
Abstract
Research conducted over the last two decades has yielded a detailed understanding of the molecular lesions that contribute to the malignant transformation of hematopoietic stem cells and committed progenitors into the various forms of acute and chronic leukemia. Although our understanding of the molecular pathology of leukemia remains incomplete, the information gained to date has had a profound impact on the way these malignancies are both diagnosed and monitored during therapy. More recently, targeted therapies have been developed against some of the identified genetic lesions. These therapies have led to significant improvements in patient outcomes while simultaneously decreasing therapy-related toxicity. With the advent of genome-wide methods to define the total complement of genetic and epigenetic lesions involved in leukemogenesis, new targeted therapies can be anticipated. This review highlights some of the targeted therapies that are presently being used to treat hematopoietic malignancies and describes some of the recent advances that should have a significant impact on the development of future target therapies.
Similar content being viewed by others
Main
A goal of modern cancer research is to define the underlying molecular genetic lesions responsible for the establishment of the malignant clone. The hope is that this information will lead to the identification of rational molecular targets against which novel therapeutic agents can be developed. Although these hopes are being realized across the spectrum of human cancers, to date their direct influence on clinical care is probably most evident in the treatment of hematopoietic malignancies. Specifically, molecular genetic data have resulted in significant changes in our understanding of the pathogenesis of both acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL), and have resulted in the identification of a number of molecular targets against which targeted therapies have been developed.1, 2 This article summarizes some of the recent advances made in our understanding of the molecular pathology of acute leukemia, and highlights how this information is being used to develop targeted therapies.
The first molecularly targeted cancer therapy: ATRA
Acute Promyelocytic Leukemia
Analysis of chromosomal abnormalities within leukemic blasts revealed somatically acquired chromosomal rearrangements in the majority of cases. The targets of these rearrangements are genes encoding proteins that play fundamental roles in normal hematopoietic cell development, differentiation, proliferation, and survival. The most common targets of these rearrangements are genes that encode DNA-binding transcription factors or transcriptional regulatory proteins. The prototypical translocation is the t(15;17) translocation of acute promyelocytic leukemia (APL). The product encoded by this translocation not only drives the formation of the leukemic clone, but also serves as a specific target for the highly effective therapy, all trans retinoic acid (ATRA).3, 4
APL consists of a clonal population of malignant myeloid cells blocked at the promyelocyte stage of differentiation. In over 95% of cases, a t(15;17)(q22;q21) chromosomal translocation is present within the leukemic cells. This translocation generates a chimeric gene between the promyelocytic leukemia gene (PML) on chromosome 15 and the retinoic acid receptor α gene (RARα) on chromosome 17, and results in the production of a PML-RARα fusion protein. Expression of PML-RARα as a transgene in mice induces acute leukemia with features of APL, confirming the central role of this fusion protein in leukemogenesis.
RARα is a ligand-dependent transcription factor and a member of the nuclear hormone receptor superfamily. In the presence of its ligand, retinoic acid, RARα regulates the expression of a large number of target genes, a subset of which are important for normal myeloid cell differentiation. RARα is organized into distinct functional domains including a DNA-binding domain and a large ligand-binding, heterodimerization, and transactivation domain. PML is a multidomain protein that contains a unique cysteine- and histidine-rich zinc-binding RING finger domain followed by two cysteine/histidine-rich regions known as B-boxes and an α-helical coiled-coil domain. In the PML-RARα chimeric product, nearly all of the key functional domains of each molecule are retained, suggesting that the fusion protein influences the biological function of both normal proteins.
RARα normally dimerizes with a member of the retinoid-X receptor (RXR) family leading to the formation of a heterodimer with high DNA-binding affinity. In the absence of retinoic acid, these RARα/RXR heterodimers bind to specific retinoic acid response elements (RAREs) in the promoter of target genes and induce transcriptional repression. This repression is mediated by the RARα/RXR heterodimer interacting with transcriptional repressors including the nuclear receptor-corepressor N-CoR/SMRT, the transcriptional corepressors Sin3A or Sin3B, and histone deacetylases (HDACs). The latter enzymes deacetylate lysine residues in nucleosomal histones, leading to a closed chromatin structure and repression. Binding of retinoic acid to RARα induces a conformational change that causes disassembly of the repression complex and the recruitment of transcriptional coactivators including CBP and p300 to RARα/RXR. CBP and p300 contain intrinsic histone acetylase (HAT) activity, which acetylates the lysine residues of histones resulting in nucleosome unfolding and an increased accessibility of the DNA to transcription factors. In addition, HATs can directly acetylate transcription factors and other cellular proteins and alter their activity.
