
Abstract
In the World Health Organization classification, one major and four minor criteria are specified for the diagnosis of systemic mastocytosis. We report our experience using these criteria to diagnose systemic mastocytosis involving bone marrow. A total of 59 patients with clinically suspected systemic mastocytosis underwent comprehensive bone marrow examination, including immunophenotyping by immunohistochemistry and/or flow cytometry and molecular studies for KIT exon 17 mutations. Serum tryptase levels were also assessed. Of these 59, 53 (90%) patients met the diagnostic criteria for systemic mastocytosis. In these patients, multifocal dense infiltrates of mast cells, the major criterion, was observed in 36 (68%) patients. Atypical mast cell morphology was observed in 53 (100%), an aberrant immunophenotype was identified in 50 of 52 (96%), KIT mutation was present in 33 of 44 (75%), and an elevated serum tryptase (>20 ng/ml) was detected in 44 of 52 (85%). In the six patients in which bone marrow examination could not confirm systemic mastocytosis, one had systemic mastocytosis involving spleen, one patient had chronic idiopathic myelofibrosis, and four had no specific diagnosis, but systemic mastocytosis was still considered most likely. Of these six patients, atypical mast cell morphology was identified in five, aberrant immunophenotype in five, KIT mutation in two, and elevated serum tryptase in two. None of these cases met the major criteria. We conclude that the World Health Organization criteria are useful for the diagnosis of systemic mastocytosis in bone marrow specimens. The results also show the relative values of traditional morphologic criteria (ie, major criterion) and the results of ancillary testing (ie, minor criteria). However, as illustrated by the case of splenic systemic mastocytosis as well as the patient with chronic idiopathic myelofibrosis, the current World Health Organization system is neither completely sensitive nor specific for systemic mastocytosis.
Similar content being viewed by others
Main
Mast cells are bone marrow derived cells that arise from agranular, rare-circulating progenitor cells that express CD13, CD34, CD117, and high-affinity IgE receptor I (FcɛRI).1, 2 Mast cells have a wide distribution throughout the human body with increased numbers in tissues with direct exposure to the external environment, ie, skin, gastrointestinal tract and respiratory tract.
Mastocytosis is defined as abnormal growth and accumulation of mast cells in one or more organ systems. Mastocytosis is not a single, well-defined entity, but rather is heterogeneous and characterized by multiple clinical variants. The current World Health Organization (WHO) classification subdivides mastocytosis into seven major categories: (1) cutaneous mastocytosis, (2) indolent systemic mastocytosis, (3) systemic mastocytosis with associated clonal, hematological non-mast-cell lineage disease, (4) aggressive systemic mastocytosis, (5) mast cell leukemia, (6) mast cell sarcoma, and (7) extracutaneous mastocytoma. Within these major categories are additional variants and subvariants, some of which are currently considered ‘provisional entities.’3, 4
Systemic mastocytosis is characterized by involvement of at least one extracutaneous organ, with or without evidence of skin infiltration. Bone marrow is the most commonly involved extracutaneous site and bone marrow aspiration and biopsy are commonly employed to establish the diagnosis.4 An algorithm for the diagnostic approach for systemic mastocytosis has been proposed. All adults with cutaneous mastocytosis or suspected systemic mastocytosis should undergo complete staging including bone marrow examination. Children with cutaneous mastocytosis should have bone marrow examination if a serum tryptase is markedly elevated (>100 ng/ml), have other signs or symptoms of systemic disease, or if the skin lesions and/or serum tryptase >20 ng/ml persist into adolescence.5
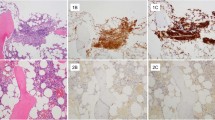
According to the WHO classification, one major and four minor criteria are useful for the diagnosis of systemic mastocytosis (Table 1). The morphological identification of multifocal, dense infiltrates of mast cells in bone marrow or other extracutaneous sites, confirmed by tryptase immunohistochemistry or special stains, is the major criterion (Figures 1a and b). In essence, this criterion is based on using the traditional morphologic approach to establish the diagnosis of systemic mastocytosis. The four minor criteria include: (1) >25% of mast cells with cytologic atypia (Figure 1c), (2) KIT mutation at codon 816, (3) CD117+ mast cells that aberrantly express CD2 and/or CD25 (Figure 1d) and (4) serum tryptase persistently >20 ng/ml. As specified in the WHO classification, systemic mastocytosis can be diagnosed if one major and one minor or three minor criteria are met. Here we report our experience using these criteria to diagnose systemic mastocytosis involving bone marrow.

