
Abstract
The intestinal epithelium must balance efficient absorption of nutrients with partitioning commensals and pathogens from the bodies’ largest immune system. If this crucial barrier fails, inappropriate immune responses can result in inflammatory bowel disease or chronic infection. Enteroendocrine cells represent 1% of this epithelium and have classically been studied for their detection of nutrients and release of peptide hormones to mediate digestion. Intriguingly, enteroendocrine cells are the key sensors of microbial metabolites, can release cytokines in response to pathogen associated molecules and peptide hormone receptors are expressed on numerous intestinal immune cells; thus enteroendocrine cells are uniquely equipped to be crucial and novel orchestrators of intestinal inflammation. In this review, we introduce enteroendocrine chemosensory roles, summarize studies correlating enteroendocrine perturbations with intestinal inflammation and describe the mechanistic interactions by which enteroendocrine and mucosal immune cells interact during disease; highlighting this immunoendocrine axis as a key aspect of innate immunity.
Similar content being viewed by others
Introduction
The intestinal epithelium represents one of the body’s most important interfaces with the environment. Not only must it act as a point of nutrient absorption, but also as a barrier against the vast amount of commensal and pathogenic microbes it constantly encounters.1, 2 As such, the gut hosts the major immune system of the body determining tolerance versus immunity and dysregulation leads to inflammatory bowel disease (IBD) in response to commensals,3 or excessive inflammation in response to infectious pathogens.1 This single layer of epithelium forms a crucial barrier, but is also believed to play important functions in regulation of the intestinal immune system. Within this epithelium reside the enteroendocrine cells (eecs), specialized trans-epithelial signal transduction conduits which respond to luminal nutrients by secreting peptide hormones to control gastrointestinal enzyme secretion, motility, and appetite regulation.4, 5 These key sensory cells comprise just 1% of the epithelium and are dispersed throughout the gut, but collectively form the largest endocrine system in humans. Their elusive nature coupled with a lack of specific surface markers had caused the biology of eecs to remain somewhat enigmatic. However, via the use of transgenic reporter mice,6, 7, 8, 9, 10, 11, 12 and the emergence of Claudin-4 as a specific surface marker,13 we are now uncovering novel concepts of eec biology and surprisingly revealing key interactions between these sensory sentinels and the intestinal mucosal immune system.
Enteroendocrine differentiation
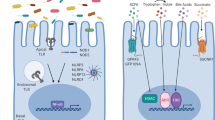
Within the intestinal crypt resides the stem-cell niche which is responsible for supplying the entire epithelial cell population. These Leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5)-positive stem cells14 regularly divide providing highly proliferative transit amplifying cells which further differentiate into absorptive or secretory cellular lineages, supplying a constant cascade of epithelial renewal every 3–5 days.15 Lineage differentiation (Figure 1) is based on Wnt, Notch, and Mitogen-activated protein kinases (MAPK)-dependent signaling16 with the transcription factor hairy and enhancer of split-1 (Hes1) required for differentiation into absorptive enterocytes, while Protein atonal homolog 1 (Atoh1) expression drives secretory cell fate.17, 18, 19, 20 Growth Factor Independent 1 Transcriptional Repressor (Gfi-1) is required for both goblet cells and Paneth cells,21 with Kruppel-like factor 4 (Klf4)22 and Sox923, 24 essential for each population, respectively. Tuft cells require the expression of the Pou domain, class 2, transcription factor 3 (Pou2f3),25, 26 while the development of Microfold (M)-cells is independent of Hes1/Atoh1, instead relying on SpiB transcription factor expression27 and TNF superfamily member receptor activator of NF-kappaB ligand (RankL) induction.28, 29 Eecs depend on the transient expression of Neurogenin3 (Neurog3)30, 31 and micro-RNA-375 (ref. 32) followed by a variety of overlapping transcription factors18, 19 (Table 1), including Neurogenic differentiation 1 (Neurod1),33, 34, 35 Paired box (Pax) 4/6,36, 37, 38 Insulin gene enhancer protein (Isl1),39 pancreatic and duodenal homeobox 1 (Pdx1),40, 41, 42 Nkx6-1 (ref. 43), and Nkx2-2,44, 45 this in turn with spatio-temporal expression of transcription factors46, 47 Pdx1, caudal type homeobox 2 (Cdx-2),48, 49 Gata4, Gata-5, Gata6,50, 51, 52, 53, 54 Hepatocyte nuclear factor-1α (Hnf-1α),55 Hnf-1β,56 and CCAAT-displacement protein (Cdp),47, 57 determines the eec subset and array of peptide hormones they can secrete. Similar to Paneth cells, in Drosophila eecs have been shown to play an important role in maintaining the stem-cell niche,58, 59 while in both man60 and mouse61 quiescent, label retaining cells, have the potential to differentiate into Paneth and eecs. Therefore eecs can potentially play important roles during the modulation of the epithelium in health and disease.

Intestinal epithelial cell differentiation. Epithelial cells arise from the same LGR5+ pluripotent stem cell found in the crypt niche and based on the expression of the Notch-dependent basic helix loop helix transcription factors Hes1 or Atoh1 develop into absorptive enterocytes or secretory epithelial lineages, or via SpiB transcription factor expression to antigen-sampling M-cells. The secretory cells further differentiate into mucin secreting goblet cells, anti-microbial peptide secreting Paneth cells, opioid and alarmin secreting Tuft cells and peptide hormone secreting enteroendocrine cells, whose peptide hormone secretome further depends on spatio-temporal expression of further transcription factors.
Enteroendocrine subsets
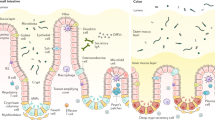
Eecs respond to luminal stimuli by secreting a variety of peptide hormones, including cholecystokinin (CCK), glucagon-like peptide 1 and 2 (GLP-1, GLP-2), glucose dependent insulinotropic peptide (GIP), peptide YY (PYY), gastrin, secretin, somatostatin, motilin, leptin, nesfatin-1, and ghrelin; as well as bioactive amines such as histamine and serotonin (5-HT). The historical dogma of distinct differentiated eec subsets secreting individual hormone peptides mediating biological function (Figure 2) has been superseded, via the analysis of eecs from transgenic reporter mice12, 62, 63, 64, 65 as well as cell ablation studies,35, 63 to reveal considerable overlap of the eec secretome. It now appears that the secretome ‘cocktail’ secreted by individual eecs is based on tissue location,66 although it is likely that certain peptide hormones remain rarely co-expressed.66, 67

Spatio-temporal expression of enteroendocrine peptide hormones. The dogma of terminally differentiated enteroendocrine cells secreting individual peptide hormones has been superseded with a secretome that contains a comprehensive array of peptide hormones altering based on their location within the gut. However, the traditional lettering nomenclature helps to demonstrate the role and function of individual peptide hormones.
Once secreted these peptide hormones can act in a traditional endocrine fashion on distant organs, or in a paracrine action to neighboring cells, including other eecs, and to vagal afferents and enteric neurons communicating at a central or local level respectively. Eecs have classically been studied for their roles in enabling efficient post-prandial assimilation of nutrients via alterations in gastrointestinal secretion, motility, pancreatic insulin release, and satiety4, 68 (Figure 2). However, it is now emerging that eecs have a huge array of chemosensory mechanisms to detect stimuli beyond nutrient intake, further indicating their importance beyond appetite and digestion.
Chemosensory pathways and peptide secretion
Eecs express a broad array of sensory machineries, mirroring their ability to respond to a diversity of ingested nutrients and other components in the gut lumen.5 Gut hormones are packaged into secretory vesicles, the release of which is mobilized by elevated concentrations of cytoplasmic calcium and enhanced by cyclic adenosine monophosphate (cAMP). Central to the detection of ingested food by eecs is a requirement that macronutrients are first digested to their component parts, including glucose, amino acids, and fatty acids. These small molecules are then detected by specific transporters and receptors located on the eecs themselves, and stimulate hormone secretion predominantly at the sites where nutrient absorption is maximal. The essential role of eecs is notably demonstrated by the impaired lipid absorption, reduced weight gain, growth retardation and high frequency of mortality in mice lacking the transcription factor Neurog3 and hence all intestinal eecs.30
Important pathways for the detection of glucose, amino acids and dipeptides by eecs are the families of brush border transporters that couple substrate absorption to ionic gradients.69, 70 Coupling of nutrient fluxes to the movement of sodium or hydrogen ions is an effective mechanism for driving absorption out of the gut lumen against a concentration gradient, but additionally has the consequence of causing small inward movements of positive charge into cells, that in turn can trigger membrane depolarization and voltage gated calcium entry. Glucose uptake by the sodium glucose cotransporter (SGLT1) is a well-studied example of this mechanism, and underlies the majority of glucose-triggered GIP and GLP-1 secretion.70
Fatty acids, bile acids, and amino acids are detected by specific G-protein coupled receptors (GPCRs) located directly on the eecs.69, 71, 72 The majority of nutrient-responsive GPCRs are coupled to Gas or Gaq signaling pathways, so their activation results in elevation of cytoplasmic cAMP and/or calcium concentrations, respectively, which in turn enhance vesicle release from the basolateral eec membrane. It has been recognized recently that fatty acids and bile acids, like glucose, must also be absorbed if they are to trigger gut hormone secretion. Thus, both the G-protein coupled bile acid receptor (GPBAR1) and the long chain free fatty acid receptor 1 (FFAR1/GPR40) appear to be localized and functional predominantly on the basolateral membrane of GLP-1 secreting cells.72, 73
Linking hormone secretion to nutrient absorption generates a robust physiological signal to the body about the quantity and quality of substrates entering the bloodstream at any time. Eecs also, however, respond to a range of non-nutrient stimuli,5 including bacterial metabolites,74, 75, 76, 77, 78, 79 hormonal,80 paracrine81. and neurotransmitter signals.12, 82 They thus form essential components of a network of complex signaling circuits, linking the gut with the rest of the body.
Enteroendocrine alterations during intestinal inflammation
Intestinal inflammation is often associated with microbial dysbiosis, be it inflammatory bowel disease, infection, colorectal cancer or food allergies.83, 84, 85, 86 Interestingly, eecs are the prime epithelial expressers of the receptors that sense bacterial metabolites,84, 87 such as GPR41/43, and therefore have the unique ability to relay dysbiosis into physiological adaptation, such as modulating energy homeostasis, glucose metabolism, gut barrier function, and mucosal immunity.77, 78, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102
Inflammatory bowel disease-human studies
Inflammatory bowel disease, typified by Crohn’s disease (CD) and ulcerative colitis (UC), is often linked to reduced appetite, anorexia and abnormal intestinal contractility.103 Eecs and the peptide hormones they secrete are now being recognized as potential instigators of these intestinal pathologies, due to their underlying role in mediating these systems during homeostasis. Indeed, genome-wide association studies for CD identified a single nucleotide polymorphism in the eec associated transcription factor Paired-like homeobox 2b (Phox2B),104 while auto-antibodies for the eec ubiquitination factor E4A (UBE4A) are seen as a biomarker for CD.105 Moreover, both Phox2B and UBE4A are seen to increase in ileal CD displaying active inflammation.106 An accumulation of studies has now begun to enumerate the alterations in eecs and secretions in clinical IBD (Table 2), aiding the ability to properly access their function in disease.
