
Abstract
Miniaturized gas chromatography (µGC) systems hold potential for the rapid analysis of volatile organic compounds (VOCs) in an extremely compact and low-power enabled platform. Here, we utilize microfabrication technology to demonstrate the single chip integration of the key components of a µGC system in a two-step planar fabrication process. The 1.5 × 3 cm microfluidic platform includes a sample injection unit, a micromachined semi-packed separation column (µSC) and a micro-helium discharge photoionization detector (µDPID). The sample injection unit consists of a T-shaped channel operated with an equally simple setup involving a single three-way fluidic valve, a micropump for sample loading and a carrier gas supply for subsequent analysis of the VOCs. The innovative sample injection technique described herein requires a loading time of only a few seconds and produces sharp and repeatable sample pulses (full width at half maximum of approximately 200 ms) at a carrier gas flow rate that is compatible with efficient chromatographic separation. Furthermore, our comprehensive characterization of the chip reveals that a wide variety of VOCs with boiling points in the range of 110–216 °C can be analyzed in less than 1 min by optimizing the flow and temperature programming conditions. Moreover, the analysis of four VOCs at the concentration level of one part per million in an aqueous sample (which corresponds to a headspace concentration in the lower parts-per-billion regime) was performed with a sampling time of only 6 s. The µDPID has demonstrated a linear dynamic range over three orders of magnitude. The system presented here could potentially be used to monitor hazardous VOCs in real time in industrial workplaces and residential settings.
Similar content being viewed by others

Introduction
Over the past several decades, the progress in microfabrication technology has revolutionized the fields of computing and signal processing as well as the automobile industry. The miniaturization of well-established analytical instruments is another paradigm in which the use of this technology has produced significant advancements. One such example is the gas chromatography (GC) system, which is used in various scientific, medical, and industrial settings to separate and analyze volatile organic compounds (VOCs). The monitoring of VOCs is of interest in various applications, including homeland security, space and fossil fuel exploration, worker exposure assessment, and biomedical diagnostics1–8. Miniaturized GCs (µGCs) are being intensely developed to enable the rapid diagnosis of VOCs in remote locations with low cost and low consumption. A typical µGC consists of three important components: an injector/pre-concentrator for sample introduction9,10, a micromachined column for VOC separation11,12 and a single detector or array of detectors13,14 located at the column exit to identify the separated compounds.
The majority of standalone µGC systems reported to date address the manual assembly of separately fabricated µGC components using commercially available off-chip fluidic interconnects15–19. This approach is commonly known as the hybrid integration method. The hybrid integration method has certain benefits, such as the optimization of components and the absence of thermal crosstalk between individual µGC components. Nevertheless, the implementation of µGC systems in a hybrid format is time consuming, cumbersome, expensive, and prone to error. In addition, the hybrid format degrades the overall performance of µGCs due to the presence of cold spots between transfer lines. The condensation of compounds in these cold spots can result in extensive peak broadening. In particular, high boilers experience peak broadening in a more profound manner, which can negatively impact their analysis. In contrast, a chip-scale GC system, in which all three components are located on a single chip, may suffer from thermal crosstalk between the individual µGC components. Thermal crosstalk can occur during two chromatographic events: (1) the desorption process of the pre-concentrator and (2) the temperature programming of the column. Thermal crosstalk could potentially change the retention time of a compound and also produce undesirable drift or noise in the detector signal. Fortunately, these issues can be avoided in the chip-scale GC platform by developing an optimal back-end detector technology.
Recently, there have been significant advancements in detector technology, and a wide variety of chemical detectors have been developed13,14,20–27. The micro-helium discharge photoionization detector (µDPID) presented by our group has demonstrated a minimum detection limit of approximately 10 pg, which is the same as that of a flame ionization detector (FID), which is mainly used in bench-top GC systems28. This high sensitivity of µDPIDs reduces the system’s reliance on the front-end pre-concentration step, which is typically performed to improve the detection limit of the detector. In addition, we have previously demonstrated that the µDPID can be easily integrated with the separation column and demonstrated its operation under the temperature and flow rate programming conditions28. These two features of µDPIDs make them a promising candidate for developing chip-scale GC systems, thereby addressing the above-mentioned issues and improving the overall cost and performance of this technology.