WHO morphologic criteria for systemic mastocytosis. (a) Bone marrow biopsy, H&E, × 200: large aggregate of mast cells (>15) with intermixed lymphocytes and eosinophils. (b) Bone marrow biopsy, tryptase immunostain, × 40: multifocal aggregates of mast cells, positive for tryptase. (c) Aspirate smear, Wright–Giemsa, × 1000: atypical morphology of mast cells including spindled, degranulated and hypogranulated forms. (d) Bone marrow biopsy, CD25 immunostain, × 400: aggregate of mast cells with positive membranous staining with CD25.
Materials and methods
Study Group
Electronic medical records at The University of Texas MD Anderson Cancer Center were searched for patients with a high clinical suspicion of systemic mastocytosis between the years 2001 and 2007. We limited our search to those patients who underwent a bone marrow aspiration and biopsy and who had ancillary testing performed at our institution. The clinical notes were reviewed as well as complete blood counts performed at time of presentation. Total serum tryptase was assessed at a reference laboratory using a fluorenzyme immunoassay.6 This test measures both forms of tryptase, α and β, and our normal reference range is 2–10 ng/ml.
Morphology and Immunophenotypic Methods
In all cases, bone marrow aspirate smears, touch imprints, aspirate clot and core biopsy specimens were reviewed. Bone marrow aspirate smears and touch imprints were stained with Wright–Giemsa. Bone marrow aspirate clot and core biopsy specimens were routinely processed and stained with hematoxylin and eosin.
Immunohistochemical stains were performed using 5 μm thick, formalin-fixed, paraffin-embedded tissue sections and heat-induced epitope retrieval as previously described.7 The following antibodies were used: CD2 (Leica/Vision Biosystems, Bannockburn, IL, USA; 1 : 75), CD25 (Leica/Vision Biosystems; 1 : 150), CD117 (Dako, Carpinteria, CA, USA; 1 : 100) and tryptase (Dako; 1 : 200). Tryptase and/or CD117 highlighted the mast cell infiltrates in bone marrow aspirate clot or biopsy sections in a cytoplasmic pattern. A positive result for CD2 and/or CD25 by immunohistochemistry was manifested by crisp membranous expression by mast cells.
Specimens were processed for flow cytometry by a stain-lyse-wash method using Pharm Lyse solution (Becton Dickinson (BD) Biosciences, San Diego, CA, USA) as previously described using (1–2) × 106 cells per staining tube.8 Each case was assessed with antibodies directed against the following: CD2, CD25, CD45, and CD117 (all from BD Biosciences). CD2 and CD25 were both conjugated to PE. Stains were performed in four-color combinations using CD45-PerCP and CD117-APC in each tube for gating. Nonspecific staining and autofluorescence was measured on mast cells using PE-conjugated isotype-matched controls. Acquisition and analysis of flow cytometry data were performed using four-color FACSCalibur instruments (BD Biosciences) with CELLQuest software (BD Biosciences). An initial tube was analyzed using open gates (Figure 2a), and then CD117-positive events were specifically acquired (Figure 2b), with 1–8 × 105 total cells collected per tube. CD117 brightly positive cells were then analyzed for expression of CD2 and CD25 (examples in Figure 2c–f).

Flow cytometry in cases of systemic mastocytosis. (a) Total bone marrow cells stained with CD117. Rare cells (0.18%) are positive for CD117. (b) Specific acquisition of CD117+ cells with further gating on mast cells (CD117+ bright with high side scatter). (c, d) Comparison of normal control (CD2−) with neoplastic mast cells (partial CD2+). (e, f) Comparison of normal control (CD25−) with neoplastic mast cells (CD25+).
Expression of either CD2 or CD25 by mast cells, shown by either immunohistochemistry or flow cytometry, was considered aberrant as defined in the WHO classification.
Molecular Testing
Over the course of time that this group of patients was seen, a variety of methods were performed for the detection of the KIT mutation on exon 17 at codon 816 including pyrosequencing, Sanger sequencing, and allele-specific PCR as previously described.9 In most cases, bone marrow aspirate material was used for assessment for KIT mutations. In a few patients, this analysis was performed using DNA extracted from paraffin-embedded bone marrow biopsy specimens.
