
Abstract
Renal cysts and clear cell renal cell carcinoma are common clinical manifestations of people with germ-line mutations of the von Hippel–Lindau tumor suppressor gene, VHL. Recent cell biological evidence suggests that the VHL gene product, pVHL, functions to maintain the primary cilium, a microtubule-based antenna-like structure whose functional integrity is believed to have an important role in cell-cycle control. As VHL mutations are common in sporadic clear cell renal cell carcinoma, but not papillary renal cell carcinoma, we asked whether there is an association between VHL status and primary cilia in vivo. VHL status was assessed in 20 cases of clear cell renal cell carcinoma and 9 cases of papillary renal cell carcinoma by DNA sequencing and by immunohistochemical staining for the hypoxia-inducible factor-α target gene products CA9 and GLUT-1. Of 20, 18 clear cell renal cell carcinomas, but only 1 of 9 papillary renal cell carcinomas, displayed evidence of VHL inactivation. In clear cell renal cell carcinoma the frequency of ciliated tumor cells ranged from 0 to 22% (median value 7.8±6.0%), whereas cilia frequency was significantly higher (P<0.0001) in papillary renal cell carcinoma (range 12–83%, median value 43.3±21.3%). There was no correlation between Ki-67 staining and cilia frequency, suggesting that the observed differences between the tumor types in cilia frequency are not accounted for by differences in cellular proliferation rates and that primary cilia degeneration in sporadic clear cell renal cell carcinoma depends on VHL inactivation. We propose that the different ciliation status of clear cell and papillary renal cell carcinoma may contribute, at least in part, to the different biological behaviors of these tumor types.
Similar content being viewed by others

Main
Primary cilia are specialized cellular organelles that project from the apical surface of almost all nondividing or interphase mammalian cells. They are thought to function in sensing various molecular and mechanical cues in the extracellular environment and to propagate signals into the cell. Defects in primary cilia structure and/or function have been causally linked to the development of various human diseases, including polycystic diseases of the kidney.1
Epithelial cells of the nephron have primary cilia, that protrude into the tubule lumen where they are believed to act in sensing the flow of urine.1 Disruption of the cilia structure or function causes uncontrolled kidney epithelial cell proliferation and cyst formation. At the core of a cilium lies the axoneme, which consists of a bundle of nine stable microtubule doublets (9+0) that are nucleated from the basal body. The composition of proteins within the cilium and hence cilium structure and function is regulated by and dependent on microtubule motor-driven transport of specific cargo proteins and vesicles. Thus, proper regulation of microtubule stability and dynamics is central for the structural and functional integrity of the cilium.1 For example, kidney-specific deletion of Kif3a coding, a subunit of the kinesin II motor protein responsible for retrograde transport in the cilium, results in loss of cilia and cyst formation in mice.2
Inactivating germ-line mutations of VHL represents the genetic hallmark of VHL disease, which, among other clinical manifestations predisposes affected individuals to develop renal cysts and clear cell renal cell carcinoma (ccRCC). The development of these kidney pathologies has been linked to the inactivation of VHL-dependent tumor-suppressive mechanisms. Prominent among these is pVHL's ability to act as an adapter protein of an E3 ubiquitin ligase complex that targets hypoxia-inducible factor-α (HIFα) subunits for ubiquitin-mediated degradation in the presence of oxygen.3 But pVHL also binds and stabilizes microtubules by protecting them from depolymerization, which is a prerequisite for cilium formation.4 Recent studies indicated that pVHL is necessary for the formation of primary cilia in cultured renal cell carcinoma cell lines.5, 6 It has been demonstrated that pVHL localizes to the primary cilium in human and mouse kidney cells and functions as an important part of a primary cilium-maintenance signaling network.7 Taken together, these observations strongly suggest that pVHL function is a critical aspect in primary cilium maintenance.
pVHL inactivation is not only restricted to the familial VHL cancer syndrome. In more than two-thirds of patients with sporadic ccRCC, which is the most frequent subtype of renal cancer, pVHL function is compromised by gene mutations or hypermethylation.8 Like ccRCC, papillary renal cell carcinoma (pRCC) arises from tubular epithelial cells.9 Morphologic, clinical, and genetic features clearly separate pRCC from ccRCC. Papillary tumors are characterized cytogenetically by trisomies/tetrasomies of chromosomes 7 and 17, and losses of chromosome Y, whereas chromosome 3p deletions and VHL mutations are common in ccRCC.10 These findings raise the possibility that ccRCC may be characterized by the absence of primary cilia and as pRCC tumorigenesis follows a VHL-independent pathway, that pRCC might represent a disease that is not characterized by loss of primary cilia. To test this hypothesis, we analyzed and compared the frequencies of primary cilia in normal kidney tissue, 20 ccRCCs, and 9 pRCC specimens.