PML is expressed in all cells and exists both as a free form that localizes within the nucleoplasm and as a conjugated form covalently linked to the ubiquitin-like protein SUMO, which localizes to the so-called PML nuclear bodies (NBs) or PML oncogenic domains. Approximately 10–30 PML NBs are present within a cell. In addition to PML, these NBs contain over 30 other proteins. Recent studies have demonstrated that PML functions in several cellular processes, including proliferation, senescence, and apoptosis. Importantly, chemical-induced tumors occur at an increased frequency in PML-deficient mice, suggesting that PML is a tumor suppressor.
Mechanism of Transformation in APL and the Role ATRA Played in Its Discovery
Clinical trials initially performed in China revealed a unique sensitivity of APL to ATRA. The most remarkable aspect of these observations were that they occurred before RARα was identified as one of the targets of the t(15;17) translocation. In fact, the results of these clinical studies were in part the driving force to assess whether the RARα gene might be involved in the pathogenesis of this acute leukemia subtype. With the cloning of the translocation, the reasons for APL's responsiveness to ATRA were quickly elucidated.
PML-RARα induces leukemia in part by inhibiting the normal biological activities of both RARα and PML. As predicted from its structure, PML-RARα continues to bind RAREs, but is unresponsive to retinoic acid, and thus instead of activating transcription, it dominantly represses transcription of ligand-bound RARα. Moreover, the PML-RARα fusion protein sequesters RXRs away from other nuclear hormone receptors, such as those for vitamin D and thyroid hormone, resulting in an inhibition of their transcriptional activity. The combined effect on the transcriptional cascade controlled by the nuclear hormone receptors contributes to the block in differentiation at the promyelocyte stage. PML-RARα also induces disruption of normal PML NBs and the redistribution of PML into a microparticulate pattern within the nucleus. This change in nuclear localization impairs the normal growth-inhibitory and proapoptotic effects of PML. Recent studies suggest that these activities of PML-RARα may result from the full-length fusion protein, as well as the cleaved product that results from proteolytic cleavage by neutrophil elastase, a neutral serine protease expressed at high levels in promyelocytes.
PML-RARα, like wild-type RARα, directly binds the nuclear corepressor complex, resulting in transcriptional repression. Unlike the wild-type receptor, however, PML-RARα is unresponsive to physiological levels of retinoic acid and remains a constitutive repressor in its presence. ATRA, by contrast, induces a conformational change in PML-RARα resulting in the release of NcoR and its replacement by transcriptional coactivators. Thus, treatment with ATRA reverses the inhibitory activity of PML-RARα on RARα. Moreover, ATRA induces cleavage of PML-RARα through a proteasome-mediated pathway reversing the effect of the fusion protein on normal PML functions. Together, these activities induce growth arrest and terminal differentiation of the malignant promyelocytes.
Although ATRA is highly effective at inducing the terminal differentiation, it is not curative as a single agent. Moreover, ATRA resistance resulting from the acquisition of mutations in the RARα ligand-binding domain of the fusion protein is a frequent occurrence. As a result, ATRA is now used along with other chemotherapeutic agents, a strategy that has been shown to be superior to either ATRA or chemotherapy alone.
Recently, studies have demonstrated APL cases to be sensitive to treatment with arsenic trioxide (As2O3). In contrast to ATRA, As2O3 does not bind to RARα, but instead binds to the PML portion of the chimeric product, inducing its degradation through a proteasome-dependent mechanism. Arsenic trioxide's primary effect on promyelocytes is thought to be induction of apoptosis. Protocols combining the use of ATRA, As2O3, and standard chemotherapeutic are showing promise for relapse disease, and may provide a survival advantage over standard ATRA plus chemotherapy for newly diagnosed patients.