Results
A total of 59 patients were included in this study. These patients included 25 men and 34 women ranging from 23 to 75 years in age (median 54). Other clinicopathologic features of the study group are reported in Table 2. The most common presenting symptoms were rash, fatigue and bone pain. A complete blood count was recorded on all patients. Hematologic abnormalities were seen in 41% of systemic mastocytosis cases and included anemia, leukopenia, leukocytosis, thrombocytopenia, and thrombocytosis. A subset of patients had a previous diagnosis of systemic mastocytosis and had received prior therapy (mostly symptomatic treatment only).
Out of 59, 53 patients had findings sufficient to fulfill the WHO criteria for the diagnosis of systemic mastocytosis (Table 3). Multifocal dense aggregates of mast cells positive for tryptase immunostain (major criterion) were seen in 36 (68%) of systemic mastocytosis patient specimens. As for the minor criteria, atypical morphology was observed in 53 of 53 (100%), KIT mutation was present in 33 of 44 (75%), and an aberrant immunophenotype was identified by immunohistochemistry or flow cytometry immunophenotyping in 50 of 52 (96%) patient specimens. In the assessment of immunophenotype, flow cytometry detected aberrancy equally as well as immunohistochemistry, 92% (44 of 48) versus 88% (29 of 33). However, mast cell numbers detected by flow cytometry immunophenotyping of bone marrow aspirate specimens were typically 10–100 × lower than that estimated by immunohistochemical examination of the biopsy specimen. Flow cytometric analysis detected an aberrant mast cell population as low as 0.002%.
The serum tryptase level was elevated in 44 of 52 (85%) patients. Serum tryptase levels were >200 ng/ml in 14 patients, 101–200 ng/ml in 6, 51–100 ng/ml in 10, 21–50 ng/ml in 16, and ≤20 in 12 (one patient did not have a serum measurement). Nine of our patients had systemic mastocytosis with an associated clonal hematological non-mast-cell lineage disease. Therefore, serum tryptase level was excluded as a minor criterion. As a result, the actual number of patients for whom this criterion could be used was 35 of 43 (81%). In two of six patients who failed to meet the diagnostic criteria for systemic mastocytosis, the serum tryptase could not be used as a minor criterion because they had an associated clonal hematological non-mast-cell lineage disease. All patients with serum tryptase greater than 50 ng/ml (n=30) met the diagnostic criteria for systemic mastocytosis.
For the 53 patients in whom the diagnosis of systemic mastocytosis was established, all 36 (68%) that met the major criterion also had ≥2 minor criteria. In 17 (32%) patients, the diagnosis of systemic mastocytosis was supported only by ≥3 minor criteria.
Conventional cytogenetics was performed on 58 patients and 88% had a diploid karyotype. A total of three patients had abnormal cytogenetics, two of which met the criteria for systemic mastocytosis. One patient had 46, XY, t(5;12)(q33;q13) in nine metaphases. The second patient had systemic mastocytosis with associated clonal hematologic non-mast-cell disease associated with a 45, X, −Y karyotype. The third patient had a neoplasm with a complex karyotype, 47, XY, +1, der(1;7)(q10;p10), +9. This patient did not meet criteria for systemic mastocytosis and was diagnosed to have chronic idiopathic myelofibrosis with progression to acute myeloid leukemia.
We further classified the 53 patients that met the criteria for systemic mastocytosis into variants using the criteria specified by the WHO classification.4 Indolent systemic mastocytosis was most common in 35 (66%). Six patients (11%) met the criteria for aggressive systemic mastocytosis, nine (17%) had systemic mastocytosis with an associated clonal hematological non-mast-cell lineage disease and three (6%) patients were diagnosed with mast cell leukemia. These findings are similar to other reports.3, 10 All six cases of aggressive systemic mastocytosis were placed in this category because of the presence of anemia (hemoglobin <10 g/100 ml) according to the WHO classification. The most common associated clonal hematological disorder found in association with systemic mastocytosis was chronic myelomonocytic leukemia in four patients (Table 4). The three cases of mast cell leukemia met the criteria for systemic mastocytosis and additionally had sheets of mast cells in the bone marrow biopsy specimen and >30% mast cells on aspirate smears.