A large number of studies have focused on measuring peptide hormone levels in the serum or plasma of IBD patients. Serum and plasma Chromogranin A (CgA) levels, a pan-eec-marker,107 strongly increases in IBD patients and correlates to tumor necrosis factor (TNF)α,108, 109 however histological analysis of CgA and mucosal healing are lacking in these studies and are required to allow differentiation between cause and effect. The level of fecal CgA, has been seen to increase in UC but is not associated with disease index,110 while other studies demonstrate differences in microscopic colitis but not in UC or CD.111 More specifically, numerous studies have measured individual peptide hormone serum and plasma levels during the course of IBD, with significant changes seen in PYY, somatostatin, ghrelin, gastrin, GLP-1, CCK, 5-HT, and motilin during IBD (Table 2).112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129 Many of these reports also correlate increases of blood detected peptide levels with active disease, with somatostatin,117 ghrelin,115, 120, 122 and gastrin125 decreasing on remission. Moreover, ghrelin119, 121 and gastrin125 correlate with levels of the pro-inflammatory cytokine TNFα and IL-6. However, once again conflicting reports exist within the literature123, 126 and this is most likely explained by the heterogeneity seen in IBD. Indeed, in studies examining gastric emptying, post-prandial plasma CCK was seen to increase in CD,128 but not in a follow-up study by the same investigators.129 Another possible explanation for these disparities is that as eec alterations are seen to correlate with markers of inflammation they may be restricted to the inflammatory niche and are hence too localized to be detected via blood sampling.
The availability of patient biopsies and resections has allowed precise examination of tissues from IBD patients and has identified actual alterations in eec peptide hormone storage granules.130 Immunohistological quantification, in parallel with blood readings, is the most direct measure of eec fluctuation and various reports of changes in PYY, somatostatin, gastrin, GLP-1/2 and 5-HT+ cells exist in the literature (Table 2).106, 112, 131, 132, 133, 134, 135, 136, 137 Interestingly, similar to Paneth cell occurrence in the colon,138 gastrin+ cells are strangely found in the repairing small intestine of CD patients131 and subsets of IBD patients have auto-antibodies to gastrin.139 GLP-1 (ref. 140) and GLP-2 (ref. 141) are epithelial growth factors, with GLP-2 also having anti-inflammatory action both direct142 and indirect via Paneth cells.102 It is therefore a possibility that increases in the glucagon-like peptides during IBD are a possible response to epithelial damage and play a direct role in repair.137 Indeed, long acting analogs of GLP-2 could potentially be used for the treatment of short-bowel syndrome following CD.143
Despite the potential benefit that some eec peptide hormones may offer during IBD, they are also likely responsible for the reduced appetite, anorexia, and nausea that accompanies inflammation. Indeed, increases in plasma GLP-1 (ref. 129) and CCK128 are thought to be responsible for changes in gastric emptying, while decreased appetite and nausea in small bowel CD correlate with increased PYY levels.115 Plasma motilin also increases in IBD and is related to altered contractility,144, 145 with a polymorphism of the motilin gene interestingly seen in subsets of patients with CD.146 Although the majority of these human studies rely on small population sizes, collectively, these data strongly correlate alterations in eec peptide release with inflammation in IBD. Going forward, future studies should report the precise location of biopsy sampling, given the spatio-temporal expression of eec peptides. There is also an urgent need for more mechanistic approaches as overall there remains a lack of human data, besides co-localization, demonstrating direct cross-talk between intestinal inflammation and eecs. The varying and often clinically unknown burden in IBD has led to the use of animal models to decipher possible pathogenic mechanisms at play.
Inflammatory bowel disease-animal models
In rodents, chemically induced and genetically prone models of IBD are well associated with reduced feeding and weight loss, which is linked to eec function (Table 3).147, 148, 149, 150, 151, 152 Similar to the observations in human IBD, eec changes are often correlative to inflammation. PYY+ cell decreases in the dextran sulfate sodium (DSS) colitis model are restored with prednisolone treatment,144 while, the interleukin (IL)-2–/– colitis model reductions in PYY+ cells occur on activation of inflammation.153 Animal models have also begun to demonstrate that alterations in eec function are likely to be key factors in disease. Blocking CCK receptors in an acetic acid model of colitis reduces TNFα levels and ameliorates pathology,154 while 2,4,6-trinitrobenzenesulfonic acid (TNBS) colitis is inhibited with a CCKB receptor antagonist.155 Interestingly CCK was a novel and verified hit in a recent zebrafish enterocolitis small molecule screen.156 In addition, the regulatory peptide nesfatin-1 (ref. 157) and somatostatin have been shown to be anti-inflammatory in the acetic acid model of colitis.158, 159 Indeed, somatostatin agonists are able to increase intestinal tight junctions in models of dextran sulfate sodium (DSS) and Citrobacter rodentium induced colitis160 and modulate the water and sodium uptake protein NHE8, associated with UC pathology, via MAPK signaling.161 Neurotensin+ cells are seen to increase in the mouse DSS model and blocking signaling via antagonists increases pathology via a cyclooxygenase (COX)-2 mediated pathway,162 indicating a protective effect. Indeed, therapeutic use of peptides or agonists has been beneficial in mouse models, GLP-2 can rescue DSS colitis163 and small intestinal enteritis164, 165 possibly by reducing bacterial translocation,166 while nanodelivery of GLP-1 is also protective.167 Taken together this suggests that eecs play an essential and varied role in the pathology of IBD and are strong candidates for therapeutic intervention.168
As is often the case, the majority of initial observations have arisen from readily available chemical models of IBD, and while these results remain valid, new scientific knowledge is likely to arise by examining the influence of eecs in more complex models which better relate to IBD. It is therefore imperative to begin to examine alterations of eec biology in models such as the T-cell transfer model and Helicobacter hepaticus induced models of colitis.169 Furthermore, given the high concentration of eecs in the small intestine, examining eec changes in the SAMP1/YitFc model,170 which most closely resembles human ileal CD, should be a priority.
Non-infectious enteropathies
Beyond IBD, there is strong evidence that eecs are involved in multiple inflammatory driven diseases of the gut and may again be potential therapeutic targets. Celiac disease is associated with changes in eec number171, 172 as well as peptide granule storage.130 Serum levels of GLP-1, GIP173 and plasma CCK, thought to be responsible for the pancreatic dysfunction seen in celiac patients,174 are seen to be reduced in celiac blood. However, increases in CgA+ cells are also observed,174 with increased ghrelin+ cells seen in the duodenum that correlate with inflammation.175, 176 Increased serum somatostatin177 and GLP-2,178 plasma oxyntomodulin,179 neurotensin180, and motilin181 are also reported despite the villous blunting seen in the disease. In particular, 5-HT+ cell increases are thought to prolong inflammation via increased IFN-γ in tissue samples of refractory celiac patients,174 again pointing to a direct role for eecs in pathology.
With their close links to intestinal function it is unsurprising that eec alterations are also linked to irritable bowel syndrome (IBS). CD remission patients with IBS-like symptoms have increased levels of markers for 5-HT biosynthesis, rather than an increase in actual enterochromaffin cells.182 Somatostatin also increases in IBS post-IBD,183 while post-infectious IBS is strongly linked to changes in nerve sensitivity to peptide hormones.184, 185, 186, 187 Interestingly, high correlations of Chlamydia antigens are associated with eecs in IBS sufferers,188 further linking eecs not only to inflammation but also to intestinal infection.
Infection models and human correlation
Helminth infections in particular show alterations in eec function, perhaps due to the close association of helminths with the epithelium. Initial correlations were revealed in the livestock industry, with increases in serum CCK levels correlating with weight loss in pigs and lambs infected with the helminths Ascaris suum and Trichostrongylus colubriformis, respectively.189, 190 Calves infected with Ostertagia ostertagi have elevated gastrin191 while sheep infected with Ostertagia circumcincta have reduced gastrin and somatostatin+ cells and this is linked to the development of hypergastrinemia in parasitized animals.192 Furthermore, helminth induced alterations are not limited to mammal livestock with increases in CCK cells seen in Eubothrium crassum infected trout193 and Anisakis simplex infected flounders;194 while CCK and gastrin+ cell increase, but GLP-1/2 reduce in Eubothrium crassum infected trout.193 Experimental murine models have been used to further dissect the association of helminth infection with alterations in eec function.
CCK+ cell hyperplasia195 and hypersecretion196 are seen during Trichinella spiralis mouse infection and this correlates with hypophagia during enteritis. Furthermore, mice lacking CCK display no period of hypophagia associated with inflammation, identifying CCK as the sole mediator of hypophagia during this infection.195 This does not seem to be the case in all helminth infections as serum CCK levels are reduced in Nippostrongylus brasilliensis infection in rats,197 while increased serum gastrin is seen during T. spiralis, but not tape worm infection;198 furthermore decreased somatostatin+ cells are seen during intestinal inflammation resulting from intestinal schistomiasis in mice.199
Importantly, alterations in eec function during infection are also reported in the clinic and are not limited to helminth infection, with increases in CCK+ cells occurring in patients with upper intestinal infection, such as Giardia lambia.200 Alterations are also seen in bacterial and viral infection with reduced CgA+ cells seen in Helicobacter pylori patients.201 In particular reductions in ghrelin are associated with disease pathology,202 with eradication of H. pylori associated with increased ghrelin which correlates with abatement of dyspepsia.203 Importantly, in mouse models, changes occur before any general epithelial damage caused by the infection,204 while reduced 5-HT and somatostatin+ cells in HIV-1 infected individuals are associated with lower survival prognosis,205 again correlating alterations in eec function to pathology. Indeed, upon sensing chlamydia infection, eecs respond via a distinct transcript alteration,206 supporting their role as innate sensors of disease.
Collectively, the specific alterations of peptide secretion during inflammation indicate an uncoupling of eec subtype differentiation in disease, which holds promising therapeutic potential given the diverse functional roles of individual eec peptide hormones. In the case of infection, it will be interesting to resolve if peptide hormone release is driven by a detection of the parasites themselves or the microbial dysbiosis that often accompanies disease.