Histone deacetylase inhibitors
The lessons learned from APL suggest that reversing constitutive transcriptional repression could lead to terminal differentiation of leukemia cells. In APL, ATRA reverses repression by directly inhibiting the ability of the oncogenic PML-RARα fusion protein to direct the repression complex to the transcriptional regulatory region of target genes. An alternative approach is to directly inhibit the enzymatic activity of the histone deacetylases within the transcriptional repression complex.5, 6, 7
The relative state of chromatin in the transcriptional regulatory regions of genes plays a key role in controlling the level of gene transcription. The nucleosome serves as the basic unit of chromatin structure and consists of approximately two turns of the DNA double helix wound around a histone octamer core. A diverse range of post-translational modifications of the amino-terminal tails of histones controls the packing of the nucleosomes, and thus the relative access of the DNA to transcriptional regulatory proteins. Modification of the histone tails includes methylation, phosphorylation, ubiquitination, sumoylation, and acetylation. Acetylation is controlled by the balance between HATs and HDACs. HATs transfer acetyl groups to the amino-terminal lysine group in histones, resulting in the elimination of the positive charge and an opening of the chromatin structure. HDACs catalyze the removal of the acetyl group and lead to a condensation of the chromatin and transcriptional repression. A relatively large number of small molecule histone deacetylase inhibitors (HDACi) have been developed and tested in preclinical and clinical trials.5, 6, 7 These agents have been shown to not only have anticancer activity, but to demonstrate a remarkable level of tumor specificity, with cancer cells showing a 10-fold greater sensitivity than normal cells. Typically, these agents induce apoptosis of cancer cells through a combination of targeted effects on the oncogenic transcription factors and through more global effects on gene transcription and non-transcriptional targets.5, 6, 7, 8, 9
In addition to PML-RARα, a large number of other translocation-encoded onco-fusion proteins function in part through transcriptional repression. These include t(8;21) (AML1-ETO), inv(16) (CBFβ-MYH11), t(1;19) (E2A-PBX1), t(8;16) (MOZ-CBP), and t(11;16) (MLL-CBP), as well as others. As predicted, HDACi have been demonstrated to have activity in these leukemias. The activity of the HDACi even within these leukemias, however, goes beyond the transcriptional repression mediated by the oncogenic transcription factors. Specifically, cancer cells appear to have global modifications of their chromatin that lead to wide ranging changes in gene transcription. This modified epigenome provides a setting where the HDACi lead to differential effects (gene activation) between normal and cancer cells. This differential in part explains the marked difference in the sensitivity of cancer cells vs normal cells to the HDACi.
Preclinical work with several HDACi has shown impressive antitumor activity for a variety of cancers, including acute leukemia. In phase I clinical trials, these agents have been well tolerated and have shown evidence of partial responses. These encouraging results are being followed in single-agent phase II studies and in combination with a variety of other agents. The theoretical underpinning of this work suggests that HDACi will likely become a stable part of the combinatorial treatment of cancer.
Tyrosine kinase inhibitors in acute leukemia
Protein kinases play a central role in the regulation of a wide variety of intracellular signaling pathways. Through reversible post-translational modifications of substrate proteins, kinases influence nearly all cellular processes, including development, differentiation, metabolism, proliferation, cell movement, and apoptosis. In addition, aberrant kinase signaling is a recurrent mechanism implicated in cellular transformation. Constitutive activation of protein kinases through oncogenic mutations, autocrine or paracrine stimulation, or overexpression as a result of genomic amplification has been documented in a wide spectrum of human cancers.
In normal hematopoiesis, both tyrosine and serine/threonine kinases play essential roles in cytokine-induced proliferation, survival, and differentiation. Thus, oncogenic activation of members of this extended family of genes would be predicted to contribute to the neoplastic transformation of hematopoietic stem cells (HSCs) and lineage-committed progenitors. In fact, it has been suggested that constitutive activation of growth factor receptors or downstream components of their signaling cascades may be an obligate step in leukemogenesis, providing a proliferative and/or survival advantage.
As described in the first lecture in this course, one of the first examples of the contribution of altered protein kinase signaling to leukemogenesis was the identification in chronic myeloid leukemia (CML) of the t(9;22)-encoded BCR-ABL fusion protein, a constitutively activated form of the ABL tyrosine kinase. A second example is the constitutive activation of the FMS-related tyrosine kinase-3 (FLT3), a member of the class III receptor tyrosine kinase family, which also includes CSFR, c-kit, PDGFRα, and PDGFRβ. FLT3 is comprised of an extracellular domain, a juxtamembrane (JM) domain just within the cytoplasmic membrane, and two tandem tyrosine kinase domains. FLT3 activates several downstream signaling molecules through direct phosphorylation or association, including the p85 subunit of phosphatidyl inositol, SHC, SHP2, GRB2, STAT 5A, and STAT 5B. Although the expression of FLT3 is normally restricted to early hematopoietic progenitors, it is also expressed in many primary hematopoietic malignancies, including the majority of AMLs, a proportion of B-cell and T-cell lineage ALLs, and CML in lymphoblast crisis. These data suggest that aberrant signaling through the FLT3 receptor may play a role in the survival or proliferation of leukemic blast. Consistent with this suggestion, stimulation of FLT3-expressing leukemias with the FLT3 ligand (FL) induced a dose-dependent proliferation of leukemic blasts. In addition, a significant proportion of leukemic cell lines coexpress both FL and FLT3, suggesting that autocrine stimulation may play a role in the proliferation of leukemia blast.