Materials and methods
Renal Cell Carcinoma Tissues
All formalin-fixed and paraffin-embedded renal tumors were from the archive of the Institute for Surgical Pathology, Zurich University Hospital, and were reviewed by one pathologist (HM). A total of 20 ccRCCs and 9 pRCCs were selected for this study and diagnosed according to the 2004 WHO (World Health Organization) classification.11 Dominant architectural patterns were recorded for all ccRCCs. In total, 8 cases were dominantly solid/nested, 10 alveolar/acinar, and 2 tumors were mixed solid, alveolar, and papillary. This study was approved by the local commission of ethics (reference no. StV 38-2005).
Sequence Analysis
The VHL sequence analysis of 29 formalin-fixed and paraffin-embedded renal cell carcinomas was performed as described.12 Sequencing was performed using the BigDye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems, Rotkreuz, Switzerland) and forward or reverse primers that were applied for amplification of the VHL exons. Cycle sequencing products were analyzed using the AbiPrism 3100 Genetic Analyzer (Applied Biosystems).
Immunohistochemistry and Immunofluorescence
After dewaxing, rehydrating, and antigen retrieval for 10 min at 96°C in 0.1 M citrate buffer, renal cell carcinoma tissue sections (4 μm) were incubated using MIB-1 (1:20 dilution) (Dako, Glostrup, Denmark), mouse anti-CA9 M75 (1:200 dilution, gift of J Zavada, Institute of Molecular Genetics, Prague, Czech Republic), and rabbit anti-GLUT-1 AB1341 (1:1000 dilution) (Lucerna-Chem AG, Lucerne, Switzerland). Standard immunohistochemical detection of bound antibody was performed using biotin-conjugated goat F(ab′)2 anti-mouse immunoglobulin G (IgG; Jackson ImmunoResearch, West Grove, PA, USA) (1:500 dilution) or goat anti-rabbit IgG secondary antibodies followed by the ABC Vectastain and peroxidase substrate kits (Vector Laboratories, Burlingame, CA, USA) (1:200 dilution). Tumors were considered CA9 and GLUT-1 positive if membranous staining was found.
For immunofluorescence detection of primary cilia,13 sections were incubated with mouse anti-acetylated tubulin (Sigma-Aldrich, Basel, Switzerland) (1:100 dilution) and goat anti-mouse secondary antibodies conjugated to Alexa 488 (Molecular Probes, Invitrogen, Basel, Switzerland) (1:1000 dilution). Nuclei were labeled with 2 μg/ml 4-6-diamidino-2-phenylindole (DAPI). Cilia counting was performed by focusing up and down on the microscope to capture cilia and nuclei that lay in different focal planes within the section. Pictures were obtained with a × 40 oil objective with samples mounted in an immersion medium. Optical sections were 0.2-μm thick and stacks were made encompassing a Z-plane depth of 2–4 μm. From each sample the end points were determined by focusing on the uppermost and lowermost visible primary cilia within the microscopy field.
Immunohistochemical and immunofluorescence images were obtained using a Zeiss Axioplan 2 microscope or an Olympus IX70 Delta Vision Spectris deconvolution microscope. Obtained stacks were deconvolved using SoftWoRx 3.3.4 (Applied Precision, Issaquah, WA, USA) and processed with Imaris 4.2 (Bitplane AG, Zurich, Switzerland) and Photoshop 7.0 (Adobe Systems Inc.).
Statistical Analysis
Mean and median values, contingency table analysis, and χ2-tests for evaluating correlations between cilia frequencies and tumor subtypes were calculated using Statview Statistic Program (SAS, USA).