The first µGC, which was developed in 1979 by Terry and co-workers29, included a monolithic integrated sample injection loop, a 1.5 m long separation column, and a separately fabricated thermal conductivity detector (TCD). Their hybrid µGC system required the TCD to be integrated after fabrication. In addition, TCDs generally have poor sensitivity (e.g., detection limit of 10 parts per million in Terry’s work) and also produce an unstable baseline when operated under flow and temperature programming conditions (the TCD temperature must be controlled to ±0.1 °C or better for baseline stability)30. Sandia National Laboratories launched µChemLabTM for homeland security applications in 199831. Their research is unique in that it was focused on the development of a truly monolithic integrated μGC system. Their system included a preconcentrator, a separation column (86 cm long, 100 μm wide, and 400 μm deep) and a magnetically actuated pivot plate resonator (PPR) fabricated on a silicon-on-insulator wafer. Although the fabrication and subsequent coating mechanism for the integrated chip were demonstrated, no chromatogram was published using an on-chip PPR detector32.
This paper is intended to demonstrate a truly compact chip-scale GC platform that has dimensions of 1.5 cm × 3 cm. This lab-on-a-chip GC has three essential elements—a sample injector, separation column, and detector—all on the same platform. Our results indicate that a loading time of a few seconds is required to introduce a VOC mixture into the chip, with the subsequent analysis performed in less than a minute for compounds with a wide range of boiling points (110–216 °C). This development will be particularly suitable for producing low-cost and efficient µGC systems for the rapid analysis of compounds in real-time situations.
System description
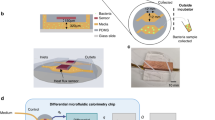
Figure 1a illustrates the configuration of fluidic connections between the chip, valve, miniaturized pump, and helium carrier gas supply. The valve position and pump are controlled by an eight-bit microcontroller interfaced with a keypad to initiate the command. The operation of the chip-scale GC platform is quite straightforward. In normal operation, the valve is connected to the carrier gas supply. The sample inlet port is manually inserted into a sealed vial containing the unknown mixture of VOCs. To avoid liquid phase injection, the sample inlet port is kept in the headspace volume above the sample. To perform an injection, a single command from the keyboard initiates a sequence of events, which is completed within a few seconds. First, the valve is switched to the pump position, and the pump is turned on. The pump applies a negative pressure (flow rate of 1 mL min−1) to load the sample into the fluidic channel (internal volume of approximately 1.2 µL) connecting the valve and chip. Then, the valve position is turned again to the carrier gas position, which injects the sample into the µSC (Figure 1b). The sample moves through the µSC with helium carrier gas and is separated into individual compounds. The separation process is based on the relative solubility of the compounds in the silane-treated alumina stationary phase (Figure 1c) and their relative vapor pressures. As the compounds separated in space and time elute from the µSC, they are ionized with high-energy (>10 eV) photons. These photons are generated through a microplasma produced in helium by applying a direct current (DC) voltage of 550 V across a pair of excitation electrodes. The gap between the excitation electrodes is approximately 20 µm. The microplasma is sustained by a constant supply of helium from the auxiliary channel. A 50 MΩ ballast resistor is connected in series with the excitation electrode to limit the current flow, which could potentially damage the electrodes. In addition to the pair of excitation electrodes, the detector has a bias and collector electrode. The bias and collector electrodes are 1.5 mm apart, and the space between them is called the collector volume. The circuit shown in Figure 1d measures the signal in the form of an electrical current, which is produced through the ionization of VOCs. Here, a pico-ammeter and the bias voltage are connected in series to the collector and bias electrode, respectively. The positive voltage applied to the bias electrode assists in capturing the electron inside the collector volume. The recombination of electrons with the ionized species can reduce the measured current. The current measured by the pico-ammeter is fed to the Keithley 2700 multimeter (Tektronix, Beaverton, OR, USA), which communicates with the LABVIEW program to plot the output signal.

Conceptual image describing the major components of the chip-scale GC platform: (a) setup showing the configuration of fluidic interconnections between the chip, valve, micropump, and carrier gas; (b) description of the injection mechanism: the top image shows the loading phase, whereas the bottom one shows the injection phase; (c) coating mechanism for the µSC; (d) electrical circuit for measuring the current signal produced by the ionization of VOCs. This figure is not drawn to scale. The size of the helium cylinder can be customized according to the requirements of the application.