More recently, activating mutations have been identified in FLT3 in a subset of leukemias.10 Specifically, an internal tandem duplication (ITD) of the JM region of FLT3 was shown to induce constitutive activation of the receptor's tyrosine kinase activity. The length of ITD varies from 3 to ≥400 base pairs, and is always in-frame. Overall, the frequency of this mutation has been estimated to be 15–25% in AML, but less than 1% in cases of ALL. A second less common type of FLT3 activating mutation is a missense point mutation in codons encoding amino acids within the activation loop of the kinase domain. The activation loop is a general component of tyrosine kinase and exerts autoinhibitory function by limited access of ATP and substrate to the catalytic domain. FLT3 activation loop mutations have been identified in approximately 7% of AML, but is again only rarely seen in cases of ALL. Among all the mutations identified, the most common is D835Y; other substitutions include D835V, D835H, D835E, D835N, and I836M. Importantly, the presence of an FLT3 ITD, and to a lesser degree the D835/836 point mutations, is predictive of a poor prognosis. The frequency of FLT3 mutations in AML is somewhat higher in adults than in children, largely due to the rarity of these mutations in children less than 10 years of age. Furthermore, the frequency of FLT3 mutations varies significantly between genetic subtypes of AML, with the highest incidence in APL.
Stable expression of either class of FLT3 mutations renders hematopoietic cells growth factor-independent. Indeed, mice receiving bone marrow cells expressing mutant FLT3 develop an oligoclonal myeloproliferative disorder characterized by splenomegaly and leukocytosis.
The high frequency of FLT3 mutations makes this an ideal candidate for a small molecule inhibitor. A number of inhibitors have been developed with varying degrees of specificity. These include CEP-701 (Cephalon), PKC-412 (Novartis), and SU11248 (Sugen). Phase I/II clinical studies have been carried out in relapse patients with AML or MDS with mutant FLT3. These studies have demonstrated significant partial responses in a proportion of the patients, although the responses were short lived. Biochemical analysis demonstrated effective inhibition of FLT3 kinase activity at the doses achieved. The limited responses are not surprising and FLT3 inhibitors in combination with other therapeutic agents will likely be required for optimal clinical effects. Phase I studies of combinations of these FLT3 inhibitors with standard AML therapies are in progress.
Monoclonal antibodies as therapeutic agents
Over the last two decades, detailed phenotypic studies have led to the identification of leukemia-specific immunophenotypic signatures consisting of lineage-specific and nonspecific antigens in the majority of ALLs and AMLs. These phenotypic signatures are now routinely used to measure the so-called minimal residual disease and to modify therapy based on these measurements. What has been less rewarding is the search for individual leukemia-specific antigens, that is surface proteins that are expressed only on leukemic blasts and not on normal HSCs or lineage-committed progenitors.
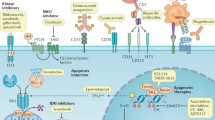
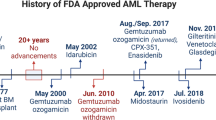
An antigen that comes close to these criteria is CD33 in AML. CD33 is a 67 kDa surface glycoprotein of unknown function that is expressed on the leukemic blasts in over 80% of cases, but is not expressed on normal HSCs. Monoclonal antibodies (mAbs) against CD33 are routinely used in the characterization of leukemias. To convert mAbs to therapeutic agents, the antibodies need to be humanized to reduce immunogenicity, enhance stability in circulation, and improve their ability to recruit cytotoxic cells and complement for Ab-mediated cell killing.11, 12 A humanized IgG4κ anti-CD33 mAb has been developed. This antibody has been conjugated to the chemotherapy agent calicheamicin through a hydrazone linker to form the therapeutic agent known as gemtuzumab ozogamicin (Mylotarg).13
Gemtuzumab ozogamicin bind to CD33 on the surface of AML blasts and is rapidly internalized by receptor-medicated endocytosis. Upon entering the acidic environment of the lysosome, the hydrozone linker is cleaved resulting in the release of calicheamicin. Calicheamicin is a highly potent antitumor antibiotic that cleaves double-stranded DNA, resulting in tumor cell killing. Importantly, calicheamicin is highly toxic and cannot be delivered systemically without significant toxicity. Thus, by targeting the agents specifically to leukemic blasts, gemtuzumab ozogamicin allows the effective use of this agent for the treatment of AML.
