
Abstract
During inflammation lymphatic vessels (LVs) are enlarged and their density is increased to facilitate the migration of activated immune cells and antigens. However, after antigen clearance, the expanded LVs shrink to maintain homeostasis. Here we show that interleukin (IL)-17A, secreted from T helper type 17 (TH17) cells, is a negative regulator of lymphangiogenesis during the resolution phase of TH17-mediated immune responses. Moreover, IL-17A suppresses the expression of major lymphatic markers in lymphatic endothelial cells and decreases in vitro LV formation. To investigate the role of IL-17A in vivo, we utilized a cholera toxin-mediated inflammation model and identified inflammation and resolution phases based on the numbers of recruited immune cells. IL-17A, markedly produced by TH17 cells even after the peak of inflammation, was found to participate in the negative regulation of LV formation. Moreover, blockade of IL-17A resulted in not only increased density of LVs in tissues but also their enhanced function. Taken together, these findings improve the current understanding of the relationship between LVs and inflammatory cytokines in pathologic conditions.
Similar content being viewed by others


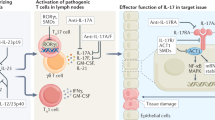
Introduction
Lymphatic vessels (LVs), which compose the circulatory system along with blood vessels (BVs), consist of a single layer of lymphatic endothelial cells (LECs) that have a blunt-ended structure specialized for draining extracellular fluids.1 Under physiological conditions LVs transport lymph fluid containing macromolecules, such as protein and dietary lipids, into the blood circulation.2 However, during inflammation LVs also deliver soluble antigens and immune cells from sites of infection. This includes antigen-presenting cells (APCs) carrying antigens that travel along the LVs and enter draining lymph nodes (LNs), which act as immunological platforms bridging innate and adaptive immunity.3 In LNs CD4+ T cells differentiate into many subsets of T helper (TH) cells (e.g., TH1, TH2, and TH17 cells) depending on the categories of pathogens4 presented through the MHC-peptide-TCR interaction with APCs, and then migrate to sites of infection through efferent LVs.5 Following the elimination of antigens, the immune system goes through a resolution phase. This process involves the reduction of inflammation in order to maintain homeostasis accompanied by the elimination of granulocytes from infection sites,6 production of anti-inflammatory cytokines, apoptotic cell death of effector cells,7 and regression of LVs.1
Vascular endothelial cell growth factors (VEGFs), are well-known growth factors for blood endothelial cells (BECs) and also control lymphangiogenesis in both normal and pathological conditions.8 During inflammation, VEGFs promote expansion and enlargement of LVs and facilitate efficient transportation of APCs and antigens.9 Immune cells are known to be a source of these growth factors as macrophages produce VEGF-A, C, and D after LPS administration,10 neutrophils enhance lymphangiogenesis by secreting bioavailable VEGF-A and increasing total VEGF-D,11 dendritic cells (DCs) secrete VEGF-C,12 and B cells express VEGF-A and C, and angiopoietins.13 In addition, several studies also showed that epithelial cells and stromal cells can produce VEGFs.14 However, little is known about anti-lymphangiogenic factors, which are essential for regression of expanded LVs and resolution of immune responses in order to maintain homeostasis. Recently it was discovered that several cytokines secreted by effector TH cells negatively regulate lymphangiogenesis. For example, interferon (IFN)-γ, a representative TH1 cell-associated cytokine, was reported to negatively control the formation of LN-LVs after LPS challenge.15 Interleukin (IL)-4 and IL-13 secreted by TH2 cells also have suppressive effects on lymphangiogenesis and play a pathogenic role in allergic diseases by interrupting antigen clearance in peripheral tissues.16, 17 Therefore, based on the previous reports, we hypothesized that other types of TH cells could also regulate LVs through cytokine production.
IL-17A, predominantly produced by TH17 cells, binds to the IL-17 receptor (IL-17R) complex composed of IL-17RA and IL-17RC,18 which are expressed on immune cells, epithelial cells, and endothelial cells.19, 20 Although TH1 and TH2 cytokines such as IFN-γ and IL-4 activate Janus kinase (JAK) and signal transducer and activator of transcription (STAT) proteins, IL-17A signaling is mediated by different molecules named NF-κB activator 1 (Act1) and TNF receptor-associated factor 6 (Traf6).19 Act1 interacts with IL-17R complex through SEFIR domain and ubiquitinates E3 ubiquitin ligase Traf6 without degradation resulting in the recruitment of kinase TAK1 and activation of classical NF-κB pathway.21, 22 In addition, there is another pathway that is accompanied by I-κB kinase (IKKi)-mediated Act1 phosphorylation and interaction with Traf2–Traf5 instead of Traf6, eventually induction of CXCL1 mRNA stabilization.23, 24
IL-17A plays a central role in TH17-associated inflammation including fungal infection and autoimmune diseases.25 A well-known function of IL-17A is the recruitment of macrophages and neutrophils to infection sites during inflammation.25, 26 In contrast, less is known about the effect of IL-17A on LECs and LV formation. To this end, we investigated the roles of IL-17A in LV formation and found that IL-17A negatively regulates lymphangiogenesis during the resolution of acute inflammation. Furthermore, we show that IL-17A supplementation inhibits gene expression of major lymphatic markers both in LECs and during in vitro lymphangiogenesis, even under lymphatic growth factor-enriched conditions. Furthermore, this role of IL-17A was confirmed using an in vivo TH17-mediated inflammation model where blockade of IL-17A was found to cause not only increased LV formation but also enhanced LV function in the lung during the resolution phase. Therefore, our results suggest that IL-17A plays a prime role in LV regression after a TH17-mediated immune response. Together these findings provide expanded insights into the control of lymphangiogenesis during immune regulation.