Results
We utilized two different methods to characterize the VHL status of our cohort of renal cell carcinoma samples. First, direct VHL DNA sequencing revealed that 9 ccRCCs carried wild-type VHL alleles, 11 ccRCCs carried VHL ‘loss-of-function’12 (frameshift) mutations, and 9 pRCCs carried wild-type VHL alleles (Table 1), consistent with previous reports about the mutation frequency of VHL in these tumor types.8, 14
CA9 as well as GLUT-1 expression is widely accepted as readout for pVHL loss and HIF stabilization.15, 16, 17 To indirectly assess pVHL status, we utilized immunohistochemical staining for the HIFα target genes CA9 and GLUT1 (Table 1; Figure 1a). Strong membranous positivity of CA9 was accompanied by GLUT-1 expression in 10 of 11 ccRCCs with VHL mutation and 1 tumor was CA9 negative but showed GLUT-1 staining. Five of nine ccRCCs with wild-type VHL expressed both CA9 and GLUT-1 and two tumors were positive for CA9 but negative for GLUT-1, suggesting VHL inactivation without VHL mutation. Two tumors were CA9 and GLUT-1 negative. Only one pRCC showed moderate CA9 and weak GLUT-1 expression. Taken together, we conclude that 18 of 20 ccRCC samples, but only 1 of 9 pRCCs, are characterized by VHL mutation or inactivation.

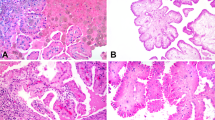
(a) Example of an immunohistochemically stained renal tumor showing membranous GLUT-1 expression in a clear cell renal cell carcinoma. (b–d) Ciliated tumor cells (white arrows) stained with antibody against acetylated tubulin and DAPI in tubular epithelial cells of normal kidney (magnification × 40) (b), one papillary renal cell carcinoma ( × 40) (c), and one clear cell renal cell carcinoma ( × 40) with the highest number of primary cilia (d).
We next assessed the frequency of primary cilia in normal kidney tubules, ccRCC, and pRCC by counting DAPI-stained cell nuclei and primary cilia marked by immunofluorescent staining using an antibody against acetylated tubulin (Table 1), a marker to study primary cilia in tumor cells.13 On average 87.3% (median value) of epithelial cells in 10 normal tubules contained a primary cilium (Figure 1b). For each of the renal cell carcinoma tissues, the frequency of primary cilia was evaluated by analyzing 60 cells within an area that contained more than 90% tumor cells. Strikingly, ccRCC samples exhibited frequencies of primary cilia that were very low (median value 7.5±6.0%) compared to normal tubules (Table 1; Figure 1c). Cilia frequency was independent of the architectural pattern in ccRCC (nested, alveolar, solid, and papillary). Compared to ccRCC, pRCC exhibited significantly higher (43.3±21.3%) frequencies of ciliated cells (P<0.0001) (Figures 1d and 2). Thus, ccRCC is characterized by very low frequencies of primary cilia compared to normal tubules and to pRCC.

Cilia frequencies (median values) in normal epithelial cells of renal tubules, papillary renal cell carcinoma, and clear cell renal cell carcinoma with and without VHL mutations.
Because primary cilia formation occurs in the quiescent phase of the cell cycle (G0) and not in mitotically active cells,18 we compared the frequency of expression of the cell proliferation marker Ki-67, which is expressed in late G1-, S-, G2-, and M-phases of the cell cycle but not in nonproliferating G0, with the frequency of ciliated cells in our renal cell carcinoma tissues (Table 1). We used the same cutoff (7%) for high and low Ki-67 labeling index (LI) as previously described.12 We observed no correlation between Ki-67 and cilia frequencies within or between groups of pRCC and ccRCC, suggesting that the ciliation frequency differences between pRCC and ccRCC do not arise because of differences in proliferation rates of these tumor types.
Discussion
We demonstrate that cilia degeneration is common in sporadic ccRCC. It seems obvious that genetic alterations affecting cilia stabilization and maintenance have a minor role in the majority of pRCC. This is supported by the fact that cytogenetic differences between pRCC and ccRCC are strikingly different.19, 20 In contrast to ccRCC, 3p loss19 and VHL mutations14 are rare in pRCC. Thus, the association between VHL inactivation and loss of cilia strongly argues for a characteristic feature of ccRCC and not of pRCC.
Besides mutations, additional molecular mechanisms may contribute to pVHL inactivation and consequently to HIFα stabilization in ccRCC carrying wild-type VHL alleles. Hypermethylation of the VHL promotor may lead to gene silencing and occurs in about 20% of ccRCCs.8, 21 As the VHL methylation status was not analyzed in our samples, possible hypermethylation in some ccRCCs with wild-type VHL cannot be excluded. E2-EPF ubiquitin carrier protein (UCP) associates with and targets pVHL for ubiquitin-mediated proteolysis in tumor cells.22 High expression of UCP supports HIF-1α stabilization in human liver, colon, and breast tumors. Whether UCP expression influences the VHL–HIF pathway in ccRCC still remains to be elucidated.
