports and review of the literature
Journal of Medical Case Reports (2022)
-
A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma
Nature Cancer (2020)
-
A machine learning based delta-radiomics process for early prediction of treatment response of pancreatic cancer
npj Precision Oncology (2019)