Results
Suppression of LEC marker expression by IL-17A in vitro
To investigate the possibility that LECs respond to T cell cytokines, we first examined the expression of IL-17RA and IL-17RC as well as other cytokine receptors on LECs. We showed mRNA expression of IL-17RA and IL-17RC in human LECs (hLECs) and protein expression of IL-17RC in mouse LECs (mLECs), indicating that LECs may respond to IL-17A directly through the expression of IL-17R (Supplementary Figure S1 online). We next investigated the effect of IL-17A on hLECs in vitro and compared with IFN-γ, a strong anti-lymphangiogenic factor.15 The level of Prospero homeobox protein 1 (Prox-1) mRNA was downregulated at both 36 h (∼0.67-fold) (Figure 1a) and 72 h (∼0.75-fold) (Figure 1b) after treatment with recombinant human IL-17A (rhIL-17A). In addition, the expression of lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), another LEC-specific surface protein, was also diminished (∼0.64-fold) at 72 h (Figure 1d) after addition of rhIL-17A but was not affected at the 36 h time point (Figure 1c). Similar to the results in a previous study,15 treatment with rhIFN-γ decreased Prox-1 (∼0.49-fold) and LYVE-1 (∼0.05-fold) mRNAs at 72 h after treatment (Figure 1b, d). LYVE-1 expression showed delayed response against cytokine treatment compared with Prox-1 as the expression of LYVE-1 is regulated by Prox-1 activity.27 Correspondingly, immunocytochemistry analysis showed that the expression of Prox-1 was reduced (∼0.73-fold) at 72 h after rhIL-17A treatment (Figure 1e, f). Although the effect of IL-17A was not strong as IFN-γ, we found that IL-17A also has a modest inhibitory effect on major lymphatic marker expression. In contrast, another TH17-related cytokine, IL-22, did not cause a decrease in expression of Prox-1 nor LYVE-1 in hLECs, thereby indicating that hLECs specifically respond to IL-17A (Supplementary Figure S2).

Suppression of major lymphatic marker expression after IL-17A treatment. 1 × 105 of hLECs were cultured for 72 h with 500 ng/ml of rhIFN-γ or rhIL-17A. (a–d) Prox-1 and LYVE-1 mRNA level at 36 h (a, c) and 72 h (b, d) after treatment with rhIFN-γ or rhIL-17A were examined by real-time RT-PCR. Each group, n=3. Data are representative of three experiments. *P<0.05, **P<0.01 was compared with the control by unpaired Student’s t-test. (e) Prox-1 protein expression was examined by confocal microscopy at 72 h after rhIFN-γ or rhIL-17A treatment. Scale bar=50 μm. (f) Prox-1 signal intensity was measured by Image J. Each group, n=5. Data are representative of three experiments. **P<0.01 versus control by non-parametric Mann–Whitney test.
We also investigated the putative signaling pathways of IL-17A which regulate gene expression in hLEC. As described before, Traf6 is a well-known downstream molecule of IL-17A,19 and we treated with Traf6 inhibitor peptide or control peptide along with IL-17A on hLECs. We found that Traf6 inhibitor peptide restored mRNA expression of LYVE-1 almost completely (∼1.12-fold) (Figure 2a). At the same time, we showed partial recover of Prox-1 expression in mRNA (∼0.85-fold) (Figure 2b) and protein level (∼0.81-fold) (Figure 2c, d), respectively.

Effects of Traf6 inhibition on major lymphatic marker expression. 1 × 105 of hLECs were cultured for 72 h with 500 ng/ml of rhIL-17A. Indicated concentration of specific inhibitor peptides was added following rhIL-17A treatment. LYVE-1 (a) and Prox-1 (b) mRNA level at 72 h after rhIL-17A and inhibitor peptide treatment were examined by real-time PCR. Each group, n=3. Data are representative of three experiments. **P<0.01 was compared with the control by unpaired Student’s t-test. (c) Prox-1 protein expression was examined by confocal microscopy at 72 h after rhIL-17A and 50 μM of inhibitor peptide treatment. Scale bar=50 μM. (d) Prox-1 signal intensity was measured by Image J. Each group, n=5. Data are representative of three experiments. *P<0.05, **P<0.01 versus control by non-parametric Mann–Whitney test.
We next performed a tube formation assay which is an in vitro model of lymphangiogenesis that reflects the ability of endothelial cells to make connections with adjacent cells. To this end, hLECs or mLECs were cultured on Matrigel-coated dishes with or without rhIL-17A or rmIL-17A and tube formation was examined at 24 h after seeding (Figure 3a, f). The results showed decreases in tube length (Figure 3b, g), tube area (Figure 3c, h), the number of junctions (Figure 3d, i) and tubes (Figure 3e, j) in the rIL-17A treated group compared with the control. Collectively, these data indicate that IL-17A inhibits gene expression of major lymphatic markers through a Traf6-dependent manner and suppresses in vitro lymphangiogenesis.

Inhibition of tube formation following IL-17A treatment. 4 × 104 of hLECs or mLECs were cultured on Matrigel for 24 h with or without 1 μg/ml of rhIL-17A or rmIL-17A, respectively. Representative images of tube formation with hLEC (a) or mLEC (f) were taken after 24 h and tube length (b, g), tube area (c, h), number of junctions (d, i) and number of tubes (e, j) of hLEC or mLEC were measured by Image J. Scale bar=100 μm. Each group, n=4–6. Data are representative of three experiments. *P<0.05, **P<0.01 versus control by non-parametric Mann–Whitney test.