In clinical trials, gemtuzumab ozogamicin has shown impressive results, with 30% of patients in first relapse achieving a complete remission, with median relapse-free survival of 7.2 months. More recent studies have suggested that gemtuzumab ozogamicin may yield a higher complete remission rate when administered with intensive chemotherapy. Phase III studies are underway in which gemtuzumab ozogamicin is being incorporated with standard chemotherapy in newly diagnosed patients. If positive results are obtained from these studies, it would result in the first change in standard induction therapy in AML in over 30 years. Treatment with gemtuzumab ozogamicin is not without side effects. Severe neutropenia and thrombocytopenia are frequently seen and this is consistent with the expression of CD33 on maturing myeloid progenitors. Transient and reversible hepatotoxicity is also seen in a proportion of patients, and occasional cases of veno-occlusive disease have been documented.
Are there new targets for directed therapy?
An important observation from modeling leukemias in mice is that individual oncogenic lesions alone are insufficient to generate a full leukemia. Thus, lesions affecting multiple different pathways are required to lead to cellular transformation. Candidate gene approaches have been used to identify contributing lesions and have led to the identification of deletions and epigenetic silencing of CDKN2A and mutations of NOTCH1 in a high percentage of T-lineage ALL cases. However, for most leukemias, the full complement of cooperating lesions remains to be defined. It is anticipated that identifying these lesions will help to define the molecular ‘Achilles heels’ against which targeted therapy can be developed.
Contributing mutations include deletions, amplifications, and point mutations, as well as epigenetic changes that influence gene expression.14 The identification of the full complement of point mutations within a population of leukemia cells is cost prohibitive with present sequencing methodology. However, rapid advances in re-sequencing approaches are being made and it is anticipated that full genome sequence of individual cases will be achievable at a reasonable cost within the next 5–10 years. A variety of genome-wide approaches have been developed to identify copy number alterations including fluorescence in situ hybridization, comparative genomic hybridization (CGH), array-CGH, and single nucleotide polymorphism (SNP) arrays. Array-based methods involve hybridizing leukemia cell DNA to arrays containing thousands of pieces of DNA either as bacterial artificial chromosome (BAC) or oligonucleotide probes. The resolution is dependent upon the platform used and can vary from a mean intermarker distance of over 1 Mb for older BAC arrays to less than 5 kb for newer oligonucleotide-based platforms. A particularly powerful platform is the 500K SNP arrays from Affymetrix. By comparing the SNP genotype of germline and tumor samples, these arrays allow the detection of copy-neutral loss of heterozygosity (or uniparental disomy), in addition to directly inferring DNA copy number from probe hybridization intensity.
As a representative example, we have recently used Affymetrix 500K SNP arrays to detect copy number alterations in pediatric ALL at a resolution of approximately 5.0 kb. This analysis is very accurate and provides a fingerprint of deletions and amplifications. The methodology was able to identify the known DNA copy number changes including the multiple whole chromosomal gains characteristic of hyperdiploid B-progenitor ALL with >50 chromosomes, the duplication of chromosome 1q in ALL with t(1;19)(q23;p13) as a result of unbalanced translocation, and deletions at 9p21.3 (harboring the CDKN2A and CDKN2B genes) and 12p13.2 (ETV6). In addition to these known lesions, this analysis identified a number of new lesions. The most striking finding was mono-allelic deletions of genes that play a critical role in regulating B-cell development and differentiation in approximately 40% of B lineage cases.15
The most frequently targeted gene within the B-cell development pathway, occurring in almost 30% of cases, was PAX5, a paired domain transcription factor located at 9p13. The PAX5 copy number alterations included mono-allelic loss in 53 cases, bi-allelic loss in 3 cases, and an internal amplification in 1 case. Importantly, these alterations were somatic in nature and were present in over 90% of blasts. Moreover, almost half of the deletions were confined to PAX5, with most resulting in the deletion of only a subset of PAX5 exons. The PAX5 lesions are predicted to result in either haploinsufficiency or the generation of hypomorphic alleles that produce proteins lacking the DNA-binding domain or the transcriptional activation domain. In addition, four cases were identified with cryptic translocations (two cases with PAX5-TEL, one case with PAX5-FOXP1, and one case with PAX5-EVI3) and 14 cases had point mutations including missense, frame-shift, and splice-site mutations that clustered in the DNA-binding paired domain and transactivation domains. In addition to PAX5, a number of other genes involved in B-cell development and differentiation were found to be deleted including EBF (8 cases), Ikaros (19 cases), Aiolos (6 cases), TCF3 (1 case), and BLNK (2 cases). Taken together, mutations in genes regulating B-cell development were identified in over 40% of pediatric B-progenitor ALL.