Patients That did NOT Meet Criteria for Systemic Mastocytosis Involving Bone Marrow
There were six patients who did not meet the criteria for systemic mastocytosis involving the bone marrow. Three of these patients had a history of cutaneous mast cell disease and one patient had a history of systemic mastocytosis involving spleen. In four patients, no major and less than three minor criteria were present: three patients had atypical mast cells and an aberrant immunophenotype, and one patient had a KIT D816V mutation and an aberrant immunophenotype. In two patients, three minor criteria were present including an elevated serum tryptase. However, the elevated serum tryptase level could not be used as a criterion because these two patients had associated myeloid disorders, chronic myelomonocytic leukemia or chronic myeloproliferative disorder, respectively. One patient had atypical mast cells and a KIT mutation, and the other patient had atypical mast cells and an aberrant immunophenotype. Interestingly, the patient with chronic myelomonocytic leukemia that did not meet the criteria for systemic mastocytosis in bone marrow had a history of systemic mastocytosis diagnosed in a splenectomy specimen (spleen not available for review).
Discussion
Using the WHO criteria, in patients with suspected systemic mastocytosis based on clinical and laboratory findings, the diagnosis of systemic mastocytosis can be rendered by examination of bone marrow with ancillary testing in 90% of patients, precluding the need for additional tissue biopsy. However, multifocal, dense infiltrates of mast cells, the major criterion in the WHO classification and the most traditional means of diagnosing systemic mastocytosis were only observed in approximately 70% of patients in this study. In an additional 30% of patients, the diagnosis of systemic mastocytosis could only be made by the use of ancillary testing as specified by the minor criteria of the WHO classification. Of these, atypical mast cell morphology was most common, in 100% of cases, followed by aberrant CD25 and/or CD2 expression (96%), elevated serum tryptase (85%), and presence of KIT mutations (75%). These data support the importance of ancillary testing to establish the diagnosis of systemic mastocytosis by bone marrow examination.
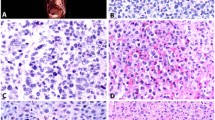
Although atypical mast cell morphology is highly sensitive, in our experience this is best assessed by examination of aspirate systemic smears or touch imprints. In this study, we attempted to judge mast cell morphology in aspirate clot and biopsy specimens but we found this to be difficult for at least two reasons. First, it is challenging to identify low-level numbers of mast cells in routinely stained sections by examination using standard high-power ( × 400) magnification. Immunohistochemical stains are extremely helpful in highlighting scattered mast cells. Second, in our opinion mast cell morphology is difficult to judge using an immunohistochemical stain for tryptase in bone marrow clot or biopsy specimens because the cells often look more atypical than is appreciated in smears and touch imprints. To further explore this idea, we assessed bone marrow aspirate smears and biopsy specimens with mast cell hyperplasia for comparison. We chose to study bone marrow specimens of five cases of lymphoplasmacytic lymphoma using a tryptase immunohistochemical stain because of the increased numbers of mast cells commonly associated with this type of lymphoma.11, 12 A number of mast cells appeared to be cytologically atypical on the tryptase immunostain and yet mast cells seen on the aspirate smears were normal (Figure 3). In two of these cases, approximately 25% of the mast cells on histologic sections were large and spindled; therefore, meeting this minor criterion. We conclude that using a tryptase immunostain on bone marrow biopsy or clot specimens to assess morphology is not completely reliable. Also, a tryptase immunostain may overestimate the number of mast cells (overstaining, bleeding over to nearby cells) making them appear as clusters of >15.

Mast cells in lymphoplasmacytic lymphoma involving bone marrow. (a) Bone marrow biopsy, tryptase immunostain, × 400. Inset: Bone marrow biopsy tryptase immunostain, × 1000. A subset of mast cells appears atypical with spindled morphology. (b) Touch imprint, Wright–Giemsa, × 1000: normal appearing mast cell in a case with discordant atypical morphology on biopsy section stained with tryptase.