Immune cell recruitment, cytokine production, and lymphangiogenesis following CTO challenge
Defining inflammation and resolution phases
Thus far, we have shown a role for IL-17A as a negative regulator of major lymphatic markers and in vitro lymphangiogenesis. Next, we adopted an acute TH17 inflammation model to show the in vivo inhibitory effects of IL-17A on LVs. As described before, intranasal immunization with cholera toxin mixed with OVA peptide (CTO) induced TH17-dominant immune responses, during which peaked TH17 cell differentiation and IL-17A production was observed for a week.28 To define the inflammation and resolution phases, we enumerated the numbers of immune cells recruited into the lung by collecting and analyzing bronchoalveolar lavage (BAL) fluid as an indication of the severity of inflammation over time. Total BAL cell counts were dramatically increased 7 days after CTO challenge, with the highest count at day 10 (∼10-fold), indicating the peak of inflammation (Figure 4a). The total BAL cell counts then started to decrease after day 10, indicating the start of the resolution phase and continued to decrease until reaching the basal level at day 21 following the initial challenge. As neutrophilia is one of the most important indicators of TH17-mediated responses, we also enumerated CD11b+Ly6G+ neutrophils and found a similar trend to that of the total BAL count, with a peak at day 10 and return to baseline by day 21 after CTO challenge (Figure 4b and Supplementary Figure S3a, b). Interestingly, the proportion of CD4+ T cells in BAL fluid was retained at ∼40% until day 21 after CTO challenge and the number of CD4+ T cells was also higher in resolution phase compared with day 7 (Figure 4c and Supplementary Figure S3c, d). Thus, the TH17-mediated acute inflammation model used in our study shows that the transition between inflammation and resolution phases occurs on day 10 following CTO challenge.

Accumulation of immune cells in BAL fluid after CTO administration. After CTO challenge, BAL fluid was collected from mice by washing their lungs with PBS on indicated day. (a) Numbers of total immune cells in BAL fluid were counted. (b, c) Numbers of CD11b+Ly6G+ neutrophils (b) and CD4+ T cells (c) were analyzed and quantified by FACS. Each group, n=5. Data are representative of three experiments. *P<0.05, **P<0.01 versus day 0 or day 7 by non-parametric Mann–Whitney test. (d, e) BAL fluid was obtained from CTO-challenged mice on day 10 and re-stimulated with PMA/Ionomycin for 5 h. Intracellular cytokine staining was performed after stimulation and IL-17A-producing CD4+ T cells were analyzed and quantified by FACS. Each group, n=3. Data are representative of three experiments. **P<0.01 versus day 0 by non-parametric Mann–Whitney test.
Cytokine production following CTO challenge
Next, we examined cytokine levels in BAL fluid following CTO challenge by ELISA. High levels of IFN-γ, IL-6, and IL-17A were detected in BAL fluid of the CTO-challenged group compared with saline-treated control group, whereas IL-4 was not detected in either group (Supplementary Figure S4a–d). Because IFN-γ is a well-known strong, negative regulator of lymphangiogenesis, we performed all following experiments with IFN-γ-knockout (IFN-γ-KO) mice as well to exclude the negative effects of IFN-γ in CTO-induced lung inflammation. In BAL fluid from IFN-γ-KO mice, higher levels of IL-6 and IL-17A, but not IFN-γ, were detected (Supplementary Figure S4e–h). To investigate the major source of IL-17A in the CTO-induced lung inflammation, we analyzed cytokine-producing immune cells in BAL fluid. A large increase in IL-17A production was detected in BAL fluid at 10 days after CTO challenge, and we found that the majority of IL-17A was produced by CD4+ T cells (∼75.9%) while γδ+ T (∼17.8%) and CD8+ T cells (∼2.25%) also produce some IL-17A (Figure 4d, e and Supplementary Figure S5). These results indicate that CD4+ T cells are the major sources of this cytokine. Next, to compare the dynamics of cytokine production between wild-type (WT) and IFN-γ-KO mice and confirm the source of IL-17A in the lung after CTO administration, we performed intracellular cytokine staining of lung cells (Figure 5a–e). In both WT and IFN-γ-KO mice, CD4+ T cells started producing IL-17A at 7 days after CTO challenge, and this production drastically increased after re-stimulation with either OVA323–339 peptide or PMA/Ionomycin, indicating that CTO administration generates specific immune responses. Interestingly, CD4+ T cells produced a considerable amount of IL-17A during resolution phase as well as inflammation phase (Figure 5d, e). Although IFN-γ-producing CD4+ T cells were detected after re-stimulation with PMA/Ionomycin in WT mice, no antigen-specific responses were apparent (Figure 5b, c). The percentage of IL-17A-producing CD4+ T cells was slightly elevated in IFN-γ-KO mice, which resulted from a well-known inhibitory function of IFN-γ on the differentiation of TH17 cells (Figure 5d, e). We also examined IL-17A and IFN-γ production in secondary lymphoid organs. Although less, we could detect IL-17A-producing CD4+ T cells after re-stimulation with PMA/Ionomycin (Supplementary Figure S6). Altogether, these data demonstrate that IL-17A is predominantly produced by TH17 cells in CTO-induced inflammation at the beginning of the resolution phase.