Intestinal neoplasia
As eec precursor cells are label retaining, Lgr5+ quiescent cells that have the potential to be recalled to the stem-cell fate, they have a potential role in neoplasia.61 Indeed, increased eec numbers in UC have been suggested to act as promoters for the neoplasia associated with IBD,207 with animal models demonstrating GLP-1 agonists as regulators of intestinal tumorigenesis.140 Moreover, at rest a subset of eecs express the cancer-associated transcription factor Brachyury208 and although rare, neuroendocrine tumors (NETs) are the most common cancer of the small intestine. Around 29% of small intestinal NETs carry amplifications or activating mutations in the PI3K/AKT mammalian target of rapamycin (mTOR) pathway209 and recent data demonstrating that EGF signaling is inhibited during eec differentiation,16 suggests it is reactivated during NET neoplasia.210 Therefore, the current targeting of the mTOR pathway in intestinal neoplasia211 is perhaps suggestive of a future focus on eecs in tumor pathology. Beyond the well-defined NETs, eecs have a long observed differentiation with sporadic colorectal cancer, occurring in 35% of colorectal carcinomas212, 213 and are often associated with the proliferative compartments of adenocarcinomas.214, 215 There is much debate regarding the clinical impact of eec differentiation on colorectal cancer, reviewed in (ref. 216). Of particular interest is the production of VEGF from eecs during cancer,212, 217 a factor whose targeting has been shown to prolong survival in colorectal cancer patients,218, 219 and promising results are again coming from drug trials blocking mTOR.220, 221 In line with the observations in IBD, the heterogeneity of intestinal neoplasia may account for some of the discrepancies seen, but beyond a strong correlation we are again in need of mechanistic studies, as well as stricter terminology within the intestinal cancer field.222
Mechanistic cross-talk between enteroendocrine cells and immune cells during intestinal inflammation
Inflammatory driven alterations in enteroendocrine cells
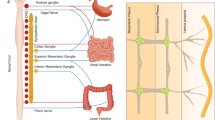
Numerous of the above studies correlate inflammation to alterations in eecs, and changes in IBD-mouse models are prevented with prior treatment of NFκβ or AP-1 inhibitors, which although not exclusively activated by immune cells, suggests the changes as immune driven.148, 223 There is a close physical association of immune cells with eecs224 and infection driven 5-HT+ cell hyperplasia observed during Citrobacter rodentium infection is absent in severe combined immunodeficiency (SCID) mice,225 as is the CCK and 5-HT+ cell hyperplasia seen in helminth infection.195, 226 5-HT+ cell increases seen during T. muris infection are also driven by specific T-helper (Th)2 CD4+ T-cell responses.227, 228 Recent studies have shown that the pro-inflammatory cytokines interferon (IFN)γ and TNFα increase CgA+ eecs in an autophagy and protein kinase B (Akt) dependent manner.229 Bromodeoxyuridine (BrdU) pulse-chase labeling of proliferative cells has demonstrated that increases in 5-HT+ cells during TNBS-colitis are due to alterations in the stem-cell niche rather than division of existing eecs.230 Collectively this points to cytokine mediated alterations of specific eec subsets via adaptation at the stem-cell niche as opposed to proliferation of existing eecs. Indeed, IL-33 derived from pericryptal fibroblasts during Salmonella infection has been shown to downregulate notch signaling in epithelial progenitors and increase CgA+ cells.231 Due to the high turnover of intestinal epithelial cells eec hyper/hypoplasia can therefore quickly influence the inflammatory state. Cytokines can also directly mediate peptide hormone secretion with TNFα decreasing GLP-2 expression by upregulating G-protein coupled receptor 120 in CD,232 IL-6 increasing GLP-1 release,233 while IL-1β has been shown to cause 5-HT secretion from CD enterochromaffin cells ex vivo.234 Immune cells and cytokines therefore directly influence eec biology and can mediate anorexia, which is now seen as a key modulator of specific immune responses.195, 235 Furthermore, eec signaling can be protective to the gut, with peptide hormones shown to modulate barrier function and therefore potentially limit antigenic load (Figure 3a). Moreover, this immunoendocrine crosstalk is unidirectional with chemosensory eecs able to mediate mucosal immunity, both direct and indirectly, acting as “cytokines” (Figure 3b) and initiating vagal anti-inflammatory pathways.

Enteroendocrine cell influence on epithelial barrier function and immune cells. (a) Enteroendocrine cells possess multiple chemosensory apparatus and are uniquely equipped to sense microbial metabolites and PAMPs. In response they secrete both peptide hormones and cytokines which directly influence barrier function. (b) Furthermore, as mucosal immune cells have numerous receptors for peptide hormones they can act as “cytokines” and can therefore be directly influenced by enteroendocrine cells. Reference numbers are indicated in superscript and black arrows indicate increase or decrease in specified cell activation.
Direct immune modulation
Enteroendocrine production of cytokines
Similar to recent findings in the chemosensory Tuft cells subset,26, 236, 237, 238 eecs are a source of cytokines and play roles in intestinal disease progression. Enteroendocrine cells have functional toll-like receptors and secrete cytokines following toll-like receptor (TLR) 1, 2, and 4 stimulation resulting in increased NF-κβ, MAPK signaling, as well as Ca2+ flux culminating in TNFα, transforming growth factor (TGF)β, macrophage inflammatory protein-2 and CCK release.74 Importantly, eecs are able to modulate their secretome in response to pathogenic detection, secreting chemokine (CXC-motif) 1/3 and IL-32 in response to flagellin and lipopolysaccharide (LPS), but not to fatty acids.239 In the case of IBD eecs are key producers of the pro-inflammatory cytokine IL-17C and therefore are involved in the pathogenesis of active disease.240 Mice lacking the exopeptidase carboxypeptidase E (CPE), an eec specific processing peptide, demonstrate reduced levels of PYY and are more susceptible to DSS-induced colitis.241 Moreover, at rest these mice display elevated IL-6 and KC levels from the epithelium as a whole, suggesting a CPE mediated immunosuppressive effect on intestinal barrier function by influencing the processing of specific neuropeptides.241
Enteroendocrine peptide modulation of barrier function
Further to producing cytokines, peptide hormones themselves have innate roles in maintaining barrier function (Figure 3a). At the most basic level they play a role in detecting toxins, with eecs releasing CCK following activation of the T2R38 bitter receptor limiting the absorption of toxic substances through modulation of gut efflux membrane transporters in neighboring epithelium.242 Moreover, chemotherapy drug induced emesis is dependent on 5-HT release and 5-HT3 receptor triggering,243 while more recently rotavirus toxin induced emesis was hypothesized to act via a similar mechanism.244 Interestingly, CCK and motilin can alter the behavior and movement of the liver fluke Fasciola hepatica,245 while ghrelin also has direct anti-parasitic246 and anti-bacterial effects,247 although the basolateral release of peptide hormones brings this suggested anti-microbial function into question. Moreover, eecs modulate production and secretion of classical anti-microbials, Drosophila have been shown to respond to Pseudomonas entomophila by expressing the peptide hormone allatostatin A which in turn regulates epithelial cell anti-microbial peptides and survival.248 The process of peptide hormones influencing anti-microbial production also extends to Paneth cells. GLP-2 receptor-null mice have increased bacterial colonization of the small intestine and reduced expression of Paneth cell anti-microbial gene products,102 although it remains to be ascertained if this is a result of other cellular phenotypes arising in the GLP-2 receptor-null mouse.102
Beyond anti-microbial effects, GLP-2 has been seen to maintain barrier function in mouse,97, 249 and human250 models, via the modulation of intestinal tight junction mechanisms and hence directly influences intestinal permeability. The most well-studied role of peptide hormones influencing barrier function is that of GLP-2,141 and more recently GLP-1,140 as potent epithelial growth factors. GLP-2s trophic affects act via myofibroblast produced insulin-like growth factor141 and keratinocyte growth factor251 as well as the ErbB signaling network in intestinal tissue;252 while GLP-1 mediates growth via fibroblast growth factor 7.140
Enteroendocrine peptides as “cytokines”
Intriguingly, immune cells express a vast array of receptors for eec secreted hormone peptides253 suggesting the potential for peptide hormones to act as “cytokines” (Figure 3b). Most notably the adipokine leptin and the amines histamine and 5-HT, although not exclusively produced from eecs, have well established direct immunomodulatory roles on numerous innate and adaptive cell types; reviewed in refs 254–256254,255,256.
Similarly to leptins role in influencing CD4+ T-cell responses, eec peptides have been shown to modulate T-cell polarization; nesfatin-1 has been linked to Th17 cell activation,257 while conversely ghrelin inhibits Th17 formation258 via mTOR,259 being beneficial in EAE models.260, 261 CCK has been shown to promote a Th2 (ref. 262) and regulatory T-cell (Treg) phenotype in vitro,262 as does GLP-1 (ref. 263) via decreased MAPK activation.264 As well as influencing T-cell differentiation, peptide hormones can also shape T-cell proliferation and migration. The orexigenic peptide hormone ghrelin increases T-cell proliferation via Phosphatidylinositol-4,5-bisphosphate 3-kinase, extracellular signal–regulated kinases and protein kinase C265 and has an anti-inflammatory effect in DSS colitis;266 with CD patients interestingly demonstrating a reduction of the ghrelin receptor GHSR-1a on T-cells.267 Somatostatin is also inhibitory to T-cell proliferation,268 downregulates LFA-1 expression269 and is ultimately involved in thymus development.270 Apart from CD4+ T-cells, GLP-1 signals to intraepithelial lymphocytes ameliorating the inflammation in DSS-induced colitis271 and signals to fat resident invariant NKT-cells mediating weight loss272 and psoriasis at the skin barrier.273 A number of these effects seem to be tissue specific with somatostatin inhibiting Peyer’s patch, but not splenic natural killer activity;274 and CCK altering lamina propria but not blood sourced cells.275
B-cells are also under the control of peptide hormones with CCK driving acetylcholine (Ach) production to recruit neutrophils independently of vagal stimulation.276 CCK277 and somatostatin278 can reduce B-cell activation, while ghrelin279 and neurotensin280 are able to enhance B-cell activation and proliferation respectively. CCK,277 somatostatin278 and GLP-2281 also influence immunoglobulin production and strikingly, the huge reduction in Immunoglobulin A production, seen during parenteral feeding can be rescued via the infusion of CCK,253, 282 although the mechanism remains undefined.
Eec peptide hormones also modulate innate immunity and hence quickly relay chemosensory detection of microbial metabolites and pathogens to the immune system. CCK has been shown to inhibit TLR9 stimulation of plasmacytoid DCs via TNF receptor associated factor 6 signaling,283 while somatostatin284 and neurotensin285 are also reported to be inhibitory to DC activation. Conversely CCK can promote IL-12 production286 and secretin acts as a chemoattractant to DCs287 suggesting more than a simple, global peptide hormone anti-inflammatory signal. Similarly, macrophages and monocytes are influenced by peptide hormones. CCK can inhibit macrophage activation,288, 289, 290 including inducible nitric oxide synthase production,291 and cause monocytes to produce inflammatory cytokines and eicosanoids.292 Several studies have importantly also deciphered the intracellular pathways involved,GLP-1 receptor agonists reduce endoplasmic reticulum stress and decrease inflammation-associated gene expression in macrophages,293, 294 while GLP-2 inhibits macrophage LPS stimulation via reduced NFκβ295 in an IL-10 independent manner.142 Discrepancies in these in vitro studies exist, with monocytes releasing IL-6 in response to somatostatin,296 while it can be anti-inflammatory in other settings,297 similar to GLP-1 (ref. 298) and ghrelin.299, 300 Peptide hormones appear to play an important role in transferring luminal signals during obesity, be it nutritional or microbial, to the immune system. GLP-1 agonists can inhibit monocyte to foam cell transition via altering autophagy, but this occurs only in obese patients,301 placing eecs under the spot light in this growing epidemic.
Granulocytes are generally inhibited by peptide hormone signaling; with basophils and eosinophils immunosuppressed by somatostatin117 and GLP-1 (ref. 302), respectively. Neutrophil phagocytosis,303, 304, 305 elastase release,306 and adhesion305, 307 are all inhibited by multiple peptide hormones, of particular interest is the role of GIP in ameliorating obesity-induced adipose tissue inflammation via modulation of neutrophil function.308 Most notably mast cells are strongly responsive to peptide hormones, with CCK,309 gastrin,310 and somatostatin311 inhibitory for degranulation, while ghrelin312 and PYY313 increase histamine release. CCK also induces intestinal contraction via mast cells during Giardia infection,314 demonstrating distinct fine tuning of mast cell function over other granulocytes. Interestingly, mast cells can populate 5-HT+ producing cells in the Neurog3 null mouse,315 and under homeostatic conditions share a transcriptome similar to mast cells,316 presenting an evolutionary link between these cellular populations. Eecs therefore have a unique ability to sense the intestinal environment and directly interact with the underlying innate and adaptive immune system through cytokines and peptide hormone signaling.