No VHL mutations were found in our pRCC samples, suggesting that other, VHL-independent, pathways contribute to the development of this tumor subtype. In contrast to ccRCC, in most of the pRCCs, the decrease of primary cilia was relatively mildly that supports pVHL's important role in maintaining the primary cilia structure in this tumor subtype. Interestingly, the pRCC with the highest cilia frequency showed moderate CA9 and weak GLUT-1 positivity. Hypermethylation of the VHL promoter or transcription factor(s) other than HIF might be responsible for the activation of the two genes in this tumor. It is of note that in another pRCC, although being CA9 and GLUT-1 negative, cilia numbers were similar to those seen in ccRCC. As VHL was not affected in this tumor mutations or deregulation of another, yet unknown, genetic factor might lead to the disruption of primary cilia.
A number of proteins localized to the primary cilia are associated with various hereditary human cystic kidney disorders. These include the autosomal dominant and recessive forms of polycystic kidney disease and several other cystic kidney diseases, such as nephrophthisis, oral–facial–digital syndrome type 1, and Bardet–Biedl syndrome.23 As primary cilia harbor pVHL7 and renal cysts are present in about 60% of VHL patients,24 this syndrome highly likely represents a ciliopathy. In fact, compared to normal tubular cells, pVHL-defective epithelial cells lining cysts of VHL patients display a reduced frequency of cilia.7 Kidneys isolated from VHL patients typically contain hundreds of VHL mutant and, consequently, HIFα activated preneoplastic lesions, the majority of which are single cells.7, 16 It appears that only a few of these single cell lesions progress further to cysts and about half of them (21–45%)24 are destined to form tumors. This observation suggests that loss of pVHL is necessary but not sufficient for promoting cyst formation in the setting of VHL disease. Recently it has been shown that in renal cysts, glycogen synthase kinase (GSK)3β that regulates pVHL-mediated microtubule stabilization by phosphorylation,25 is subjected to inhibitory phosphorylation through the PI(3)K signaling pathway. Importantly, these cysts exhibit reduced frequencies of primary cilia.7 Thus, in VHL patients combined VHL mutation and GSK3β inactivation is required to disrupt primary cilia, which in turn cause renal cyst formation and possibly ccRCC.
Patients with sporadic ccRCC also frequently have intratumoral cysts.26 Interestingly, 5 of 20 (25%) of our ccRCCs displayed detectable phospho-serine 9-GSK3β (data not shown). Therefore, our findings suggest that cilia degeneration driven by VHL dysfunction and GSK3β inactivation is also an important mechanism of renal carcinogenesis in a significant fraction of non-VHL patients. It also indicates that in a larger subset of sporadic ccRCC, the inactivation of proteins other than GSK3β is required to destabilize primary cilia.
The reduction in primary ciliation could be the result, rather than the cause of the renal carcinogenesis and therefore be reflective of its morphology. For example, pRCC typically has papillary or tubular structures that are lined with tumor cells with apical cell membrane where cilia are found. In contrast, ccRCC is frequently composed of nests or solid sheets of tumor cells with less apical cell membranes. We have therefore looked at whether the number of cilia correlates with the architectural patterns (solid/nested, alveolar/acinar, and papillary). Importantly, a majority of ccRCC showed an acinar or alveolar pattern with apical cell membranes. There was no correlation between the number of cilia and the architectural pattern. We conclude that cilia degeneration is definitely driven by pVHL dysfunction and is not just a reflection of morphology.
In conclusion, this is the first study demonstrating that the frequencies of primary cilia differ significantly between sporadic clear cell and papillary subtypes of renal cell carcinoma in vivo. Low numbers of ciliated tumor cells correlated with the upregulation of HIF targets CA9 and GLUT-1 in ccRCC. In contrast, high cilia frequencies were found in the papillary subtype in which functioning pVHL exists. Our results suggest that loss of primary cilia may contribute to the development of sporadic ccRCC that is characterized by loss of pVHL function. In contrast, pRCC can progress without dismantling their primary cilia. This difference may explain at least in part the different biological behavior and phenotype of ccRCC and pRCC.