Dynamics of IL-17A production by CD4+ T cells and lymphangiogenesis in CTO-induced lung inflammation. (a) Total lung cells from CTO-challenged WT and IFN-γ-KO mice were re-stimulated with OVA peptide or PMA/Ionomycin for 5 h. Intracellular cytokine staining was performed following stimulation and analyzed by FACS. Representative flow cytometric plots pre-gated on CD4+ T cells were depicted. (b–e) Quantification of IFN-γ- or IL-17A-producing CD4+ T cells with OVA323–339 peptide (b, d), or PMA/Ionomycin stimulation (c, e). Each group, n=3–5. Data are representative of three experiments. *P<0.05, **P<0.01 versus day 0 by unpaired t-test. (f) LVs in lungs of CTO-challenged WT and IFN-γ-KO mice were stained with specific antibodies and imaged by confocal microscopy. (g–j) LVD (g, h) and BVD (i, j) were quantified by Image J. Scale bar=500 μm. Each group, n=5. Data are representative of three experiments. *P<0.05 versus day 0 by non-parametric Mann–Whitney test.
Dynamic LV formation and regression
As LVs are a potential target of IL-17A during the resolution phase of inflammation, we investigated the dynamics of lymphatic and blood vessels in lungs after CTO challenge. To do this, we performed immunohistochemistry with specific antibodies and showed increased LV density (LVD) after CTO challenge, which was maintained until day 10 after CTO administration. Also, we found that many new and enlarged chaotic LVs formed around tertiary bronchi in both WT and IFN-γ-KO mice (Figure 5f). This suggests immune cell extravasation from BVs or direct secretion of VEGFs from epithelial cells. Moreover, these LVs were strikingly remodeled by day 14, and most had disappeared by day 21 (Figure 5g, h). As we described before, Prox-1 is the most important transcription factor of LECs, thus we tried to identify LVs by using IFN-γ-KO-Prox-1-GFP mouse system. Similar to VEGFR3 staining, we found dynamic lymphangiogenesis until day 10 and regression of LVs after day 10 (Supplementary Figure S7). In addition to immunohistochemistry, we quantified the expression of Prox-1 mRNA in lung tissues during CTO-induced inflammation. In agreement with GFP expression in IFN-γ-KO-Prox-1-GFP mice, we found Prox-1 mRNA level was also upregulated after CTO challenge and reduced after the peak of inflammation (Supplementary Figure S8). On the other hand, no changes in BV density (BVD) were observed in either WT or IFN-γ-KO mice (Figure 5i, j). Thus, CTO-induced lymphangiogenesis occurs during inflammation and the increased LVD is reduced to baseline during the resolution phase regardless of IFN-γ signaling.
Blockade of IL-17A increases LVD and enhances lymphatic function
Based on the above findings, we next examined the suppressive effects of IL-17A on in vivo lymphangiogenesis by utilizing an IL-17A-neutralizing antibody. As previously shown, CTO-induced immune responses gradually diminished after day 10. Therefore, we administered 25 μg of IL-17A blocking or isotype control antibodies intratracheally daily from day 11 to 15, and LVD was measured in the lung on day 16 by confocal microscopy. The results show significantly increased LVD after blockade of IL-17A (∼1.64-fold) in the lung (Figure 6a, b), as there was no difference in BVD (Figure 6a, c). In addition, we checked LVD after blockade of IL-17A in IFN-γ-KO-Prox-1-GFP mice. Consistently, the results show increased Prox-1+ LVD after inhibition of IL-17A (∼2.38-fold) in the lung (Figure 6d, e).

Increased LVD after anti-IL-17A treatment. (a) CTO-challenged IFN-γ-KO mice were treated with anti-IL-17A antibody or isotype control antibody intratracheally on day 11–15. Lungs were dissected on day 16, stained with specific antibodies and imaged by confocal microscopy. (b, c) LVD (b) and BVD (c) were quantified by Image J. Scale bar=1 mm. Each group, n=5. (d) CTO-challenged IFN-γ-KO-Prox-1-GFP mice were treated with anti-IL-17A antibody or isotype control antibody intratracheally on day 11–15. Lungs were dissected on day 16, stained with specific antibodies and imaged by confocal microscopy. (e) LVD was quantified by Image J. Scale bar=200 μm. Each group, n=5. Data are representative of two experiments. *P<0.05 versus isotype control treatment (Iso) by the non-parametric Mann–Whitney test.
Lastly, we examined the function of LVs in the lung as evaluated by antigen clearance after IL-17A neutralization. For this, fluorescein isothiocyanate (FITC)-conjugated dextran (FITC-dextran), which should be transported through LVs due to its high-molecular weight (two million Daltons), was administered intratracheally 1 day after the final administration of IL-17A-neutralizing antibody. The total amount of remaining FITC-dextran in the lung was quantified after 6 h with an in vivo imaging system (IVIS). In agreement with the increase in LVD, the remaining quantity of FITC-dextran was reduced (∼0.47-fold) in IL-17A-neutralizing antibody-treated mice compared with the isotype antibody-treated group (Figure 7a, b). Furthermore, we examined antigen transport from the lung to mediastinal LN by intratracheal administration of red fluorescence protein (RFP)-conjugated beads (RFP-bead) 1 day after final administration of IL-17A-neutralizing antibody. The number of RFP-beads in the mediastinal LN was increased (∼2.01-fold) compared with the control following IL-17A blockade (Figure 7c, d). In addition, the LVD in the mediastinal LN was elevated (∼1.64-fold) in the IL-17A-neutralizing antibody-treated group, which is consistent with the increased number of RFP-bead (Figure 7c, e). Therefore, our results show that blockade of IL-17A increases LV formation and improves their function in vivo.