The purest evidence of peptide hormone immune cell influence is via in vitro assays, especially given the numerous pathways and tissues these hormones may affect. However, older studies may have been susceptible to endotoxin contamination317 and cell specific peptide receptor-null studies are required to fully decipher the overall importance of the immunoendocrine axis.
Indirect immune modulation
Vagal anti-inflammatory reflex
Eec released peptide hormones may also influence immunity via signaling to vagal afferents and influence the intestinal cholinergic anti-inflammatory pathway318 via the release of Ach from vagal efferents. Recent evidence has demonstrated that eecs possess a direct contact with neurons and this “neuropod” allows direct neuroepithelial communication,319 a portal that pathogens may have evolved to target infection of the nervous system.320, 321, 322 This anti-inflammatory pathway was originally highlighted in an LPS model of hemorrhagic shock; prior nutritional stimulation of mice with a high-fat diet induced a vagal reflex and Ach release which inhibited LPS-induced cytokine secretion and reduced pathology. This was seen to be dependent on vagal CCK stimulation and resulting Ach stimulation of macrophage alpha7-nAch receptor.323 This pathway is also dependent on post-absorptive chylomicron formation, lipoprotein formations shown to release endogenous CCK324 and also requires GLP-1 receptor activation325 and potentially ghrelin.326 GLP-2 also acts via enteric nerves to increase the secretion of immunomodulatory vasoactive intestinal peptide during animal models of IBD.327
Others have demonstrated similar CCK-induced vagal anti-inflammatory pathways in a variety of inflammatory settings, such as post-operative ileus328 and lung damage during endotoxemia.329 Furthermore, interfering with the vagal reflex has also been shown to exacerbate DSS colitis.330 Although not thought to be B- or T-cell dependent,331 CCK-induced Ach release has also been shown to influence other innate cells such as mast cells.332 Recently vagally released Ach has been shown to influence the level of a key host-protective mediator, PCTR1, in group 3 innate lymphoid cells (ILCs) regulating tissue resolution tone and myeloid cell responses in an Escherichia coli peritonitis model.333 However, it remains to be seen if eec peptides can influence Ach production to effect intestinal specific cell types or directly modulate ILC function. This anti-inflammatory role of the vagus nerve, and therefore eec peptide hormone stimulation, is an exciting and growing area of research.334
Control of appetite
Beyond the vagal reflex response is the concept of altered feeding itself as an immune modulator. This is not a new concept with the adage “starve a fever, feed a cold” familiar to many, however growing evidence has demonstrated that anorexia is an essential aspect of certain,335, 336, 337 but not all,338 acute infections. Most recently, Medzhitov and colleagues have confirmed that although anorexia is beneficial in Listeria monocytogenes infection, it is detrimental during influenza. This was shown to be due to the differing stress pathways elicited during the distinct immunopathology associated with each disease, and therefore explains why anorexia does not always supply the correct metabolic requirements for tolerance in each disease setting.235 This offers the intriguing hypothesis that feeding behavior induced by altered eec dynamics is an attempt to influence immunity and minimize immunopathology.
Utilizing the helminth T. spiralis model of T-cell induced eec driven hypophagia,196 Worthington et al.195 investigated the possible molecular mechanisms and actual purpose of the hypophagia seen during this parasitic infection. During infection CD4+ T-cells hijack classical cholecystokinin feeding pathways to reduce food intake during enteritis.196 Increased c-Fos brain expression during helminth infection,339, 340 supports that hypophagia relies on increased gut-brain axis signaling, as opposed to intestinal hypomotility. This hypophagia results in significant weight loss and visible reduction of visceral fat pads, which are a key source of adipokines such as leptin.255, 341 As T-cells express functional leptin receptors342 and leptin stimulation polarizes T-cells toward a pro-inflammatory Th1 state,343 it was postulated that the immune driven reduction in leptin driven by CCK during T. spiralis infection, would be beneficial in allowing a helminth expelling Th2 immune response to develop. Indeed, delayed expulsion of Heligmosomoides bakeri is seen in protein deficient mice and is linked to higher levels of leptin.344 Restoration via recombinant leptin treatment, resulted in a significant reduction in CD4+ Th2 cytokine production and accompanying mastocytosis, which is essential for worm expulsion.345 This restoration of basal leptin levels and shift in immune response culminated in a significant delay in parasite expulsion. Hence, identifying immune driven alterations in eec mediated feeding mechanisms, as a novel mechanism in helminth expulsion.195
Conclusions
In summary, the eec secretome encompasses cytokines as well as peptide hormones that have the ability to directly and indirectly influence the majority of the intestinal mucosal immune system. Novel transgenic reporter models are now allowing the scientific community to fully investigate this exciting crosstalk between our intestinal endocrine and immune systems, opening up the possibility to repurpose current drugs used for metabolic syndromes in wider immune inflammatory settings such as IBD, infection and cancer. Indeed, as eecs transpose microbial signals it may be possible to utilize eec peptide agonist/antagonists over and above microbial interventions in the treatment of disease. Moreover, the expression and role of epithelial endocrine cells at other mucosal sites such as the lung is hugely understudied. Indeed, this potential may go beyond diseases of the intestine with peptide agonists showing potential in models of psoriasis, multiple sclerosis and rheumatoid arthritis,260, 273, 286 highlighting the huge therapeutic potential of the immunoendocrine axis.
References
Artis, D. & Grencis, R.K. The intestinal epithelium: sensors to effectors in nematode infection. Mucosal Immunol. 1, 252–264 (2008).
Peterson, L.W. & Artis, D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14, 141–153 (2014).
Pastorelli, L., De Salvo, C., Mercado, J.R., Vecchi, M. & Pizarro, T.T. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front. Immunol. 4, 280 (2013).
Begg, D.P. & Woods, S.C. The endocrinology of food intake. Nat. Rev. Endocrinol. 9, 584–597 (2013).
Gribble, F.M. & Reimann, F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu. Rev. Physiol. 78, 277–299 (2016).
Liou, A.P. et al. The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140, 903–912 (2011).
Bohorquez, D.V., Chandra, R., Samsa, L.A., Vigna, S.R. & Liddle, R.A. Characterization of basal pseudopod-like processes in ileal and colonic PYY cells. J. Mol. Histol. 42, 3–13 (2011).
Parker, H.E., Habib, A.M., Rogers, G.J., Gribble, F.M. & Reimann, F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 52, 289–298 (2009).
Reimann, F., Habib, A.M., Tolhurst, G., Parker, H.E., Rogers, G.J. & Gribble, F.M. Glucose sensing in L cells: a primary cell study. Cell Metab. 8, 532–539 (2008).
Engelstoft, M.S. et al. Research resource: a chromogranin a reporter for serotonin and histamine secreting enteroendocrine cells. Mol. Endocrinol. 29, 1658–1671 (2015).
Sakata, I. et al. Characterization of a novel ghrelin cell reporter mouse. Regul. Pept. 155, 91–98 (2009).
Adriaenssens, A. et al. A transcriptome-led exploration of molecular mechanisms regulating somatostatin-producing D-Cells in the gastric epithelium. Endocrinology 156, 3924–3936 (2015).
Nagatake, T., Fujita, H., Minato, N. & Hamazaki, Y. Enteroendocrine cells are specifically marked by cell surface expression of claudin-4 in mouse small intestine. PLoS ONE 9, e90638 (2014).
Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 (2007).
Barker, N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 15, 19–33 (2014).
Basak, O., Beumer, J., Wiebrands, K., Seno, H., van Oudenaarden, A. & Clevers, H. Induced quiescence of Lgr5+ stem cells in intestinal organoids enables differentiation of hormone-producing enteroendocrine cells. Cell Stem Cell 20, e174 (2017).
Shroyer, N.F., Helmrath, M.A., Wang, V.Y.C., Antalffy, B., Henning, S.J. & Zoghbi, H.Y. Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology 132, 2478–2488 (2007).
Li, H.J., Ray, S.K., Singh, N.K., Johnston, B. & Leiter, A.B. Basic helix-loop-helix transcription factors and enteroendocrine cell differentiation. Diabetes Obes. Metab. 13 (Suppl 1), 5–12 (2011).
May, C.L. & Kaestner, K.H. Gut endocrine cell development. Mol. Cell Endocrinol. 323, 70–75 (2010).
Yang, Q., Bermingham, N.A., Finegold, M.J. & Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294, 2155–2158 (2001).
Shroyer, N.F., Shultz, D.W., Venken, K.J., Bellen, H.J. & Zoghbi, H.Y. Gfi1 functions downstream of Math1 to control intestinal secretory cell differentiation. Gastroenterology 128, A127–A127 (2005).
Katz, J.P. et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 129, 2619–2628 (2002).
Bastide, P. et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol. 178, 635–648 (2007).
Mori-Akiyama, Y. et al. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology 133, 539–546 (2007).
Gerbe, F. et al. Distinct ATOH1 and Neurog3 requirements define tuft cells as a new secretory cell type in the intestinal epithelium. J. Cell Biol. 192, 767–780 (2011).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016).
Kanaya, T. et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat. Immunol. 13, 729–736 (2012).
Knoop, K.A. et al. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 183, 5738–5747 (2009).
de Lau, W. et al. Peyer's patch M cells derived from Lgr5(+) stem cells require SpiB and are induced by RankL in cultured "miniguts". Mol. Cell Biol. 32, 3639–3647 (2012).
Mellitzer, G. et al. Loss of enteroendocrine cells in mice alters lipid absorption and glucose homeostasis and impairs postnatal survival. J. Clin. Invest. 120, 1708–1721 (2010).
Jenny, M. et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J. 21, 6338–6347 (2002).
Knudsen, L.A., Petersen, N., Schwartz, T.W. & Egerod, K.L. The MicroRNA repertoire in enteroendocrine cells: identification of miR-375 as a potential regulator of the enteroendocrine lineage. Endocrinology 156, 3971–3983 (2015).
Mutoh, H., Fung, B.P., Naya, F.J., Tsai, M.J., Nishitani, J. & Leiter, A.B. The basic helix-loop-helix transcription factor BETA2/NeuroD is expressed in mammalian enteroendocrine cells and activates secretin gene expression. Proc. Natl Acad. Sci. USA 94, 3560–3564 (1997).
Naya, F.J. et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 11, 2323–2334 (1997).
Rindi, G., Ratineau, C., Ronco, A., Candusso, M.E., Tsai, M. & Leiter, A.B. Targeted ablation of secretin-producing cells in transgenic mice reveals a common differentiation pathway with multiple enteroendocrine cell lineages in the small intestine. Development 126, 4149–4156 (1999).
Larsson, L.I., St-Onge, L., Hougaard, D.M., Sosa-Pineda, B. & Gruss, P. Pax 4 and 6 regulate gastrointestinal endocrine cell development. Mech. Dev. 79, 153–159 (1998).
Trinh, D.K., Zhang, K., Hossain, M., Brubaker, P.L. & Drucker, D.J. Pax-6 activates endogenous proglucagon gene expression in the rodent gastrointestinal epithelium. Diabetes 52, 425–433 (2003).
Hill, M.E., Asa, S.L. & Drucker, D.J. Essential requirement for Pax6 in control of enteroendocrine proglucagon gene transcription. Mol. Endocrinol. 13, 1474–1486 (1999).