References
Singla V, Reiter JF . The primary cilium as the cell′s antenna: signaling at a sensory organelle. Science 2006;313:629–633.
Lin F, Hiesberger T, Cordes K, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 2003;100:5286–5291.
Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271–275.
Hergovich A, Lisztwan J, Barry R, et al. Regulation of microtubule stability by the von Hippel–Lindau tumour suppressor protein pVHL. Nat Cell Biol 2003;5:64–70.
Esteban MA, Harten SK, Tran MG, et al. Formation of primary cilia in the renal epithelium is regulated by the von Hippel–Lindau tumor suppressor protein. J Am Soc Nephrol 2006;17:1801–1806.
Lutz MS, Burk RD . Primary cilium formation requires von Hippel–Lindau gene function in renal-derived cells. Cancer Res 2006;66:6903–6907.
Thoma CR, Frew IJ, Hoerner CR, et al. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol 2007;9:588–595.
Banks RE, Tirukonda P, Taylor C, et al. Genetic and epigenetic analysis of von Hippel–Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res 2006;66:2000–2011.
Thoenes W, Storkel S, Rumpelt HJ . Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas). The basic cytological and histopathological elements and their use for diagnostics. Pathol Res Pract 1986;181:125–143.
Moch H, Mihatsch MJ . Genetic progression of renal cell carcinoma. Virchows Arch 2002;441:320–327.
Eble JN, Sauter G, Epstein JI, et al (eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press: Lyon, 2004.
Schraml P, Struckmann K, Hatz F, et al. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J Pathol 2002;196:186–193.
Alieva IB, Gorgidze LA, Komarova YA, et al. Experimental model for studying the primary cilia in tissue culture cells. Membr Cell Biol 1999;12:895–905.
Brauch H, Weirich G, Brieger J, et al. VHL alterations in human clear cell renal cell carcinoma: association with advanced tumor stage and a novel hot spot mutation. Cancer Res 2000;60:1942–1948.
Kondo K, Klco J, Nakamura E, et al. Inhibition of HIF is necessary for tumor suppression by the von Hippel–Lindau protein. Cancer Cell 2002;1:237–246.
Mandriota SJ, Turner KJ, Davies DR, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002;1:459–468.
Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 2005;25:5675–5686.
Wheatley DN, Wang AM, Strugnell GE . Expression of primary cilia in mammalian cells. Cell Biol Int 1996;20:73–81.
Jiang F, Richter J, Schraml P, et al. Chromosomal imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol 1998;153:1467–1473.
Moch H, Presti Jr JC, Sauter G, et al. Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res 1996;56:27–30.
Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 1994;91:9700–9704.
Jung CR, Hwang KS, Yoo J, et al. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat Med 2006;12:809–816.
Badano JL, Mitsuma N, Beales PL, et al. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 2006;7:125–148.
Choyke PL, Glenn GM, Walther MM, et al. von Hippel–Lindau disease: genetic, clinical, and imaging features. Radiology 1995;194:629–642.
Hergovich A, Lisztwan J, Thoma CR, et al. Priming-dependent phosphorylation and regulation of the tumor suppressor pVHL by glycogen synthase kinase 3. Mol Cell Biol 2006;26:5784–5796.
Neumann HP, Bender BU, Berger DP, et al. Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 1998;160:1248–1254.
Acknowledgements
We thank Silvia Behnke, Susanne Steu, and Martina Storz for technical support. The study was supported by UBS AG (made possible by an anonymous donor), the Swiss National Science Foundation (HM, WK) and the Zurich Cancer League, Switzerland.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article
Cite this article
Schraml, P., Frew, I., Thoma, C. et al. Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod Pathol 22, 31–36 (2009). https://doi.org/10.1038/modpathol.2008.132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.132
Keywords
This article is cited by
-
Modelling ciliopathy phenotypes in human tissues derived from pluripotent stem cells with genetically ablated cilia
Nature Biomedical Engineering (2022)
-
Group III phospholipase A2 downregulation attenuated survival and metastasis in ovarian cancer and promotes chemo-sensitization
Journal of Experimental & Clinical Cancer Research (2021)
-
Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice
Nature Medicine (2017)
-
Ror2 signaling regulates Golgi structure and transport through IFT20 for tumor invasiveness
Scientific Reports (2017)
-
The cilia-regulated proteasome and its role in the development of ciliopathies and cancer
Cilia (2016)