Enhanced LV function by anti-IL-17A blockade. (a) After CTO challenge and anti-IL-17A antibody or isotype control antibody treatment, 1 mg of FITC-dextran was administered intratracheally on day 16. The amount of remaining FITC-dextran in the lung was quantified after 6 h by IVIS Lumina Imaging System. (b) The total flux of FITC-dextran was quantified. Each group, n=5. Data are representative of three experiments. *P<0.05 versus Iso by the non-parametric Mann–Whitney test. (c) 5 × 107 of RFP-conjugated beads were administered intratracheally on day 15 following last antibody treatment. LNs were dissected on day 16. RFP-bead area (d) and LVD (e) were quantified by Image J. Scale bar=200 μm. Each group, n=5. Data are representative of two experiments. *P<0.05, **P<0.01 versus Iso by non-parametric Mann–Whitney test.
Discussion
In this study, we demonstrate that IL-17A produced by TH17 cells negatively regulates LV formation. Further, we show the suppressive effects of IL-17A on the expression of major lymphatic markers in LECs and on in vitro lymphangiogenesis. Particularly, our data prove that IL-17A directly regulates LECs and lymphangiogenesis through its cognate receptor. Furthermore, by investigating CTO-induced acute lung inflammation, which is modulated by TH17 cells, we assessed the inhibitory effects of an IL-17A-neutralizing antibody. Through these studies, we confirmed that IL-17A negatively regulates lymphangiogenesis during the resolution phase of inflammation, and blockade of IL-17A increases the density of LV and enhances lymphatic function.
The lymphatic system is one of the essential elements of the immune system and functions in the transport of immune cells and antigens, thus mediating adaptive immune responses through recognition of specific antigens.1, 3 Inflammation-induced increment and enlargement of LVs accelerate the migration of antigens and effector cells, but these structures rapidly regress as inflammatory responses subside.2 To date, several cytokines from TH cells, such as IFN-γ produced by TH1 cells,15 and IL-4 and IL-13 produced by TH2 cells,16 have been shown to inhibit LV formation during immune responses. Here, we show that blockade of IL-17A with a specific antibody not only increases LVD but also enhances LV function in the lung during the resolution phase of TH17-mediated inflammation. These results indicate that IL-17A negatively regulates LV formation during the resolution of inflammation, supporting a reduction in the immune response. In addition to its well-known inflammatory function, we propose that IL-17A has a role in negative regulation of LVs for immune resolution and maintenance of homeostasis.
Many cytokines from CD4+ TH effector cells have been broadly studied for their roles in immune cell activation and regulation.29 In pathological conditions, pro-inflammatory cytokines stimulate innate immune cells and modulate immune responses. For example, IFN-γ activates macrophages and induces direct antimicrobial and antitumor functions in addition to antigen processing and presentation.30 Likewise, IL-4 and IL-13 mediate allergic inflammatory responses by modulating airway hyper-responsiveness and eosinophil recruitment.31, 32, 33 Transforming growth factor (TGF)-β which is a prototypical cytokine produced by regulatory T (Treg) cells control inflammation response.34 However, the identification of novel functions of these cytokines in negatively regulating lymphangiogenesis during inflammation implies that they play important roles not only in inflammation but also in its resolution and homeostasis.15, 16, 17 IL-17A, another well-known pro-inflammatory cytokine secreted from TH17 cells, mediates immunity against fungus and extracellular bacterial infections as well as autoimmune diseases.35 Moreover, IL-17A promotes expression of chemokines in non-immune cells, recruitment of innate immune cells, and the phagocytic function of neutrophils.36, 37 In this study, we demonstrate a previously unknown, important function of IL-17A, which is the negative regulation of lymphangiogenesis and the maintenance of homeostasis during CTO-induced inflammation. We found that the majority of IL-17A was produced by TH17 cells during CTO-induced lung inflammation, but other populations (γδ+ T and CD8+ T cells) also secrete a little IL-17A. Following intranasal CTO administration, there was no remarkable increase in the number of immune cells, especially neutrophils, recruited into the BAL fluid of IL-17A KO mice compared with WT mice (Supplementary Figure S9), suggesting that IL-17A plays a critical role in mediating inflammatory responses as previously reported. However, more importantly, our results show that IL-17A is required for LV remodeling in order to maintain homeostasis through the reduction of immune responses. Therefore, IL-17A is necessary for both initiation and resolving immune responses.
Previously, Chauhan et al.38 reported that IL-17A promotes the production of VEGFs from corneal epithelial cells, thus enhancing lymphangiogenesis in the chronic dry eye disease (DED) model. Although this study showed IL-17A induced increases in both the VEGF mRNA and protein levels, they failed to show any direct effects of IL-17A on lymphangiogenesis. Furthermore, several inflammatory cytokines are known to induce VEGF production indirectly from other types of cells. For example, IFN-γ, IL-4, and IL-17A induce VEGF-C secretion in epithelial cells after allergen stimulation.39 IFN-γ also activates DCs and induces biologically active VEGF-C production resulting in inflammation-associated lymphangiogenesis.40 However, we and others showed that LECs express receptors not only for growth factors such as VEGFs but also for inflammatory cytokines.1, 20 Moreover, they directly respond to these growth factors and cytokines during development and inflammation.15, 16 Here we demonstrate that IL-17A directly regulates expression of major lymphatic markers in LECs and inhibits lymphangiogenesis both in vitro and in vivo.
IL-17A binds to IL-17R complex and activates Act1-Traf6 pathway resulting in the activation of classical NF-κB signaling.21 Here we showed that IL-17A suppressed the expression of major lymphatic markers such as Prox-1 and LYVE-1 in mRNA and protein levels. In addition, we discovered that negative regulation of Prox-1 and LYVE-1 was induced via Traf6 dependent signaling pathway by using a specific inhibitor peptide. Although Prox-1 expression was partially recovered, LYVE-1 expression was rescued almost completely. As it is known that the expression of LYVE-1 is induced by Prox-1 expression,27 we assumed that the restoration of LYVE-1 expression was caused by increased expression of Prox-1. Thus, we suggest IL-17A-Traf6 signaling pathway as a novel regulator of Prox-1. Nonetheless, there is a possibility that IL-17A also acts indirectly on LECs since we did not use LEC-specific IL-17R-deficient mouse model.