Materials and methods
Materials
All VOCs listed in Table 1 were purchased from Sigma-Aldrich (St. Louis, MO, USA) with >99% purity. Silicon wafers (n-type, 4 in diameter, 500 µm thickness, single side polished) and Borofloat wafers (4 in diameter, 700 µm thickness, double side polished) were purchased from University Wafers (Boston, MA, USA) and Coresix Precision Glass (Williamsburg, VA, USA), respectively. Ultra-high-purity helium was purchased from Airgas (Christiansburg, VA, USA). Fused silica capillary tubes (100 µm Inner Diameter and 200 µm Outer Diameter) were purchased from Polymicro Technologies (Phoenix, AZ, USA). The miniaturized pump (P/N SP 270 EC-LC-L) and three-way latching solenoid valve (LHLA0521111H) were purchased from Schwarzer Precision (Essen, Germany) and Lee Company (Westbrook, CT, USA), respectively.
Chip fabrication
The chip fabrication follows similar processing steps as reported in our previous publication28. Each branch of the T-shaped sample injection unit is 2 mm long and 250 µm wide. The T-shaped injection unit, separation stage (1 m long, 190 µm wide, and 240 µm deep with 20 µm embedded circular pillars), fluidic ports and cavity for the detector were fabricated in silicon using the bulk micromachining technique. Atomic layer deposition was used to coat a thin layer of alumina (approximately 10 nm) at 250 °C, which serves as a stationary phase. The detector electrodes were fabricated on the Borofloat wafer by evaporating 700 nm/40 nm of Ti/Au. The silicon and Borofloat wafers were diced, and the individual devices were bonded together. Next, the detector cavity was sealed with epoxy, electrical wires were soldered to the bond pads and capillary tubes were inserted into the four ports of the chip. A solution containing 10 mM chlorodimethyloctadecylsilane in toluene was filled in the µSC and left at room temperature for 24 h. Finally, the solution was purged with nitrogen for 30 min. The µSC was conditioned inside the conventional GC system for approximately 1 h (35 °C, ramped at 2 °C min−1to 140 °C) at a constant helium pressure of 10 psi.
Results and discussion
The top and cross-sectional images of the µSC using scanning electron microscopy (SEM) are shown in Figure 2a and b, respectively. The optical image of the packaged device is shown in Figure 2c with the capillaries attached to all four ports of the chip and the electrical connections in place. We first explain the performance evaluation of the sample injection unit and µDPID. Both components were diced from the integrated chip for that purpose. Then, the chromatographic analysis performed using the integrated chip will be demonstrated.

SEM images of the semi-packed µSC column showing (a) a top view of the channel with embedded 20 µm circular micropillars; (b) a cross-sectional view showing high-aspect-ratio pillars; (c) an optical image of the packaged chip.
Evaluating sample injection unit
The performance of the sample injection unit was characterized in terms of the peak parameters (full width at half maximum(FWHM), peak height, and area). Five compounds—heptane, toluene, chlorobenzene, ethylbenzene, and p-xylene—were used in these tests. Each compound was separately injected at flow rates set to five discrete values of 0.3, 0.45, 0.72, 1, and 1.4 mL min−1. These flow rates correspond to inlet pressures of 10, 15, 20, 25, and 30 psi. The loading time of 500 ms was used for all VOCs. The outlet of the injector was connected to the FID installed in the conventional GC system (HP 7890). Triplicate runs were performed for each value, and the average values of the FWHM and peak height were plotted, as shown in Figure 3.

(a) Effect of the carrier gas flow rate on the FWHM value of the injection plug. The curves were approximated with a polynomial trend line of order two. The R-squared values were 0.88, 0.96, 0.91, 0.98, and 0.97 for heptane, toluene, chlorobenzene, ethylbenzene, and p-xylene, respectively. The standard error calculated for each point over triplicate runs were <10%, showing that the sample injection unit produces repeatable injections. (b) Effect of the carrier gas flow rate on the peak height of the injection plug. The curves were approximated with a linear trend line. The R-squared values were 0.96, 0.96, 0.99, 0.96, and 0.97 for heptane, toluene, chlorobenzene, ethylbenzene, and p-xylene, respectively. The standard errors calculated for each point over triplicate runs were <10%.