Terry, N.A., Walp, E.R., Lee, R.A., Kaestner, K.H. & May, C.L. Impaired enteroendocrine development in intestinal-specific Islet1 mouse mutants causes impaired glucose homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G979–G991 (2014).
Larsson, L.I., Madsen, O.D., Serup, P., Jonsson, J. & Edlund, H. Pancreatic-duodenal homeobox 1 -role in gastric endocrine patterning. Mech. Dev. 60, 175–184 (1996).
Miller, C.P., McGehee, R.E. Jr. & Habener, J.F. IDX-1: a new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J. 13, 1145–1156 (1994).
Offield, M.F. et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983–995 (1996).
Lee, E., Ryu, G.R., Moon, S.D., Ko, S.H., Ahn, Y.B. & Song, K.H. Reprogramming of enteroendocrine K cells to pancreatic beta-cells through the combined expression of Nkx6.1 and Neurogenin3, and reaggregation in suspension culture. Biochem. Biophys. Res. Commun. 443, 1021–1027 (2014).
Desai, S. et al. Nkx2.2 regulates cell fate choice in the enteroendocrine cell lineages of the intestine. Dev. Biol. 313, 58–66 (2008).
Gross, S. et al. Nkx2.2 is expressed in a subset of enteroendocrine cells with expanded lineage potential. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G975–G987 (2015).
Middendorp, S. et al. Adult stem cells in the small intestine are intrinsically programmed with their location-specific function. Stem Cells 32, 1083–1091 (2014).
Fang, R., Olds, L.C. & Sibley, E. Spatio-temporal patterns of intestine-specific transcription factor expression during postnatal mouse gut development. Gene Exp. Patterns 6, 426–432 (2006).
Grainger, S., Savory, J.G. & Lohnes, D. Cdx2 regulates patterning of the intestinal epithelium. Dev. Biol. 339, 155–165 (2010).
Mutoh, H. et al. The intestine-specific homeobox gene Cdx2 induces expression of the basic helix-loop-helix transcription factor Math1. Differentiation 74, 313–321 (2006).
Beuling, E. et al. GATA factors regulate proliferation, differentiation, and gene expression in small intestine of mature mice. Gastroenterology 140, e1211–e1212 (2011).
Bosse, T. et al. Gata4 is essential for the maintenance of Jejunal-Ileal identities in the adult mouse small intestine. Mol. Cell Biol. 26, 9060–9070 (2006).
Dimaline, R., Campbell, B.J., Watson, F., Sandvik, A.K., Struthers, J. & Noble, P.J. Regulated expression of GATA-6 transcription factor in gastric endocrine cells. Gastroenterology 112, 1559–1567 (1997).
Dusing, M.R. & Wiginton, D.A. Epithelial lineages of the small intestine have unique patterns of GATA expression. J. Mol. Histol. 36, 15–24 (2005).
Gao, X.P., Sedgwick, T., Shi, Y.B. & Evans, T. Distinct functions are implicated for the GATA-4, -5, and -6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol. Cell Biol. 18, 2901–2911 (1998).
Lussier, C.R. et al. Loss of hepatocyte-nuclear-factor-1alpha impacts on adult mouse intestinal epithelial cell growth and cell lineages differentiation. PLoS ONE 5, e12378 (2010).
D'Angelo, A. et al. Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development 137, 1573–1582 (2010).
Boudreau, F. et al. A novel colonic repressor element regulates intestinal gene expression by interacting with Cux/CDP. Mol. Cell Biol. 22, 5467–5478 (2002).
Amcheslavsky, A. et al. Enteroendocrine cells support intestinal stem-cell-mediated homeostasis in Drosophila. Cell Rep. 9, 32–39 (2014).
Radford, I.R. & Lobachevsky, P.N. An enteroendocrine cell-based model for a quiescent intestinal stem cell niche. Cell Prolif. 39, 403–414 (2006).
Jung, P. et al. Isolation of human colon stem cells using surface expression of PTK7. Stem Cell Rep. 5, 979–987 (2015).
Buczacki, S.J. et al. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495, 65–69 (2013).
Sykaras, A.G., Demenis, C., Case, R.M., McLaughlin, J.T. & Smith, C.P. Duodenal enteroendocrine I-cells contain mRNA transcripts encoding key endocannabinoid and fatty acid receptors. PLoS ONE 7, e42373 (2012).
Egerod, K.L. et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology 153, 5782–5795 (2012).
Habib, A.M. et al. Overlap of endocrine hormone expression in the mouse intestine revealed by transcriptional profiling and flow cytometry. Endocrinology 153, 3054–3065 (2012).
Grunddal, K.V. et al. Neurotensin is coexpressed, coreleased, and acts together with GLP-1 and PYY in enteroendocrine control of metabolism. Endocrinology 157, 176–194 (2016).
Svendsen, B. et al. An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology 156, 847–857 (2015).
Svendsen, B. et al. GLP1- and GIP-producing cells rarely overlap and differ by bombesin receptor-2 expression and responsiveness. J. Endocrinol. 228, 39–48 (2016).
Psichas, A., Reimann, F. & Gribble, F.M. Gut chemosensing mechanisms. J. Clin. Invest. 125, 908–917 (2015).
Diakogiannaki, E. et al. Oligopeptides stimulate glucagon-like peptide-1 secretion in mice through proton-coupled uptake and the calcium-sensing receptor. Diabetologia 56, 2688–2696 (2013).
Gorboulev, V. et al. Na(+)-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 61, 187–196 (2012).
Gribble, F.M., Diakogiannaki, E. & Reimann, F. Gut hormone regulation and secretion via FFA1 and FFA4. Handb. Exp. Pharmacol. 236, 181–203 (2016).
Brighton, C.A. et al. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology 156, 3961–3970 (2015).
Christensen, L.W., Kuhre, R.E., Janus, C., Svendsen, B. & Holst, J.J. Vascular, but not luminal, activation of FFAR1 (GPR40) stimulates GLP-1 secretion from isolated perfused rat small intestine. Physiol. Rep. 3, e12551 (2015).
Bogunovic, M. et al. Enteroendocrine cells express functional Toll-like receptors. Am. J. Physiol.-Gastrointest. Liver Physiol. 292, G1770–G1783 (2007).
Chimerel, C., Emery, E., Summers, D.K., Keyser, U., Gribble, F.M. & Reimann, F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 9, 1202–1208 (2014).
Nguyen, A.T. et al. Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: involvement of the GLP-1 pathway. Diabetes 63, 471–482 (2014).
Samuel, B.S. et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl Acad. Sci. USA 105, 16767–16772 (2008).
Tolhurst, G. et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61, 364–371 (2012).
Psichas, A. et al. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. 39, 424–429 (2015).
Gabellec, M.M., Griffais, R., Fillion, G. & Haour, F. Expression of interleukin 1 alpha, interleukin 1 beta and interleukin 1 receptor antagonist mRNA in mouse brain: regulation by bacterial lipopolysaccharide (LPS) treatment. Mol. Brain Res. 31, 122–130 (1995).
Saffouri, B., Weir, G., Bitar, K. & Makhlouf, G. Stimulation of gastrin secretion from the perfused rat stomach by somatostatin antiserum. Life Sci. 25, 1749–1753 (1979).
Engelstoft, M.S. et al. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol. Metab. 2, 376–392 (2013).
Houlden, A. et al. Chronic trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS ONE 10, e0125945 (2015).
Karaki, S. et al. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res. 324, 353–360 (2006).
Zeng, M.Y., Inohara, N. & Nunez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 10, 18–26 (2017).
Wesemann, D.R. & Nagler, C.R. The microbiome, timing, and barrier function in the context of allergic disease. Immunity 44, 728–738 (2016).
Nohr, M.K. et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 154, 3552–3564 (2013).
Cani, P.D., Dewever, C. & Delzenne, N.M. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br. J. Nutr. 92, 521–526 (2004).
Delzenne, N.M., Cani, P.D., Daubioul, C. & Neyrinck, A.M. Impact of inulin and oligofructose on gastrointestinal peptides. Br. J. Nutr. 93 (Suppl 1), S157–S161 (2005).
Cani, P.D., Knauf, C., Iglesias, M.A., Drucker, D.J., Delzenne, N.M. & Burcelin, R. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes 55, 1484–1490 (2006).
Ropert, A. et al. Colonic fermentation and proximal gastric tone in humans. Gastroenterology 111, 289–296 (1996).
Cani, P.D. et al. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am. J. Clin. Nutr. 90, 1236–1243 (2009).
Parnell, J.A. & Reimer, R.A. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am. J. Clin. Nutr. 89, 1751–1759 (2009).
Breton, J. et al. Gut commensal E. coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metab. 23, 324–334 (2016).
Cani, P.D., Everard, A. & Duparc, T. Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 13, 935–940 (2013).
Cani, P.D. & Knauf, C. How gut microbes talk to organs: the role of endocrine and nervous routes. Mol. Metab. 5, 743–752 (2016).
Cani, P.D. et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58, 1091–1103 (2009).
Everard, A. et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60, 2775–2786 (2011).
Plaisancie, P., Dumoulin, V., Chayvialle, J.A. & Cuber, J.C. Luminal glucagon-like peptide-1(7-36) amide-releasing factors in the isolated vascularly perfused rat colon. J. Endocrinol. 145, 521–526 (1995).
Lin, H.V. et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 7, e35240 (2012).
Cani, P.D. et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50, 2374–2383 (2007).
Lee, S.J. et al. Disruption of the murine Glp2r impairs Paneth cell function and increases susceptibility to small bowel enteritis. Endocrinology 153, 1141–1151 (2012).
Harrison, E., Lal, S. & McLaughlin, J.T. Enteroendocrine cells in gastrointestinal pathophysiology. Curr. Opin. Pharmacol. 13, 941–945 (2013).
Rioux, J.D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 (2007).
Sakiyama, T., Fujita, H. & Tsubouchi, H. Autoantibodies against ubiquitination factor E4A (UBE4A) are associated with severity of Crohn's disease. Inflamm. Bowel Dis. 14, 310–317 (2008).
Moran, G.W., Pennock, J. & McLaughlin, J.T. Enteroendocrine cells in terminal ileal Crohn's disease. J. Crohns Colitis 6, 871–880 (2012).
Rindi, G., Leiter, A.B., Kopin, A.S., Bordi, C. & Solcia, E. The "normal" endocrine cell of the gut: changing concepts and new evidences. Ann. N Y. Acad. Sci. 1014, 1–12 (2004).
Zissimopoulos, A. et al. Chromogranin A as a biomarker of disease activity and biologic therapy in inflammatory bowel disease: a prospective observational study. Scand. J. Gastroenterol. 49, 942–949 (2014).
Sciola, V. et al. Plasma chromogranin a in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 15, 867–871 (2009).
Strid, H., Simren, M., Lasson, A., Isaksson, S., Stridsberg, M. & Ohman, L. Fecal chromogranins and secretogranins are increased in patients with ulcerative colitis but are not associated with disease activity. J. Crohns Colitis 7, e615–e622 (2013).
Wagner, M., Stridsberg, M., Peterson, C.G., Sangfelt, P., Lampinen, M. & Carlson, M. Increased fecal levels of chromogranin A, chromogranin B, and secretoneurin in collagenous colitis. Inflammation 36, 855–861 (2013).