Mice null for PAX5 show a complete arrest in B-cell development at the pro-B-cell stage of development prior to completing immunoglobulin heavy chain rearrangement. Moreover, loss of a single EBF allele leads to a partial arrest in differentiation at this stage of B-cell development. Thus, the simplest interpretation of the data is that the identified mutations lead to a reduction in the functional level of PAX5 and/or other key regulators of B-cell development and differentiation and contribute to the block in differentiation at the pro-B-cell stage of development. In addition to contributing to the block in differentiation, the mutations are likely to also collaborate with other known genetic lesions in leukemogenesis.
The most exciting possibility raised by these data is to use the genetic defects as a way of developing leukemia-specific therapy. For example, can a small molecule inducer of differentiation (agonist) bypass the block resulting from the identified lesions, and will this in turn trigger a leukemia cell-specific apoptotic response. With recent advances in high-throughput screening methodology and the availability of small molecule libraries, such screens can be readily accomplished. The combination of genome-wide screening approaches to identify the complete complement of genetic lesions within a cancer, coupled with high-throughput screens for new small molecules targeted toward these newly identified lesions, should lead to a rapid expansion in our understanding of the molecular pathology of leukemia and in the number of drugs at our disposal to effectively treat these cancers.
Summary
Significant advances have been made in our understanding of the molecular pathology of acute leukemias. These data are beginning to have a direct impact on our ability to accurately diagnose, classify, and treat these cancers so that each patient has the best chance for a cure. The recent use of the so-called targeted therapies directed toward specific genetic lesions within leukemic cells has further advanced cure rates while simultaneously reducing treatment-associated toxicities. This is best exemplified by the use of ATRA as part of front-line therapy for newly diagnosed patients with APL. The recent advances in our understanding of the molecular pathology of acute leukemia coupled with methodological advances in genome-wide scans for mutations and high-throughput screens for drugs should lead to a significant increase in the number of targeted agents over the next decade.
References
Tallman MS, Gilliland G, Rowe JM . Drug therapy for acute myeloid leukemia. Blood 2005;4:1154–1163.
Estey E, Dohner H . Acute myeloid leukaemia. Lancet 2006;368:1894–1907.
Chou WC, Dang CV . Acute promyelocytic leukemia: recent advances in therapy and molecular basis of response to arsenic therapies. Curr Opin Hematol 2004;12:1–6.
Douer D, Tallman MS . Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol 2005;23:2396–2410.
Johnstone RW . Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 2002;1:287–299.
Marks PA, Richon VM, Miller T, et al. Histone deacetylase inhibitors. Adv Cancer Res 2004;91:137–168.
Bolden JE, Peart MJ, Johnstone RW . Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006;5:769–784.
Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med 2005;11:71–76.
Nebbioso A, Clarke N, Voltz E, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med 2005;11:77–84.
Levis M, Small D . FLT3 tyrosine kinase inhibitors. Int J Hematol 2005;82:100–107.
Harris M . Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol 2004;5:292–302.
Wu AM, Senter PD . Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 2005;23:1137–1146.
Linenberger ML . CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia 2005;19:176–182.
Veltman JA, de Vries BB . Diagnostic genome profiling: unbiased whole genome or targeted analysis? J Mol Diagn 2006;8:534–537.
Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007;446:758–764.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Downing, J. Targeted therapy in leukemia. Mod Pathol 21 (Suppl 2), S2–S7 (2008). https://doi.org/10.1038/modpathol.2008.13
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.13