The results illustrate that for all VOCs, the FWHM depends on the injector flow rate condition. All compounds experienced a sharp decrease in FWHM between 0.3 and 0.72 mL min−1followed by a more gradual decrease. A similar trend has been observed previously14 and in our experiments when a 20 cm long uncoated capillary tube was connected between a conventional GC injector and the FID. The inverse proportionality of the FWHM with the flow rate can be attributed to different factors. First, increasing the carrier gas flow rate will increase the sample injection rate from the sample loop to the detector, which reduces the extra-column band broadening. Second, the solutes are swept faster, and the longitudinal diffusion in the mobile phase is decreased as a result. Molecules diffuse in the carrier gas from the region of high concentration to that of lower concentration over time33. Increasing the flow rate decreases the time that molecules spend in the injector and connecting tubes, and therefore, the diffusion of molecules in the carrier gas decreases, which results in lower FWHM values. Furthermore, the on-chip injector tested herein operates at room temperature, which can contribute to the differences in the FWHM of high- and low-volatility compounds. In summary, the overall decreases in the FWHM for heptane, toluene, chlorobenzene, ethylbenzene, and p-xylene over the entire flow rate range were 40%, 59%, 59%, 50%, and 64%, respectively.
Because the FID is sensitive to the mass flow rate, the decrease in the FWHM was compensated for by a corresponding increase in the peak height to maintain uniform peak area. Figure 3b shows that the peak height increases linearly over the entire flow rate range (0.3–1.4 mL min−1) for all VOCs. This increase in the peak height is attributed to the increase in mass over time, as measured by the FID. The peak area for all compounds remained approximately constant for all flow rate conditions. Peak areas were calculated to be approximately 20 pA × s for these compounds except for ethylbenzene, which had a peak area of 35 pA × s.
Furthermore, the repeatability of the sample injection unit was investigated by monitoring the change in the peak parameters (FWHM, peak height, and area) over multiple headspace injections. For this purpose, toluene was chosen as a test compound, and multiple injections were performed in succession approximately every 30 s. The flow rate in this experiment was set to 1.4 mL min−1. The sample injection unit produced highly repeatable results, with less than 5% variation in the standard error values for peak parameters over six injections. The FWHMs for these injections were approximately 200 ms.
Linearity of the µDPID
The previous characterization of the µDPID has provided encouraging results, indicating a minimum detection limit of approximately 10 pg, a response time of approximately 200 ms and highly stable excitation electrodes over a long period of time28. Next, we experimentally analyzed the linear detection range of the detector.
To evaluate the linear range of the µDPID, the inlets of the detector were connected to injectors A and B of the conventional GC system. The pressures of injectors A and B were set to 15 and 10 psi, respectively. A direct current (DC) voltage of 550 V was applied (using PS-310, Stanford Research Systems, Sunnyvale, CA, USA) to create the discharge, and the bias voltage was set to 30 V. This value of the bias voltage was selected based on our previous work, which showed the enhanced sensitivity of the detector at 30 V28. Four test compounds—toluene, tetrachloroethylene, chlorobenzene, and ethylbenzene—were used. The different headspace volumes of the test compounds sealed in a vial were sampled using an autosampler (7359A) module to ensure repeatable injections. Assuming ideal gas law behavior, the mass of the compound injected from a saturated vapor above the pure liquid could be calculated from the injection volume and split ratio used for the injection. The injected mass was in the range of 10 pg to 10 ng. The response of the detector was measured in terms of the peak area for each injection. The peak area was selected as an indicator for the quantitative data analysis (instead of peak height) based on our previous results, which indicated that the µDPID is a mass flow rate-sensitive detector (MSD)28. For any MSD, variations in the flow rate could give rise to changes in the peak height or width; however, negligible changes are observed in the peak area. Therefore, the quantification of a compound in terms of peak area is more accurate. Each data point was repeated three times, and the average peak area was plotted against the injected mass of each test compound (Figure 4a). Because the injected mass extends over several orders of magnitude, these curves have been plotted on a log–log scale for clarity (Figure 4b).

(a) Calibration curves for the µDPID produced by injecting different masses (10 pg–13 ng) of four test compounds and calculating the response (peak area). Each point was repeated in triplicate, and the average value was plotted against the injected mass. Linear regression (forced zero Y-intercept) R2values are shown for each compound. The slopes (A.s g−1) were 0.0361, 0.0488, 0.0841, and 0.2171 for tetrachloroethylene, toluene, chlorobenzene, and ethylbenzene, respectively. (b) Corresponding curves plotted on a log-scale for clarity.