Immunophenotyping by either immunohistochemistry or flow cytometry to detect an aberrant immunophenotype was also a very sensitive minor criterion in this study. We detected aberrant CD25 expression by mast cells using immunohistochemistry in 88% of tested cases. CD25 expression by neoplastic mast cells using similar methods has been reported by others to be present in 80–98% of cases.5, 13, 14 CD2 can also be used for the detection of aberrancy by immunohistochemistry; however, the sensitivity and specificity of CD2 are reportedly low.13 Only 9 of 23 cases (39%) assessed had positive staining with CD2 by immunohistochemistry in this study, supporting the results of others and emphasizing the utility of CD25 immunostaining to assess aberrancy. Similar to the immunohistochemical results, CD2 was less robust than CD25 for detecting aberrancy by flow cytometric analysis. CD2 expression was often dim or negative compared with CD25 expression. For this reason, we suggest staining with CD2 conjugated to a bright fluorochrome, such as PE.
KIT is a member of the receptor tyrosine kinase family and is expressed by mast cells, hematopoietic stem cells, melanocytes, germ cells, and gastrointestinal stromal cells. Accordingly, KIT mutations are not specific for mastocytosis.1, 15, 16, 17 KIT is a known oncogene and gain-of-function mutations have been identified in systemic mastocytosis, by far the most common (>80%) being found in exon 17 where a somatic point mutation, A>T in nucleotide 2468 of KIT cDNA, results in the replacement of an aspartic acid by valine at codon 816 (D816V).18 This mutation leads to a ligand-independent constitutive activation and autophosphorylation of KIT. We detected KIT mutations in approximately two-thirds of patients with systemic mastocytosis. Thus, detection KIT mutations was less sensitive than immunophenotyping for establishing the diagnosis of systemic mastocytosis. This is most likely related to the sensitivity of our methods to detect KIT mutation, approximately 10%, and the low numbers of mast cells in bone marrow aspirate specimens of many patients in this study. Mutations other than D816V are not uncommon in systemic mastocytosis with associated clonal hematologic non-mast-cell disease; however, these mutations were not tested in our study group.19 In addition to testing for the KIT mutation, we also screened for the presence of FIP1L1-PDGFRα either by FISH or RT–PCR, as these mutations are associated with eosinophilia and often sensitive to treatment with imatinib.20, 21, 22 The FIP1L1-PDGFRα was not identified in any case.
An elevated serum tryptase level is a helpful finding for the diagnosis of systemic mastocytosis, and was present in 46 of 58 (79%) patients in this study. However, serum tryptase levels can be transiently elevated in severe allergic reactions, as well as in patients with myeloid neoplasms.23 Serum tryptase levels >20 ng/ml have been reported in 39% of de novo acute myeloid leukemia, 44% of secondary acute myeloid leukemia, and in patients with myelodysplastic and myeloproliferative diseases. One study reported that approximately 5% of acute myeloid leukemia cases had serum tryptase levels >200 ng/ml.24 It seems possible that some of these cases had associated, undetectable systemic mastocytosis. Serum tryptase levels in patients with myelodysplastic syndromes tend to be lower than acute myeloid leukemia, with a mean measurement of 13.8±10.5 ng/ml.25 For these reasons, the WHO classification states that this criterion cannot be used reliably in patients with an associated myeloid disorder. It is not completely clear whether an elevated serum tryptase is valid in other examples of systemic mastocytosis with associated clonal hematologic non-mast-cell lineage disease, specifically lymphoid neoplasms. In subsequent publications by the same authors, the phrase concerning the validity of serum tryptase in the diagnostic work-up has been changed from ‘associated myeloid disorder’ to ‘systemic mastocytosis with associated clonal hematologic non-mast-cell disease’ which by definition includes other entities beyond myeloid disorders.3, 23 Myeloid disorders account for 80–90% of cases of systemic mastocytosis with associated clonal hematologic non-mast-cell disease myelodysplastic syndrome/myeloprolifertive disorder being most common. Of the associated lymphoid malignancies, plasma cell myeloma is most frequent.3 Two patients in this study had associated lymphoid neoplasms, follicular lymphoma, and plasma cell dyscrasia.