In this study, we shed light upon the relationship between inflammatory cytokines and the regression of LVs during the resolution phase of inflammation. Chronic inflammation is known to result from a failure of resolution, and this is often associated with abundant lymphangiogenic growth factor production and enhanced lymphangiogenesis.41, 42 Indeed, the LV area was increased while IL-17A and IFN-γ mRNAs were found to be decreased in chronic DED compared with acute DED,43 suggesting that IL-17A functions in a context-dependent manner. Recently, LVs have been considered to have a critical role in the induction of not only adaptive immune responses but also chronic inflammation.42 In this report, we showed that blockade of IL-17A leads to increased LVD in lungs and LNs during the resolution phase of lung inflammation presumably enhancing lymph fluid drainage from infection sites to draining LNs. Although we injected anti-IL-17A antibody intratracheally for IL-17A neutralization, the role of systemic administration of anti-IL-17A antibody needs to elucidate. Nonetheless, we found increased antigen clearance and transport following blockade of IL-17A, suggesting that IL-17A blockade could bring about continuous antigen influx from sites of infection resulting in chronic inflammation.
In conclusion, we demonstrate the role of IL-17A as a negative regulator of lymphangiogenesis and show that it participates in the management of LV formation during TH17 responses. We also discovered dual functions of IL-17A in immune responses, in that it not only amplifies inflammation but also inhibits lymphangiogenesis and contributes to the remodeling of LVs during the resolution phase. Collectively with previous findings, we show that subsets of CD4+ TH cells have their own methods of regulating LV formation during the resolution phase and that the cytokines they secrete support the resolution of inflammation and maintenance of homeostasis.
Methods
Mice
Specific pathogen-free C57BL/6J WT and IFN-γ KO mice were purchased from Jackson Laboratory (Bar Harbor, ME) and bred in our pathogen-free animal facilities. Prox-1-GFP BAC transgenic was described previously.44 Male mice, 8–10 weeks old, were used for this study. Animal care and experimental procedures were performed under approval from the Animal Care Committees of KAIST (KA2015-25).
Reagents
For a generation of acute TH17 inflammation in the lung, a single injection of cholera toxin (CT, 2 μg/mouse; List Biological Laboratories, Campbell, CA, USA) mixed with OVA323–339 peptide (40 μg/mouse; Peptron, Daejeon, Korea) in PBS (CTO) was administered intranasally. For neutralization of IL-17A, anti-IL-17A antibody (TC11-18H10), or isotype control antibody (BD biosciences, San Diego, CA, USA) were administrated intratracheally on indicated days. For antigen clearance experiments, 1 mg of FITC-conjugated dextran (2 million Daltons; Sigma-Aldrich, St Louis, MO, USA) and 5 × 107 of RFP-conjugated beads (Molecular Probes, Eugene, OR) were administrated intratracheally on indicated days.
Histological analyses
These analyses were done as previously described.15 On the indicated days, mice were anesthetized by intraperitoneal injection of pentobarbital sodium (60 mg/kg, Hanlim Pharma Co., Seoul, Korea). Lungs were dissected, fixed, dehydrated, and embedded in tissue freezing medium (Leica, Wetzlar, Germany) and sectioned. Sections were blocked with 5% donkey serum in PBST (0.03% Triton X-100 in PBS) for 1 h and incubated overnight at 4 °C with the following primary antibodies: anti-Prox-1 (rabbit polyclonal, ReliaTech GmbH, Wolfenbuttel, Germany), anti-LYVE-1 (11-034, rabbit monoclonal, AngioBio, Lake Placid, NY, New York), anti-VEGFR3 (goat polyclonal, R&D Systems, Minneapolis, MN, USA), anti-CD31 (2H8, hamster monoclonal, Millipore, Billerica, MA, USA), anti-CD3e (145-2C11, hamster monoclonal, BD biosciences), and anti-B220 (RA3-6B2, rat monoclonal, BD biosciences). After washing, the samples were incubated for 2 h at room temperature (RT) with the following secondary antibodies: Cy3-conjugated anti-goat IgG, Cy3- or Cy5-conjugated anti-hamster IgG and Cy5-conjugated anti-rat IgG (Jackson ImmunoResearch, West Grove, PA, USA). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen, Grand Island, NY, New York). After incubation, the samples were mounted in fluorescent mounting medium (DAKO, Glostrup, Denmark) and immunofluorescent images were acquired using a Zeiss LSM780 confocal fluorescence microscope (Carl Zeiss, Oberkochen, Germany). Morphometric measurements of LVs were made by photographic analysis using Image J software (http://rsb.inifo.nih.gov/ij). LV or BV density was measured in 3–4 mice per group by calculating the total sectioned area density of VEGFR3+, Prox-1+, or CD31+ vessel structures on digital fluorescence images using empirically determined threshold values. Values were obtained per millimeter square and expressed as relative densities.