These results illustrate that the response of the detector remains linear to the injected mass of the test compounds over three orders of magnitude. The R-squared values and their respective slopes obtained by linear regression analysis (forced zero Y-intercept) have been indicated for each curve. Moreover, as evident from Figure 4b, as the injected mass increased by three orders of magnitude, the sensing signal also increased by 1,000-fold, showing that the linear response of the µDPID spans over three orders of magnitude. The response of µDPID is lower for tetrachloroethylene due to its high ionization potential, which makes it difficult to ionize. The ionization potentials of the remaining VOCs are reasonably close. The ionization potentials for all VOCs used in this report are provided in Table 1.
The sensitivity of the detector is defined as the signal output per unit mass of the compound in the carrier gas. For MSDs, the sensitivity S, is defined as:
where A is the integrated peak area, and W is the mass of the compound. Therefore, the slopes of the curves in Figure 4a indicate the sensitivity of the µDPID for the particular compounds under investigation. The slopes for tetrachloroethylene, toluene, chlorobenzene, and ethylbenzene were 0.036, 0.0488, 0.0841, and 0.2171 (A.s g−1), respectively, showing the higher sensitivity of the µDPID toward ethylbenzene compared to the other test compounds.
Chip-scale GC: Chromatographic analysis
The chip-scale GC platform was tested following the characterization of the sample injection unit and µDPID. Twelve VOCs—toluene, tetrachloroethylene, chlorobenzene, ethylbenzene, p-xylene, n-nonane, bromobenzene, n-decane, 1,2-dichlorobenzene, n-undecane, 1,2,4-trichlorobenzene, and n-dodecane—were selected to evaluate the performance of the chip. The following experiments were performed to demonstrate the sampling, separation and detection of compounds with boiling points in the range of 110–216 °C as quickly as possible through the optimization of the flow and temperature programming conditions.
The chip was configured as discussed in the system description section (Figure 1), with the carrier gas and auxiliary helium supplied by the conventional GC system. The pressures on these supplies were set to 22 and 10 psi, respectively, corresponding to a flow rate of 0.77 mL min−1 at the chip outlet port. The chip temperature was maintained at 40 °C. During sampling, the pump was turned on to load the sample into the fluidic connection between the chip and the valve. A sampling time of 3 s was considered sufficient for testing the compounds. The chromatogram in Figure 5 shows the successful identification of nine compounds in 2.5 min, providing good resolution and retention of the compounds. The compounds were eluted in order of decreasing vapor pressure (the pv values are provided in Table 1). The compounds with high vapor pressure tend to remain in the vapor state and thus have more affinity for the mobile phase compared to the stationary phase, which results in their early elution from the µSC. In Figure 5, n-nonane and bromobenzene were not resolved because their vapor pressures were relatively close. A similar observation was made for n-decane and 1,2-dichlorobenzene.The air peak detected by the µDPID was removed from the chromatogram for clarity. The peak width at the base (wb = 4σ) and the retention time (tr) were calculated for every compound from the chromatograms generated by the µDPID. The wb values for p-xylene, C9/bromobenzene, and C10/1,2-dichlorobenzene,all of which have high boiling points, were 7.2, 10, and 27 s, respectively. The resolution (Rs) is defined as
where d is the distance between the peak maxima for the two compounds, A and B. The resolution between p-xylene and C9/bromobenzene was 2.16, and that between C9/bromobenzene and C10 was 4.60.

(a) Response of the μDPID to a 3 s headspace sampling of a nine-compound mixture under isothermal conditions at 40 °C and a flow rate of 0.77 mL min−1 at the chip outlet. (b) Corresponding chromatogram generated by the FID.