The limitation of using serum tryptase levels as stated in the WHO classification is appreciable as seven patients in this study had systemic mastocytosis with associated clonal hematologic non-mast-cell disease with an associated myeloid disorder, representing 13% of all systemic mastocytosis patients. In a total of nine patients with systemic mastocytosis with associated clonal hematologic non-mast-cell disease (myeloid plus lymphoid), the serum tryptase levels were assessed in eight patients and the results were >200 ng/ml in four patients (chronic myelomonocytic leukemia in two, myelodysplastic syndrome/myeloproliferative disorder, and myelodysplastic syndrome), 123 ng/ml (chronic myelomonocytic leukemia), 119 ng/ml (follicular lymphoma), 94 ng/ml (chronic myelomonocytic leukemia and hairy cell leukemia), and 2 ng/ml (plasma cell dyscrasia). In our opinion, clarification of this criterion is needed. Elevated serum tryptase levels are likely more reliable in patients with systemic mastocytosis associated with lymphoma or plasma cell myeloma. Possibly, the threshold of the serum tryptase level could be raised for patients with systemic mastocytosis with associated clonal hematologic non-mast-cell disease, thereby improving specificity and allowing greater utility of this criterion in this setting.
In the patients that did not meet the criteria for systemic mastocytosis involving bone marrow, one had systemic mastocytosis involving spleen, one had chronic idiopathic myelofibrosis, and four had no specific diagnosis, but systemic mastocytosis was still thought likely. Of these six patients, atypical mast cell morphology was identified in five, aberrant immunophenotype in five, KIT mutation in two patients, and elevated serum tryptase level in two. No case had the major criteria. One patient had chronic myelomonocytic leukemia in bone marrow, systemic mastocytosis involving the spleen and three minor criteria (atypical mast cell morphology, KIT mutation, and elevated serum tryptase). This patient undoubtedly has systemic mastocytosis; however, by only examining the bone marrow and strictly using these criteria, this diagnosis could not be made because the serum tryptase level could not be used according to the WHO criteria. Another patient had chronic idiopathic myelofibrosis; however, atypical morphology was present on aspirate smears, an aberrant immunophenotype (CD25) was detected by immunohistochemistry, and the patient had an elevated serum tryptase level. The other four patients did not have a specific diagnosis; however three of them were placed on treatment protocols for systemic mastocytosis.
In summary, our study supports the value of the WHO criteria for diagnosis of systemic mastocytosis by bone marrow examination with ancillary studies. However, bone marrow examination using WHO criteria is neither completely specific nor sensitive for systemic mastocytosis. In six patients with suspected systemic mastocytosis, bone marrow examination did not confirm the diagnosis. One patient in this study with a history of systemic mastocytosis, who did not meet the criteria for systemic mastocytosis, had a splenectomy performed before bone marrow examination that showed systemic mastocytosis. The ability to diagnose systemic mastocytosis appears to be particularly difficult in patients with an associated clonal hematologic neoplasm because serum tryptase levels can be elevated as a result, and therefore serum tryptase level cannot be reliably used as a minor criterion for systemic mastocytosis. Further study of serum tryptase levels in patients with lymphoma or plasma cell neoplasms may be helpful for further assessing the utility of using serum tryptase levels for diagnosis in this setting. It seems likely that particularly high serum tryptase levels in patients with suspected systemic mastocytosis may be useful, and thus setting a higher cutoff level for serum tryptase level in patients with systemic mastocytosis with associated clonal hematologic non-mast-cell lineage disease may be helpful.
References
Robyn J, Metcalfe DD . Systemic mastocytosis. Adv Immunol 2006;89:169–243.
Escribano L, Garcia Montero AC, Nunez R, et al. Flow cytometric analysis of normal and neoplastic mast cells: role in diagnosis and follow-up of mast cell disease. Immunol Allergy Clin North Am 2006;26:535–547.
Horny HP, Sotlar K, Valent P . Mastocytosis: state of the art. Pathobiology 2007;74:121–132.
Valent P, Horny H-P, Li CY, et al. Mastocytosis (mast cell disease). In: Jaffe E, Harris N, Stein H, Vardiman J (eds). World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissue. IARC Press: Lyon, France, 2001, pp 291–302.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest 2007;37:435–453.
Schwartz LB, Bradford TR, Rouse C, et al. Development of a new, more sensitive immunoassay for human tryptase: use in systemic anaphylaxis. J Clin Immunol 1994;14:190–204.
Valbuena JR, Rassidakis GZ, Lin P, et al. Expression of heat-shock protein-90 in non-Hodgkin's lymphomas. Mod Pathol 2005;18:1343–1349.
Ravandi F, Jorgensen JL, O'Brien SM, et al. Eradication of minimal residual disease in hairy cell leukemia. Blood 2006;107:4658–4662.