Human LEC culture and reagent treatment
Human LECs were purchased from Sciencell (Cat# 2500, Carlsbad, CA, USA) and grown on a 0.1% gelatin-coated plate in 2% fetal bovine serum (FBS)-containing endothelial cell basal medium-2 supplemented with growth factors (Lonza, Verviers, Belgium) at 37 °C. 80–90% confluent LECs (passaged fewer than eight times) were used for all experiments. Recombinant human IL-17A, recombinant mouse IL-17A, recombinant human IFN-γ (eBiosciences, San Diego, CA, USA), recombinant human IL-22 (Peprotech, Rocky Hill, NJ, USA) or Traf6 inhibitor peptide (NovusBio, Littleton, CO, USA) were added to the complete media at indicated time points.
Purification of LECs from mouse LNs
Briefly, LNs were dissected and digested in 10 ml of enzyme mixture containing 0.2 mg/ml type-II collagenase, 0.1 mg/ml DNAse I (Worthington Biochem, Freehold, NJ, USA) and 0.8 mg/ml dispase (Gibco, Grand Island, NY, New York) at 37 °C for 1 h. Tissues were gently agitated and pipetted during digestion to disrupt any cell clumps. When LNs were completely digested, cell suspensions were filtered through a 40 μm nylon cell strainer and cultured. After 4–5 days, cells were sorted by flow cytometry (FACS Aria II, BD Biosciences).
LEC tube formation assay
Tube formation assay was done as previously described.16 rhIL-17A or rmIL-17A (1 μg/ml) was added at the indicated time after seeding of 4 × 104 hLECs or mLECs. Tube length, tube area, the number of junctions, and tubes were measured and quantified at the indicated time points.
Quantitative real-time PCR
Total RNA was extracted from cultured LECs using the total RNA purification kit and cDNA was synthesized using the first strand cDNA synthesis kit (Bioassay Systems, Hayward, CA, USA). Quantitative RT-PCR was performed with the indicated primers using qPCR-Kit (Bioassay Systems) in an iCycler (Bio-Rad, Hercules, CA, USA) equipped with iQTM5 (Bio-Rad). RT-PCR data were analyzed using the Bio-Rad iQTM5 Optical System Software (Bio-Rad). The average threshold cycle (Ct) was determined from triplicate reactions and levels of gene expression relative to β-actin were determined. The following primers were used: hLYVE-1 5′-GCAAGGACCAAGTTGAAACAGCCT TG-3′ (forward), 5′-CTGGAATGCACGAGTTAGTCCAAGTA-3′ (reverse), hProx-1 5′-GCTCCAATATGCTGAAGACC-3′ (forward), 5′-ATCGTTGATGGCTTGACGTG-3′ (reverse), hβ-actin 5′-CTTCTACAATGAGCTGCGTGTGGCTC-3′ (forward), 5′-GTACATGGCTGGGGTGTTGAAGGTC-3′ (reverse), mProx-1 5′-CAATCACTTGAAAAAGGCAAAAC-3′ (forward), 5′-AAATTGCTGAACCACTTGATGAG-3′ (reverse), mβ-actin 5′-GCTCTTTTCCAGCCTTCCTT-3′ (forward), 5′-CTTCTGCATCCTGTCAGCAA-3′ (reverse).
Surface staining, intracellular cytokine staining, and flow cytometric analysis
Lungs, spleens, and LNs were collected and gently pulverized against a 40 μm nylon cell strainer into FACS buffer (HBSS+2% FBS). Cells were stained at 4 °C with one or more of the following antibodies: FITC-conjugated anti-mouse B220 (RA3-6B2), FITC-conjugated anti-mouse CD11b (M1/70), APC-conjugated anti-mouse Ly6G (R86-8C5), APC-Cy7-conjugated anti-mouse CD8 (53–6.7) (eBiosciences), V450-conjugated anti-mouse CD3e (500A2), PE-Cy7-conjugated anti-mouse CD4 (RM4-5) (BD biosciences), FITC-conjugated anti-mouse IL-17RC (AAH04759), or FITC-conjugated goat IgG isotype control (R&D Systems). For intracellular cytokine staining, cells were re-stimulated with a PMA (50 ng/ml) and ionomycin (500 ng/ml) mixture or OVA323–339 peptide for 5 h at 37 °C in the presence of Brefeldin A (10 μg/ml). Cells were treated with Fix-Perm and Perm-Wash solutions (eBiosciences) and then stained with cytokine antibodies: FTIC-conjugated anti-mouse IFN-γ (XMG1.2), FITC-conjugated rat IgG1κ isotype control, APC-conjugated anti-mouse IFN-γ (XMG1.2), APC-conjugated rat IgG1κ isotype control (BD Biosciences), Alexa Fluor 700-conjugated anti-mouse IL-17A (TC11-18H10.1), Alexa Fluor 700-conjugated rat IgG1κ isotype control (BioLegend, San Diego, CA, USA), APC-conjugated anti-mouse IL-17A (eBio17B7), or APC-conjugated IgG2aκ isotype control (eBiosciences). Cells were analyzed by flow cytometry (MACSQuant Analyzer 10, Miltenyi Biotec, Bergisch Gladbach, Germany or FACS Aria II, BD biosciences). Dead cells were excluded by staining with the Fixable Viability Dye eFluor 506 (eBiosciences) before surface staining. Data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Statistical analyses
Data are presented as mean±s.e.m. P-values were calculated using unpaired Student’s t-test or non-parametric Mann–Whitney test, and are indicated with *(P<0.05), **(P<0.01), #(P<0.05). All analyses were performed using GraphPad Prism Software V. 6.0 for Windows (GraphPad Software, La Jolla, CA).
References
Tammela, T. & Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 140, 460–476 (2010).
Schulte-Merker, S., Sabine, A. & Petrova, T.V. Lymphatic vascular morphogenesis in development, physiology, and disease. J. Cell Biol. 193, 607–618 (2011).