Tari, A. et al. Peptide YY abnormalities in patients with ulcerative colitis. Jpn. J. Med. 27, 49–55 (1988).
Adrian, T.E., Savage, A.P., Bacarese-Hamilton, A.J., Wolfe, K., Besterman, H.S. & Bloom, S.R. Peptide YY abnormalities in gastrointestinal diseases. Gastroenterology 90, 379–384 (1986).
Koch, T.R., Roddy, D.R. & Go, V.L. Abnormalities of fasting serum concentrations of peptide YY in the idiopathic inflammatory bowel diseases. Am. J. Gastroenterol. 82, 321–326 (1987).
Moran, G.W., Leslie, F.C. & McLaughlin, J.T. Crohn's disease affecting the small bowel is associated with reduced appetite and elevated levels of circulating gut peptides. Clin. Nutr. 32, 404–411 (2013).
Payer, J. et al. Plasma somatostatin levels in ulcerative colitis. Hepatogastroenterology 41, 552–553 (1994).
Binimelis, J. et al. Circulating immunoreactive somatostatin in gastrointestinal diseases. Decrease after vagotomy and enhancement in active ulcerative colitis, irritable bowel syndrome, and duodenal ulcer. Scand. J. Gastroenterol. 22, 931–937 (1987).
Karmiris, K., Koutroubakis, I.E., Xidakis, C., Polychronaki, M., Voudouri, T. & Kouroumalis, E.A. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm. Bowel Dis. 12, 100–105 (2006).
Peracchi, M. et al. Circulating ghrelin levels in patients with inflammatory bowel disease. Gut 55, 432–433 (2006).
Ates, Y., Degertekin, B., Erdil, A., Yaman, H. & Dagalp, K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig. Dis. Sci. 53, 2215–2221 (2008).
Cekic, C. et al. Evaluation of the relationship between serum ghrelin, C-reactive protein and interleukin-6 levels, and disease activity in inflammatory bowel diseases. Hepatogastroenterology 61, 1196–1200 (2014).
Nishi, Y. et al. Plasma leptin and ghrelin concentrations in patients with Crohn's disease. World J. Gastroenterol. 11, 7314–7317 (2005).
Triantafillidis, J.K. et al. Serum gastrin levels in patients with inflammatory bowel disease. Hepatogastroenterology 50 (Suppl 2), cccxv–cccxvii (2003).
Essop, A.R., Segal, I. & Ming, R. High serum gastrin in ulcerative colitis. N. Engl. J. Med. 307, 192 (1982).
Hopman, W.P., de Jong, D.J., Naber, A.H. & Jansen, J.B. Tumour necrosis factor alpha antibody affects gastrin release in Crohn disease. Scand. J. Gastroenterol. 38, 522–525 (2003).
Bendet, N. et al. Enhanced glucose-dependent glucagon-like peptide-1 and insulin secretion in Crohn patients with terminal ileum disease is unrelated to disease activity or ileal resection. Scand. J. Gastroenterol. 39, 650–656 (2004).
Vu, M.K., Gielkens, H.A., van Hogezand, R.A., van Oostayen, J.A., Lamers, C.B. & Masclee, A.A. Gallbladder motility in Crohn disease: influence of disease localization and bowel resection. Scand. J. Gastroenterol. 35, 1157–1162 (2000).
Keller, J., Beglinger, C., Holst, J.J., Andresen, V. & Layer, P. Mechanisms of gastric emptying disturbances in chronic and acute inflammation of the distal gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G861–G868 (2009).
Keller, J. et al. Gastric emptying and disease activity in inflammatory bowel disease. Eur. J. Clin. Invest. 45, 1234–1242 (2015).
Dawson, J., Bryant, M.G., Bloom, S.R. & Peters, T.J. Gastrointestinal regulatory peptide storage granule abnormalities in jejunal mucosal diseases. Gut 25, 636–643 (1984).
Yokoyama, I., Kozuka, S. & Takagi, H. Gastrin producing cells in the regenerating mucosa of the small intestine. Jpn. J. Surg. 18, 54–60 (1988).
El-Salhy, M., Danielsson, A., Stenling, R. & Grimelius, L. Colonic endocrine cells in inflammatory bowel disease. J. Intern. Med. 242, 413–419 (1997).
Schmidt, P.T., Ljung, T., Hartmann, B., Hare, K.J., Holst, J.J. & Hellstrom, P.M. Tissue levels and post-prandial secretion of the intestinal growth factor, glucagon-like peptide-2, in controls and inflammatory bowel disease: comparison with peptide YY. Eur. J. Gastroenterol. Hepatol. 17, 207–212 (2005).
Magro, F. et al. Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Dig. Dis. Sci. 47, 216–224 (2002).
El-Salhy, M., Gundersen, D., Hatlebakk, J.G. & Hausken, T. High densities of serotonin and peptide YY cells in the colon of patients with lymphocytic colitis. World J. Gastroenterol. 18, 6070–6075 (2012).
Watanabe, T., Kubota, Y., Sawada, T. & Muto, T. Distribution and quantification of somatostatin in inflammatory disease. Dis. Colon Rectum 35, 488–494 (1992).
Xiao, Q., Boushey, R.P., Cino, M., Drucker, D.J. & Brubaker, P.L. Circulating levels of glucagon-like peptide-2 in human subjects with inflammatory bowel disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R1057–R1063 (2000).
Tanaka, M. et al. Spatial distribution and histogenesis of colorectal Paneth cell metaplasia in idiopathic inflammatory bowel disease. J. Gastroenterol. Hepatol. 16, 1353–1359 (2001).
Ardesjo, B. et al. Immunoreactivity against Goblet cells in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 14, 652–661 (2008).
Koehler, J.A. et al. GLP-1 R agonists promote normal and neoplastic intestinal growth through mechanisms requiring Fgf7. Cell Metab. 21, 379–391 (2015).
Dube, P.E., Forse, C.L., Bahrami, J. & Brubaker, P.L. The essential role of insulin-like growth factor-1 in the intestinal tropic effects of glucagon-like peptide-2 in mice. Gastroenterology 131, 589–605 (2006).
Ivory, C.P., Wallace, L.E., McCafferty, D.M. & Sigalet, D.L. Interleukin-10-independent anti-inflammatory actions of glucagon-like peptide 2. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1202–G1210 (2008).
Kochar, B. et al. Safety and efficacy of teduglutide (Gattex) in patients with Crohn's Disease and need for parenteral support due to short bowel syndrome-associated intestinal failure. J. Clin. Gastroenterol. 51, 508–511 (2016).
Hirotani, Y., Mikajiri, K., Ikeda, K., Myotoku, M. & Kurokawa, N. Changes of the peptide YY levels in the intestinal tissue of rats with experimental colitis following oral administration of mesalazine and prednisolone. Yakugaku Zasshi 128, 1347–1353 (2008).
Zlatkina, A.R., Bezzubik, K.V., Peeters, T.L. & Kuznetsova, O.G. Plasma motilin and diarrhea in ulcerative colitis. Sov. Med. 53, 14–18 (1990).
Annese, V. et al. Polymorphism of motilin gene in patients with Crohn's disease. Dig. Dis. Sci. 43, 715–719 (1998).
McHugh, K., Castonguay, T.W., Collins, S.M. & Weingarten, H.P. Characterization of suppression of food intake following acute colon inflammation in the rat. Am. J. Physiol. 265 (5 Pt 2), R1001–R1005 (1993).
El-Salhy, M. & Hatlebakk, J.G. Changes in enteroendocrine and immune cells following colitis induction by TNBS in rats. Mol. Med. Rep. 14, 4967–4974 (2016).
O'Hara, J.R., Lomax, A.E., Mawe, G.M. & Sharkey, K.A. Ileitis alters neuronal and enteroendocrine signalling in guinea pig distal colon. Gut 56, 186–194 (2007).
Schmidt, P.T., Hartmann, B., Bregenholt, S., Hoist, J.J. & Claesson, M.H. Deficiency of the intestinal growth factor, glucagon-like peptide 2, in the colon of SCID mice with inflammatory bowel disease induced by transplantation of CD4+ T cells. Scand. J. Gastroenterol. 35, 522–527 (2000).
Bang-Berthelsen, C.H. et al. GLP-1 induces barrier protective expression in brunner's glands and regulates colonic inflammation. Inflamm. Bowel Dis. 22, 2078–2097 (2016).
Rubin, D.C., Zhang, H.Y., Qian, P.Q., Lorenz, R.G., Hutton, K. & Peters, M.G. Altered enteroendocrine cell expression in T cell receptor alpha chain knock-out mice. Microsc. Res. Tech. 51, 112–120 (2000).
Qian, B.F., El-Salhy, M., Melgar, S., Hammarstrom, M.L. & Danielsson, A. Neuroendocrine changes in colon of mice with a disrupted IL-2 gene. Clin. Exp. Immunol. 120, 424–433 (2000).
Al Moutaery, A. Proglumide attenuates experimental colitis in rats. Exp. Toxicol. Pathol. 56, 327–332 (2005).
Barbier, M. et al. Proinflammatory role of leptin in experimental colitis in rats benefit of cholecystokinin-B antagonist and beta3-agonist. Life Sci. 69, 567–580 (2001).
Oehlers, S.H. et al. A whole animal chemical screen approach to identify modifiers of intestinal neutrophilic inflammation. FEBS J. 284, 402–441 (2016).
Palasz, A. et al. Nesfatin-1, a unique regulatory neuropeptide of the brain. Neuropeptides 46, 105–112 (2012).
Ozturk, C.C., Oktay, S., Yuksel, M., Akakin, D., Yarat, A. & Kasimay Cakir, O. Anti-inflammatory effects of nesfatin-1 in rats with acetic acid - induced colitis and underlying mechanisms. J. Physiol. Pharmacol. 66, 741–750 (2015).
Eliakim, R., Karmeli, F., Okon, E. & Rachmilewitz, D. Octreotide effectively decreases mucosal damage in experimental colitis. Gut 34, 264–269 (1993).
Li, X. et al. Somatostatin regulates tight junction proteins expression in colitis mice. Int. J. Clin. Exp. Pathol. 7, 2153–2162 (2014).
Li, X. et al. Somatostatin regulates NHE8 protein expression via the ERK1/2 MAPK pathway in DSS-induced colitis mice. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G954–g963 (2016).
Brun, P. et al. Neuropeptide neurotensin stimulates intestinal wound healing following chronic intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G621–G629 (2005).
Drucker, D.J., Yusta, B., Boushey, R.P., DeForest, L. & Brubaker, P.L. Human [Gly2]GLP-2 reduces the severity of colonic injury in a murine model of experimental colitis. Am. J. Physiol. 276 (1 Pt 1), G79–G91 (1999).
Boushey, R.P., Yusta, B. & Drucker, D.J. Glucagon-like peptide 2 decreases mortality and reduces the severity of indomethacin-induced murine enteritis. Am. J. Physiol. 277 (5 Pt 1), E937–E947 (1999).
Alavi, K., Schwartz, M.Z., Palazzo, J.P. & Prasad, R. Treatment of inflammatory bowel disease in a rodent model with the intestinal growth factor glucagon-like peptide-2. J. Pediatr. Surg. 35, 847–851 (2000).
Halaclar, B., Agac, A.y.A., Akcan, A.C., Ay, A., Oz, B. & Arslan, E. Effects of glucagon-like peptide-2 on bacterial translocation in rat models of colitis. Turk. J. Gastroenterol. 23, 691–698 (2012).