Zhao W, Bueso-Ramos CE, Verstovsek S, et al. Quantitative profiling of codon 816 KIT mutations can aid in the classification of systemic mast cell disease. Leukemia 2007;21:1574–1576.
Horny HP, Sotlar K, Sperr WR, et al. Systemic mastocytosis with associated clonal haematological non-mast cell lineage diseases: a histopathological challenge. J Clin Pathol 2004;57:604–608.
Brunning R, McKenna R . Atlas of tumor pathology. In: Rosai J (ed). Tumors of The Bone Marrow, Third Series. Armed Fores Institue of Pathology: Washington, DC, 1994, pp 353–358.
Pangalis GA, Kyrtsonis MC, Kontopidou FN, et al. Differential diagnosis of Waldenstrom's macroglobulinemia from other low-grade B-cell lymphoproliferative disorders. Semin Oncol 2003;30:201–205.
Sotlar K, Horny HP, Simonitsch I, et al. CD25 indicates the neoplastic phenotype of mast cells: a novel immunohistochemical marker for the diagnosis of systemic mastocytosis (SM) in routinely processed bone marrow biopsy specimens. Am J Surg Pathol 2004;28:1319–1325.
Baumgartner C, Sonneck K, Krauth MT, et al. Immunohistochemical assessment of CD25 is equally sensitive and diagnostic in mastocytosis compared to flow cytometry. Eur J Clin Invest 2008;38:326–335.
Hoshida Y, Hongyo T, Jia X, et al. Analysis of p53, K-ras, c-kit, and beta-catenin gene mutations in sinonasal NK/T cell lymphoma in northeast district of China. Cancer Sci 2003;94:297–301.
Corless CL, Fletcher JA, Heinrich MC . Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813–3825.
Kemmer K, Corless CL, Fletcher JA, et al. KIT mutations are common in testicular seminomas. Am J Pathol 2004;164:305–313.
Akin C . Molecular diagnosis of mast cell disorders: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn 2006;8:412–419.
Pullarkat V, Bueso-Ramos CE, Lai R, et al. Systemic mastocytosis with associated clonal hematological non-mast-cell lineage disease: analysis of clinicopathologic features and activating c-kit mutations. Am J Hematol 2003;73:12–17.
Patnaik MM, Tefferi A, Pardanani A . Kit: molecule of interest for the diagnosis and treatment of mastocytosis and other neoplastic disorders. Curr Cancer Drug Targets 2007;7:492–503.
Patnaik MM, Rindos M, Kouides PA, et al. Systemic mastocytosis: a concise clinical and laboratory review. Arch Pathol Lab Med 2007;131:784–791.
Tefferi A, Verstovsek S, Pardanani A . How we diagnose and treat WHO-defined systemic mastocytosis in adults. Haematologica 2008;93:6–9.
Valent P, Akin C, Sperr WR, et al. Mastocytosis: pathology, genetics, and current options for therapy. Leuk Lymphoma 2005;46:35–48.
Sperr WR, Jordan JH, Baghestanian M, et al. Expression of mast cell tryptase by myeloblasts in a group of patients with acute myeloid leukemia. Blood 2001;98:2200–2209.
Sperr WR, Stehberger B, Wimazal F, et al. Serum tryptase measurements in patients with myelodysplastic syndromes. Leuk Lymphoma 2002;43:1097–1105.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
All authors declared no conflicts of interest.
Rights and permissions
About this article
Cite this article
Johnson, M., Verstovsek, S., Jorgensen, J. et al. Utility of the World Heath Organization classification criteria for the diagnosis of systemic mastocytosis in bone marrow. Mod Pathol 22, 50–57 (2009). https://doi.org/10.1038/modpathol.2008.141
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.141
Keywords
This article is cited by
-
Bone fragility in patients affected by congenital diseases non skeletal in origin
Orphanet Journal of Rare Diseases (2021)
-
Smouldering systemic mastocytosis with lymph node involvement mimicking malignant lymphoma
Annals of Hematology (2014)
-
Isolated splenomegaly as the only presentation of systemic mastocytosis
Annals of Hematology (2013)
-
Evaluation of the WHO criteria for the classification of patients with mastocytosis
Modern Pathology (2011)
-
Die systemische Mastozytose – Standortbestimmung einer internistischen Erkrankung
Medizinische Klinik (2010)