Alvarez, D., Vollmann, E.H. & von Andrian, U.H. Mechanisms and consequences of dendritic cell migration. Immunity 29, 325–342 (2008).
Weaver, C.T., Hatton, R.D., Mangan, P.R. & Harrington, L.E. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25, 821–852 (2007).
Card, C.M., Yu, S.S. & Swartz, M.A. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J. Clin. Invest. 124, 943–952 (2014).
Bannenberg, G.L. et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 (2005).
Serhan, C.N. et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–332 (2007).
Kim, H., Kataru, R.P. & Koh, G.Y. Regulation and implications of inflammatory lymphangiogenesis. Trends Immunol. 33, 350–356 (2012).
Kim, H., Kataru, R.P. & Koh, G.Y. Inflammation-associated lymphangiogenesis: a double-edged sword? J. Clin. Invest. 124, 936–942 (2014).
Kataru, R.P. et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood 113, 5650–5659 (2009).
Tan, K.W. et al. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 122, 3666–3677 (2013).
Hamrah, P., Chen, L., Zhang, Q. & Dana, M.R. Novel expression of vascular endothelial growth factor receptor (VEGFR)-3 and VEGF-C on corneal dendritic cells. Am. J. Pathol. 163, 57–68 (2003).
Angeli, V. et al. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity 24, 203–215 (2006).
Senger, D.R. & Van De Water, L. VEGF expression by epithelial and stromal cell compartments: resolving a controversy. Am. J. Pathol. 157, 1–3 (2000).
Kataru, R.P. et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 34, 96–107 (2011).
Shin, K. et al. TH2 cells and their cytokines regulate formation and function of lymphatic vessels. Nat. Commun. 6, 6196 (2015).
Savetsky, I.L. et al. Th2 cytokines inhibit lymphangiogenesis. PloS ONE 10, e0126908 (2015).
Toy, D. et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J. Immunol. 177, 36–39 (2006).
Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9, 556–567 (2009).
Ge, D. & You, Z. Expression of interleukin-17RC protein in normal human tissues. Int. Arch. Med. 1, 19 (2008).
May, M.J. IL-17R signaling: new players get in on the Act1. Nat. Immunol. 12, 813–815 (2011).
Liu, C. et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci. Signal 2, ra63 (2009).
Bulek, K. et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat. Immunol. 12, 844–852 (2011).
Sun, D., Novotny, M., Bulek, K., Liu, C., Li, X. & Hamilton, T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat. Immunol. 12, 853–860 (2011).
Ouyang, W., Kolls, J.K. & Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28, 454–467 (2008).
Ferretti, S., Bonneau, O., Dubois, G.R., Jones, C.E. & Trifilieff, A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170, 2106–2112 (2003).
Karunamuni, G. et al. Expression of lymphatic markers during avian and mouse cardiogenesis. Anat. Rec. 293, 259–270 (2010).
Lee, J.B., Jang, J.E., Song, M.K. & Chang, J. Intranasal delivery of cholera toxin induces th17-dominated T-cell response to bystander antigens. PloS ONE 4, e5190 (2009).
Wan, Y.Y. & Flavell, R.A. How diverse—CD4 effector T cells and their functions. J. Mol. Cell Biol. 1, 20–36 (2009).
Schroder, K., Hertzog, P.J., Ravasi, T. & Hume, D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 (2004).
Brusselle, G.G. et al. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin. Exp. Allergy 24, 73–80 (1994).
Grunig, G. et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 282, 2261–2263 (1998).
Corry, D.B. et al. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J. Exp. Med. 183, 109–117 (1996).
Oka, M. et al. Inhibition of endogenous TGF-beta signaling enhances lymphangiogenesis. Blood 111, 4571–4579 (2008).
Peck, A. & Mellins, E.D. Precarious balance: Th17 cells in host defense. Infect. Immun. 78, 32–38 (2010).
Laan, M. et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 162, 2347–2352 (1999).
Ye, P. et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194, 519–527 (2001).
Chauhan, S.K. et al. A novel pro-lymphangiogenic function for Th17/IL-17. Blood 118, 4630–4634 (2011).
Bae, O.N. et al. Chemical allergens stimulate human epidermal keratinocytes to produce lymphangiogenic vascular endothelial growth factor. Toxicol. Appl. Pharmacol. 283, 147–155 (2015).
Gagliostro, V. et al. Pro-lymphangiogenic properties of IFN-gamma-activated human dendritic cells. Immunol. Lett. 173, 26–35 (2016).
Baluk, P. et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Invest. 115, 247–257 (2005).
Kunstfeld, R. et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 104, 1048–1057 (2004).
Chen, Y., Chauhan, S.K., Lee, H.S., Saban, D.R. & Dana, R. Chronic dry eye disease is principally mediated by effector memory Th17 cells. Mucosal Immunol. 7, 38–45 (2014).
Choi, I. et al. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-bassed transgenic mouse. Blood 117, 362–365 (2011).
Acknowledgements
We thank J-R Ha and SJ Lah for technical assistance, and the Hyewha Forum and Dr TW Kim for helpful discussion. This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2016R1A2B3013656).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Author contributions
H.J.P., C.M.Y., and K.S. performed experiments; H.J.P. analyzed data and made the figures; H.J.P and S.-H.L. contributed to the underlying hypothesis, designed the experiments and wrote the manuscript.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Park, H., Yuk, C., Shin, K. et al. Interleukin-17A negatively regulates lymphangiogenesis in T helper 17 cell-mediated inflammation. Mucosal Immunol 11, 590–600 (2018). https://doi.org/10.1038/mi.2017.76
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.76