To reduce the analysis time and increase the peak width of the high-boiling-point compounds, a series of experiments was performed under flow programming conditions. One of the best chromatographic results is shown in Figure 6a. Here, the pressure was initially set to 22 psi (0.77 mL min−1) for 0.7 min and then ramped up to 35 psi (1 mL min−1) at a rate of 35 psi min−1. The holdup time was necessary to avoid poor resolution between the first five eluting compounds. In Figure 6a, the first nine compounds eluted in 1.6 min, which shows a reduction in the analysis time of 36% compared to the previous run. In addition, the C11 peak was observed at 2.8 min. The increased pressure also had a profound impact on the wb value for high-boiling-point compounds, including p-xylene, C9/bromobenzene, and C10/1,2-dichlorobenzene. Reductions in wb of 33%, 40%, and 69% were observed for these compounds, respectively. These reductions occur because at increased pressure, less time is available for the longitudinal diffusion of molecules inside the µSC, which results in a narrower peak width. The Rs values between p-xylene and C9/bromobenzene, between C9/bromobenzene and C10/1,2-dichlorobenzene,and between C10/1,2-dichlorobenzene and C11 were reduced to 2, 4.08, and 8.16, respectively.

(a) Response of the μDPID to a 3 s headspace sampling of a 10-compound mixture under the flow programming and isothermal conditions at 40 °C. The initial and final flow rates through the column outlet were 0.77mL min−1 and 1 mL min−1, respectively. (b) Response of the μDPID to a 3 s headspace sampling of a 10-compound mixture under the flow and temperature programming conditions. The initial and final flow rates through the column were 0.77 and 0.9 mL min−1, respectively. (c) Response of the μDPID to a 6 s headspace sampling of a seven-compound mixture containing high-boiling-point compounds under isothermal conditions of 70 °C and a flow rate of 0.9 mL min−1 at the chip outlet. (d) Response of the μDPID to a 6 s headspace sampling of a four-compound mixture diluted to a concentration of 1 ppm in the aqueous phase. The corresponding concentrations in the headspace calculated using Henry’s Law are 270 (toluene), 152 (chlorobenzene), 323 (ethylbenzene), and 314 ppb (p-xylene). The flow rate at the chip outlet was set to 0.77 mL min−1.
Temperature is one of the most important variables in GCs and an effective way of optimizing the analysis time. The superposition of temperature (Tinitial = 40 °C, ramp = 30 °C min−1, Tfinal = 65 °C) and flow programming (Pinitial = 22 psi, ramp = 35 psi min−1, Pfinal = 35 psi) has been demonstrated in the chromatogram shown in Figure 6b. As a result, the analysis was completed in 1.8 min (56% reduction in the analysis time). As expected, further decreases in wb of 41%, 58%, and 86% were observed for p-xylene, C9/bromobenzene, and C10/1,2-dichlorobenzene, respectively.
A separate sample containing high-boiling-point compounds, including n-nonane, bromobenzene, n-decane, 1,2-dichlorobenzene, n-undecane, 1,2,4-trichlorobenzene, and n-dodecane, was also analyzed. The carrier gas pressure was increased to 35 psi, whereas the auxiliary helium pressure was maintained at 10 psi respectively, which corresponds to a flow rate of 0.9 mL min−1 at the chip outlet port. The chip temperature was increased to 70 °C. The loading time was increased to 6 s because of the low vapor pressure of these compounds. The resulting chromatogram is shown in Figure 6c. The analysis was completed in 0.8 min, with significantly lower values of wb. wb values of 900 ms, 1.5 s, 2.4 s, and 5.4 s were obtained for C9/bromobenzene, C10/1,2-dichlorobenzene, C11, and 1,2,4-trichlorobenzene/C12, respectively. The data for retention time (tr) and peak width (wb) for these compounds in Figures 5a and 6a–c, respectively, have been compiled in Table 2. The resolutions (Rs) for the high-boiling-point compounds are listed in Table 3.
Aqueous analysis
The chip performance was also evaluated for the analysis of VOCs in aqueous media. Four VOCs—toluene, chlorobenzene, ethylbenzene, and p-xylene—were diluted to a concentration of 1 ppm (1 mg L−1) in deionized water. A small aliquot (500 µL) of the prepared sample was transferred into a 1.8 mL vial. The vial was subsequently sealed to avoid compromising the sample integrity. A sampling time of 6 s was considered sufficient for extracting VOCs from the headspace of the sample. The concentration in the headspace was calculated using Henry’s Law. The headspace concentrations for toluene, chlorobenzene, ethylbenzene, and p-xylene were 270, 152, 323, and 314 ppb, respectively. The representative chromatogram is shown in Figure 6d, where the separation and detection were completed within 0.6 min. The United States Environmental Protection Agency (EPA) has specified maximum contamination levels for toluene, chlorobenzene, ethylbenzene, and p-xylene of 1, 0.1, 0.7 and 10 ppm, respectively.