Anbazhagan, A.N. et al. GLP-1 nanomedicine alleviates gut inflammation. Nanomed.: Nanotechnol., Biol., Med. 13, 659–665 (2016).
Moran, G.W., Leslie, F.C., Levison, S.E. & McLaughlin, J.T. Enteroendocrine cells: neglected players in gastrointestinal disorders? Ther. Adv. Gastroenterol. 1, 51–60 (2008).
Kiesler, P., Fuss, I.J. & Strober, W. Experimental models of inflammatory bowel diseases. Cell. Mol. Gastroenterol. Hepatol. 1, 154–170 (2015).
Matsumoto, S. et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut 43, 71–78 (1998).
el-Salhy, M. The nature and implication of intestinal endocrine cell changes in coeliac disease. Histol. Histopathol. 13, 1069–1075 (1998).
Domschke, S., Bloom, S.R., Adrian, T.E., Lux, G., Bryant, M.G. & Domschke, W. Coeliac sprue: abnormalities of the hormone profile of gastroduodenal mucosa. Scand. J. Gastroenterol. Suppl. 167, 86–89 (1989).
Papastamataki, M. et al. Incretins, amylin and other gut-brain axis hormones in children with coeliac disease. Eur. J. Clin. Invest. 44, 74–82 (2014).
Di Sabatino, A. et al. Increase in neuroendocrine cells in the duodenal mucosa of patients with refractory celiac disease. Am. J. Gastroenterol. 109, 258–269 (2014).
Jarocka-Cyrta, E., Kasacka, I. & Kaczmarski, M. The ghrelin-positive cells number is increased in duodenum in children with celiac disease. J. Endocrinol. Invest. 33, 165–170 (2010).
Rocco, A. et al. Tissue ghrelin level and gastric emptying rate in adult patients with celiac disease. Neurogastroenterol. Motility 20, 884–890 (2008).
Fraquelli, M., Bardella, M.T., Peracchi, M., Cesana, B.M., Bianchi, P.A. & Conte, D. Gallbladder emptying and somatostatin and cholecystokinin plasma levels in celiac disease. Am. J. Gastroenterol. 94, 1866–1870 (1999).
Caddy, G.R. et al. Plasma concentrations of glucagon-like peptide-2 in adult patients with treated and untreated coeliac disease. Eur. J. Gastroenterol. Hepatol. 18, 195–202 (2006).
Le Quellec, A., Kervran, A., Blache, P., Ciurana, A.J. & Bataille, D. [Oxyntomodulin, a new hormonal marker of intestinal malabsorption syndromes]. La Revue de Med. Interne 14, 982 (1993).
Bardella, M.T., Fraquelli, M., Peracchi, M., Cesana, B.M., Bianchi, P.A. & Conte, D. Gastric emptying and plasma neurotensin levels in untreated celiac patients. Scand. J. Gastroenterol. 35, 269–273 (2000).
Sjolund, K. & Ekman, R. Plasma motilin in untreated celiac disease. Peptides 24, 483–486 (2003).
Minderhoud, I.M., Oldenburg, B., Schipper, M.E., ter Linde, J.J. & Samsom, M. Serotonin synthesis and uptake in symptomatic patients with Crohn's disease in remission. Clin. Gastroenterol. Hepatol. 5, 714–720 (2007).
Keszthelyi, D. et al. Alterations in mucosal neuropeptides in patients with irritable bowel syndrome and ulcerative colitis in remission: a role in pain symptom generation? Eur. J. Pain 17, 1299–1306 (2013).
Serna, H., Porras, M. & Vergara, P. Mast cell stabilizer ketotifen 4-(1-methyl-4-piperidylidene)-4H-benzo 4,5 cyclohepta 1,2-b thiophen-10(9H)-one fumarate prevents mucosal mast cell hyperplasia and intestinal dysmotility in experimental Trichinella spiralis inflammation in the rat. J. Pharmacol. Exp. Ther. 319, 1104–1111 (2006).
Torrents, D. & Vergara, P. In vivo changes in the intestinal reflexes and the response to CCK in the inflamed small intestine of the rat. Am. J. Physiol.-Gastrointest. Liver Physiol. 279, G543–G551 (2000).
Aerssens, J. et al. Alterations in the brain-gut axis underlying visceral chemosensitivity in Nippostrongylus brasiliensis-infected mice. Gastroenterology 132, 1375–1387 (2007).
Kalia, N. et al. Intestinal secretory and absorptive function in Trichinella spiralis mouse model of postinfective gut dysfunction: role of bile acids. Gut 57, 41–48 (2008).
Dlugosz, A. et al. Chlamydia trachomatis antigens in enteroendocrine cells and macrophages of the small bowel in patients with severe irritable bowel syndrome. BMC Gastroenterol. 10, 19 (2010).
Dynes, R.A., Poppi, D.P., Barrell, G.K. & Sykes, A.R. Elevation of feed intake in parasite-infected lambs by central administration of a cholecystokinin receptor antagonist. Br. J. Nutr. 79, 47–54 (1998).
Yang, S., Gaafar, S.M. & Bottoms, G.D. Effects of multiple dose infections with ascaris-suum on blood gastrointestinal hormone levels in pigs. Vet. Parasitol. 37, 31–44 (1990).
Forbes, A.B. et al. Associations between blood gastrin, ghrelin, leptin, pepsinogen and Ostertagia ostertagi antibody concentrations and voluntary feed intake in calves exposed to a trickle infection with O. ostertagi. Vet. Parasitol. 162, 295–305 (2009).
Scott, I. et al. Infection of sheep with adult and larval Ostertagia circumcincta: gastrin. Int. J. Parasitol. 28, 1393–1401 (1998).
Bosi, G., Shinn, A.P., Giari, L., Simoni, E., Pironi, F. & Dezfuli, B.S. Changes in the neuromodulators of the diffuse endocrine system of the alimentary canal of farmed rainbow trout, Oncorhynchus mykiss (Walbaum), naturally infected with Eubothrium crassum (Cestoda). J. Fish Dis. 28, 703–711 (2005).
Dezfuli, B.S., Pironi, F., Shinn, A.P., Manera, M. & Giari, L. Histopathology and ultrastructure of Platichthys flesus naturally infected with Anisakis simplex s.l. larvae (Nematoda: anisakidae). J. Parasitol. 93, 1416–1423 (2007).
Worthington, J.J., Samuelson, L.C., Grencis, R.K. & McLaughlin, J.T. Adaptive immunity alters distinct host feeding pathways during nematode induced inflammation, a novel mechanism in parasite expulsion. PLoS Pathog. 9, e1003122 (2013).
McDermott, J.R., Leslie, F.C., D'Amato, M., Thompson, D.G., Grencis, R.K. & McLaughlin, J.T. Immune control of food intake: enteroendocrine cells are regulated by CD4(+) T lymphocytes during small intestinal inflammation. Gut 55, 492–497 (2006).
Ovington, K.S., Bacaresehamilton, A.J. & Bloom, S.R. Nippostrongylus-Brasiliensis - changes in plasma-levels of gastrointestinal hormones in the infected-rat. Exp. Parasitol. 60, 276–284 (1985).
Castro, G.A., Copeland, E.M., Dudrick, S.J. & Johnson, L.R. Serum and antral gastrin levels in rats infected with intestinal parasites. Am. J. Trop. Med. Hyg. 25, 848–853 (1976).
De Jonge, F. et al. Effects of Schistosoma mansoni infection on somatostatin and somatostatin receptor 2 A expression in mouse ileum. Neurogastroenterol. Motility 15, 149–159 (2003).
Leslie, F.C., Thompson, D.G., McLaughlin, J.T., Varro, A., Dockray, G.J. & Mandal, B.K. Plasma cholecystokinin concentrations are elevated in acute upper gastrointestinal infections. QJM 96, 870–871 (2003).
Roma, S.M. et al. Enteroendocrine cells modifications in Helicobacter pylori gastritis. Acta Gastroenterol. Latinoam. 31, 377–381 (2001).
Jeffery, P.L., McGuckin, M.A. & Linden, S.K. Endocrine impact of Helicobacter pylori: focus on ghrelin and ghrelin o-acyltransferase. World J. Gastroenterol. 17, 1249–1260 (2011).
Choi, Y.J. et al. Increase in plasma acyl ghrelin levels is associated with abatement of dyspepsia following Helicobacter pylori eradication. J. Gastroenterol. 51, 548–559 (2016).
Khosravi, Y. et al. Helicobacter pylori and gut microbiota modulate energy homeostasis prior to inducing histopathological changes in mice. Gut Microb. 7, 48–53 (2016).
van Marle, G., Sharkey, K.A., Gill, M.J. & Church, D.L. Gastrointestinal viral load and enteroendocrine cell number are associated with altered survival in HIV-1 infected individuals. PLoS ONE 8, e75967 (2013).
Dlugosz, A., Muschiol, S., Zakikhany, K., Assadi, G., D'Amato, M. & Lindberg, G. Human enteroendocrine cell responses to infection with Chlamydia trachomatis: a microarray study. Gut Pathog. 6, 24 (2014).
Gledhill, A., Hall, P.A., Cruse, J.P. & Pollock, D.J. Enteroendocrine cell hyperplasia, carcinoid tumours and adenocarcinoma in long-standing ulcerative colitis. Histopathology 10, 501–508 (1986).
Jezkova, J. et al. Brachyury identifies a class of enteroendocrine cells in normal human intestinal crypts and colorectal cancer. Oncotarget 7, 11478–11486 (2016).
Banck, M.S. et al. The genomic landscape of small intestine neuroendocrine tumors. J. Clin. Invest. 123, 2502–2508 (2013).
Leedham, S.J. MAP(K)ing the path to stem cell quiescence and the elusive enteroendocrine cell. Cell Stem Cell 20, 153–154 (2017).
Kunz, P.L. Carcinoid and neuroendocrine tumors: building on success. J. Clin. Oncol. 33, 1855–1863 (2015).
Gulubova, M. & Vlaykova, T. Chromogranin A-, serotonin-, synaptophysin- and vascular endothelial growth factor-positive endocrine cells and the prognosis of colorectal cancer: an immunohistochemical and ultrastructural study. J. Gastroenterol. Hepatol. 23, 1574–1585 (2008).
Swatek, J. & Chibowski, D. Endocrine cells in colorectal carcinomas. Immunohistochemical study. Polish J. Pathol. 51, 127–136 (2000).
Smith, D.M. Jr. & Haggitt, R.C. The prevalence and prognostic significance of argyrophil cells in colorectal carcinomas. Am. J. Surg. Pathol. 8, 123–128 (1984).
Cho, Y.B. et al. The clinical significance of neuroendocrine differentiation in T3-T4 node-negative colorectal cancer. Int. J. Surg. Pathol. 18, 201–206 (2010).
Kleist, B. & Poetsch, M. Neuroendocrine differentiation: the mysterious fellow of colorectal cancer. World J. Gastroenterol. 21, 11740–11747 (2015).
La Rosa, S., Chiaravalli, A.M., Capella, C., Uccella, S. & Sessa, F. Immunohistochemical localization of acidic fibroblast growth factor in normal human enterochromaffin cells and related gastrointestinal tumours. Virchows Archiv.: Int. J. Pathol. 430, 117–124 (1997).