Conclusion
This study presents an integrated µGC analysis system that has all of the important features of a lab-on-a-chip platform. It has a sample injection unit, a microseparation column, and a detector, all of which are integrated on a 1.5 cm × 3 cm platform. The integration of the µGC components on a common substrate reduces the footprint of this technology, enhances the overall performance and allows for a lower fabrication cost. Furthermore, each component can perform its role effectively and rapidly; for example, the sample injection unit requires a loading time of a few seconds to produce sharp injections (FWHM of approximately 200 ms). Similarly, the subsequent operation of the system under the temperature and flow programming conditions allows for a rapid analysis of complex VOC mixtures. In addition, the excellent detection limit (approximately 10 pg), response time (approximately 200 ms), and linearity of the µDPID (three orders of magnitude) demonstrates its potential for use in µGC technology. Further improvements in these properties through modifications in the design of the µDPID are also being considered. Although the miniaturization of the µGC system through the integration of microelectromechanical system components has been demonstrated in this work, we are also exploring its integration with electronic components to further miniaturize this technology.
References
Wilson A. Electronic-nose applications in forensic science and for analysis of volatile biomarkers in the human breath. Journal of Forensic Science and Criminology 2014; 1: doi: 10.15744/2348-9804.1.S103.
Dorman FL, Whiting JJ, Cochran JW et al. Gas chromatography. Analytical Chemistry 2010; 82: 4775–4785.
Boots A, Smolinska A, van Berkel J et al. Identification of microorganisms based on headspace analysis of volatile organic compounds by gas chromatography–mass spectrometry. Journal of Breath Research 2014; 8: doi:10.1088/1752-7155/8/2/027106.
Pietrogrande MC, Zampolli MG, Dondi F et al. In situ analysis of the Martian soil by gas chromatography: Decoding of complex chromatograms of organic molecules of exobiological interest. Journal of Chromatography A 2005; 1071: 255–261.
Oliveira CR, Ferreira AA, Oliveira CJ et al. Biomarkers in crude oil revealed by comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry: Depositional paleoenvironment proxies. Organic Geochemistry 2012; 46: 154–164.
Song G, Qin T, Liu H et al. Quantitative breath analysis of volatile organic compounds of lung cancer patients. Lung Cancer 2010; 67: 227–231.
Sottani C, Porro B, Comelli M et al. An analysis to study trends in occupational exposure to antineoplastic drugs among health care workers. Journal of Chromatography B 2010; 878: 2593–2605.
Giannoukos S, Brkić B, Taylor S et al. Membrane inlet mass spectrometry for homeland security and forensic applications. Journal of the American Society for Mass Spectrometry 2014; 8: 1–9.
James F, Breuil P, Pijolat C et al. Development of a MEMS preconcentrator for micro-gas chromatography analyses. Procedia Engineering 2014; 87: 500–503.
Camara M, Breuil P, Briand D et al. Preconcentration modeling for the optimization of a micro gas preconcentrator applied to environmental monitoring. Analytical Chemistry 2015; 87: 4455–4463.
Haudebourg R, Vial J, Thiebaut D et al. Temperature-programmed sputtered micromachined gas chromatography columns: An approach to fast separations in oilfield applications. Analytical Chemistry 2012; 85: 114–120.
Sun J, Cui D, Guan F et al. High resolution microfabricated gas chromatography column with porous silicon acting as support. Sensors and Actuators B: Chemical 2014; 201: 19–24.
Scholten K, Fan X, Zellers ET. A microfabricated optofluidic ring resonator for sensitive, high-speed detection of volatile organic compounds. Lab on a Chip 2014; 14: 3873–3880.
Zhong Q, Steinecker WH, Zellers ET. Characterization of a high-performance portable GC with a chemiresistor array detector. Analyst 2009; 134: 283–293.
Collin WR, Serrano G, Wright LK et al. Microfabricated gas chromatograph for rapid, trace-level determinations of gas-phase explosive marker compounds. Analytical Chemistry 2013; 86: 655–663.
Chen C, Tsow F, Campbell KD et al. A wireless hybrid chemical sensor for detection of environmental volatile organic compounds. IEEE Sensors Journal 2013; 13: 1748.
Zampolli S, Elmi I, Mancarella F et al. Real-time monitoring of sub-ppb concentrations of aromatic volatiles with a MEMS-enabled miniaturized gas-chromatograph. Sensors and Actuators B: Chemical 2009; 141: 322–328.
Mohsen Y, Lahlou H, Sanchez J-B et al. Development of a micro-analytical prototype for selective trace detection of orthonitrotoluene. Microchemical Journal 2014; 114: 48–52.
Garg A, Akbar M, Vejerano E et al. Zebra GC: A mini gas chromatography system for trace-level determination of hazardous air pollutants. Sensors and Actuators B: Chemical 2015; 212: 145–154.
Lewis P, Manginell R, Adkins D et al. RG manley and GC Frye-Mason. IEEE Sensors Journal 2006; 6: 784–795.
Snow E, Perkins F, Houser E et al. Chemical detection with a single-walled carbon nanotube capacitor. Science 2005; 307: 1942–1945.
Janasek D, Franzke J, Manz A. Scaling and the design of miniaturized chemical-analysis systems. Nature 2006; 442: 374–380.
Chapman PJ, Vogt F, Dutta P et al. Facile hyphenation of gas chromatography and a microcantilever array sensor for enhanced selectivity. Analytical Chemistry 2007; 79: 364–370.
Bulbul A, Kim H. A bubble-based microfluidic gas sensor for gas chromatographs. Lab on a Chip 2015; 15: 94–104.
Bae B, Kim J, Yeom J et al. Development of a portable gas analyzer using a micro-gas chromatograph/flame ionization detector (micro-GC/FID) for NASA’s environmental missions. 42nd International Conference on Environmental Systems; 15-19 Jul 2012; San Diego, CA, USA; 2012: AIAA 2012-3435.
Chen F-Y, Chang W-C, Jian R-S et al. Novel gas chromatographic detector utilizing the localized surface plasmon resonance of a gold nanoparticle monolayer inside a glass capillary. Analytical Chemistry 2014; 86: 5257–5264.
Li M, Myers E, Tang H et al. Nanoelectromechanical resonator arrays for ultrafast, gas-phase chromatographic chemical analysis. Nano Letters 2010; 10: 3899–3903.
Akbar M, Shakeel H, Agah M. GC-on-chip: Integrated column and photoionization detector. Lab on a Chip 2015; 15: 1748–1758.
Terry SC, Jerman JH, Angell JB. A gas chromatographic air analyzer fabricated on a silicon wafer. IEEE Transactions on Electron Devices, 1979; 26: 1880–1886.
McNair HM, Miller JM. Basic Gas Chromatography. John Wiley & Sons, New York, 2011.
Manginell RP, Bauer JM, Moorman MW et al. A monolithically-integrated μGC chemical sensor system. Sensors 2011; 11: 6517–6532.
Kaanta BC, Chen H, Zhang X. A monolithically fabricated gas chromatography separation column with an integrated high sensitivity thermal conductivity detector. Journal of Micromechanics and Microengineering 2010; 20: 055016.
Robards K, Robards K, Haddad PR et al. Principles and Practice of Modern Chromatographic Methods. Academic Press, Waltham, Massachusetts, 1994.
Acknowledgements
The authors would like to thank Prof. Kang Xia (Department of Crop and Soil Environmental Sciences, Virginia Tech) and Professor Emeritus Dr. Harold McNair (Chemistry Department, Virginia Tech) for their technical discussions regarding the aqueous sample analysis. This work was partially supported by the National Institute for Occupational Safety and Health (NIOSH) under award No. 1R21OH010330 and Virginia Tech's Institute for Critical Technology and Applied Science (ICTAS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0
About this article

Cite this article
Akbar, M., Restaino, M. & Agah, M. Chip-scale gas chromatography: From injection through detection. Microsyst Nanoeng 1, 15039 (2015). https://doi.org/10.1038/micronano.2015.39
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/micronano.2015.39
This article is cited by
-
Chip-based ion chromatography (chip-IC) with a sensitive five-electrode conductivity detector for the simultaneous detection of multiple ions in drinking water
Microsystems & Nanoengineering (2020)
-
Compact prototype microfabricated gas chromatographic analyzer for autonomous determinations of VOC mixtures at typical workplace concentrations
Microsystems & Nanoengineering (2018)