Okines, A. & Cunningham, D. Current perspective: bevacizumab in colorectal cancer—a time for reappraisal? Eur. J. Cancer 45, 2452–2461 (2009).
Kohne, C.H. & Lenz, H.J. Chemotherapy with targeted agents for the treatment of metastatic colorectal cancer. Oncologist 14, 478–488 (2009).
Ciunci, C.A. et al. Phase 1 and pharmacodynamic trial of everolimus in combination with cetuximab in patients with advanced cancer. Cancer 120, 77–85 (2014).
McRee, A.J. et al. A phase I trial of everolimus in combination with 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 74, 117–123 (2014).
Drucker, D.J. Evolving concepts and translational relevance of enteroendocrine cell biology. J. Clin. Endocrinol. Metab. 101, 778–786 (2016).
El-Salhy, M. & Umezawa, K. Effects of AP1 and NFkappaB inhibitors on colonic endocrine cells in rats with TNBSinduced colitis. Mol. Med. Rep. 14, 1515–1522 (2016).
Zhang, W.J., Duan, J.Z., Lei, N., Xing, H., Shao, Y. & Yang, G.B. Cellular bases for interactions between immunocytes and enteroendocrine cells in the intestinal mucosal barrier of rhesus macaques. Cell Tissue Res. 350, 135–141 (2012).
O'Hara, J.R., Skinn, A.C., MacNaughton, W.K., Sherman, P.M. & Sharkey, K.A. Consequences of Citrobacter rodentium infection on enteroendocrine cells and the enteric nervous system in the mouse colon. Cell Microbiol. 8, 646–660 (2006).
Motomura, Y., Verma-Gandhu, M., El-Sharkawy, R.T., McLaughlin, J., Grencis, R.K. & Khan, W.I. Colonic 5-HT and muscle responses to the same infectious agent differ in Th1 and Th2 dominant environments. Gastroenterology 126, A216–A216 (2004).
Wang, H.Q. et al. CD4(+) T cell-mediated immunological control of enterochromaffin cell hyperplasia and 5-hydroxytryptamine production in enteric infection. Gut 56, 949–957 (2007).
Motomura, Y. et al. Enterochromaffin cell and 5-hydroxytryptamine responses to the same infectious agent differ in Th1 and Th2 dominant environments. Gut 57, 475–481 (2008).
Hernandez-Trejo, J.A. et al. The pro-inflammatory cytokines IFNgamma/TNFalpha increase chromogranin A-positive neuroendocrine cells in the colonic epithelium. Biochem. J. 473, 3805–3818 (2016).
O'Hara, J.R. & Sharkey, K.A. Proliferative capacity of enterochromaffin cells in guinea-pigs with experimental ileitis. Cell Tissue Res. 329, 433–441 (2007).
Mahapatro, M. et al. Programming of intestinal epithelial differentiation by IL-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep. 15, 1743–1756 (2016).
Tsukahara, T. et al. Tumor necrosis factor alpha decreases glucagon-like peptide-2 expression by up-regulating G-protein-coupled receptor 120 in Crohn disease. Am. J. Pathol. 185, 185–196 (2015).
Ellingsgaard, H. et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 17, 1481–1489 (2011).
Kidd, M., Gustafsson, B.I., Drozdov, I. & Modlin, I.M. IL1beta- and LPS-induced serotonin secretion is increased in EC cells derived from Crohn's disease. Neurogastroenterol. Motility 21, 439–450 (2009).
Wang, A. et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166, e1512 (2016).
Howitt, M.R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016).
von Moltke, J., Ji, M., Liang, H.E. & Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225 (2016).
Grencis, R.K. & Worthington, J.J. Tuft cells: a new flavor in innate epithelial immunity. Trends Parasitol. 32, 583–585 (2016).
Selleri, S. et al. Induction of pro-inflammatory programs in enteroendocrine cells by the Toll-like receptor agonists flagellin and bacterial LPS. Int. Immunol. 20, 961–970 (2008).
Friedrich, M., Diegelmann, J., Schauber, J., Auernhammer, C.J. & Brand, S. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17 A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunol. 8, 943–958 (2014).
Hougaard, D.M. & Larsson, L.I. Carboxypeptidase E in rat antropyloric mucosa: distribution in progenitor and mature endocrine cell types. Histochem. Cell Biol. 121, 55–61 (2004).
Jeon, T.I., Seo, Y.K. & Osborne, T.F. Gut bitter taste receptor signalling induces ABCB1 through a mechanism involving CCK. Biochem. J. 438, 33–37 (2011).
Higgins, G.A., Kilpatrick, G.J., Bunce, K.T., Jones, B.J. & Tyers, M.B. 5-HT3 receptor antagonists injected into the area postrema inhibit cisplatin-induced emesis in the ferret. Br. J. Pharmacol. 97, 247–255 (1989).
Hagbom, M. et al. Rotavirus stimulates release of serotonin (5-HT) from human enterochromaffin cells and activates brain structures involved in nausea and vomiting. PLoS Pathog. 7, e1002115 (2011).
Sukhdeo, M.V. & Sukhdeo, S.C. Gastrointestinal hormones: environmental cues for Fasciola hepatica? Parasitology 98 (Pt 2), 239–243 (1989).
Delgado, M., Anderson, P., Garcia-Salcedo, J.A., Caro, M. & Gonzalez-Rey, E. Neuropeptides kill African trypanosomes by targeting intracellular compartments and inducing autophagic-like cell death. Cell Death Differ. 16, 406–416 (2009).
Chorny, A., Anderson, P., Gonzalez-Rey, E. & Delgado, M. Ghrelin protects against experimental sepsis by inhibiting high-mobility group box 1 release and by killing bacteria. J. Immunol. 180, 8369–8377 (2008).
Beebe, K., Park, D., Taghert, P.H. & Micchelli, C.A. The drosophila prosecretory transcription factor dimmed is dynamically regulated in adult enteroendocrine cells and protects against gram-negative infection. G3 5, 1517–1524 (2015).
Dong, C.X. et al. The intestinal epithelial insulin-like growth factor-1 receptor links glucagon-like peptide-2 action to gut barrier function. Endocrinology 155, 370–379 (2014).
Moran, G.W., O'Neill, C. & McLaughlin, J.T. GLP-2 enhances barrier formation and attenuates TNFalpha-induced changes in a Caco-2 cell model of the intestinal barrier. Regul. Pept. 178, 95–101 (2012).
Orskov, C., Hartmann, B., Poulsen, S.S., Thulesen, J., Hare, K.J. & Holst, J.J. GLP-2 stimulates colonic growth via KGF, released by subepithelial myofibroblasts with GLP-2 receptors. Regul. Pept. 124, 105–112 (2005).
Yusta, B. et al. ErbB signaling is required for the proliferative actions of GLP-2 in the murine gut. Gastroenterology 137, 986–996 (2009).
Genton, L. & Kudsk, K.A. Interactions between the enteric nervous system and the immune system: role of neuropeptides and nutrition. Am. J. Surg. 186, 253–258 (2003).
Shajib, M.S. & Khan, W.I. The role of serotonin and its receptors in activation of immune responses and inflammation. Acta Physiol. 213, 561–574 (2015).
Abella, V. et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 13, 100–109 (2017).
O'Mahony, L., Akdis, M. & Akdis, C.A. Regulation of the immune response and inflammation by histamine and histamine receptors. J. Allergy Clin. Immunol. 128, 1153–1162 (2011).
Chung, Y., Kim, H., Im, E., Kim, P. & Yang, H. Th 17 cells and Nesfatin-1 are associated with spontaneous abortion in the CBA/j x DBA/2 mouse model. Dev. Reprod. 19, 243–252 (2015).
Dixit, V.D., Yang, H., Cooper-Jenkins, A., Giri, B.B., Patel, K. & Taub, D.D. Reduction of T cell-derived ghrelin enhances proinflammatory cytokine expression: implications for age-associated increases in inflammation. Blood 113, 5202–5205 (2009).
Xu, Y. et al. Ghrelin inhibits the differentiation of T helper 17 cells through mTOR/STAT3 signaling pathway. PLoS ONE 10, e0117081 (2015).
Souza-Moreira, L., Delgado-Maroto, V., Morell, M., O'Valle, F., Del Moral, R.G. & Gonzalez-Rey, E. Therapeutic effect of ghrelin in experimental autoimmune encephalomyelitis by inhibiting antigen-specific Th1/Th17 responses and inducing regulatory T cells. Brain Behav. Immun. 30, 54–60 (2013).
Dixit, V.D. et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Invest. 114, 57–66 (2004).
Zhang, J.G., Liu, J.X., Jia, X.X., Geng, J., Yu, F. & Cong, B. Cholecystokinin octapeptide regulates the differentiation and effector cytokine production of CD4(+) T cells in vitro. Int. Immunopharmacol. 20, 307–315 (2014).
Hadjiyanni, I., Siminovitch, K.A., Danska, J.S. & Drucker, D.J. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia 53, 730–740 (2010).
He, L. et al. Anti-inflammatory effects of exendin-4, a glucagon-like peptide-1 analog, on human peripheral lymphocytes in patients with type 2 diabetes. J. Diabetes Investig. 4, 382–392 (2013).
Lee, J.H. et al. Ghrelin augments murine T-cell proliferation by activation of the phosphatidylinositol-3-kinase, extracellular signal-regulated kinase and protein kinase C signaling pathways. FEBS Lett. 588, 4708–4719 (2014).
Pamukcu, O., Kumral, Z.N., Ercan, F., Yegen, B.C. & Ertem, D. Anti-inflammatory effect of obestatin and ghrelin in dextran sulfate sodium-induced colitis in rats. J. Pediatr. Gastroenterol. Nutr. 57, 211–218 (2013).
Hosomi, S. et al. Phenotypical and functional study of ghrelin and its receptor in the pathogenesis of Crohn's disease. Inflamm. Bowel Dis. 14, 1205–1213 (2008).
Tang, S.C., Braunsteiner, H. & Wiedermann, C.J. Regulation of human T lymphoblast growth by sensory neuropeptides: augmentation of cholecystokinin-induced inhibition of Molt-4 proliferation by somatostatin and vasoactive intestinal peptide in vitro. Immunol. Lett. 34, 237–242 (1992).
Yoon, W.K. et al. Somatostatin down-regulates LFA-1 activation by modulating Rap1 expression in CD4+ and CD8+ T cells. Regul. Pept. 124, 151–156 (2005).
Petrovic-Djergovic, D.M., Rakin, A.K., Kustrimovic, N.Z., Ristovski, J.S., Dimitrijevic, L.A. & Mileva, M.V. Somatostatin modulates T cells development in adult rat thymus. Regul. Pept. 142, 101–110 (2007).
Yusta, B. et al. GLP-1 receptor (GLP-1 R) agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte (IEL) GLP-1 R. Diabetes 64, 2537–2549 (2015).
Lynch, L. et al. iNKT cells induce FGF21 for thermogenesis and are required for maximal weight loss in GLP1 therapy. Cell Metab. 24, 510–519 (2016).
Hogan, A.E. et al. Glucagon-like peptide-1 (GLP-1) and the regulation of human invariant natural killer T cells: lessons from obesity, diabetes and psoriasis. Diabetologia 54, 2745–2754 (2011).
Agro, A., Padol, I. & Stanisz, A.M. Immunomodulatory activities of the somatostatin analogue BIM 23014c: effects on murine lymphocyte proliferation and natural killer activity. Regul. Pept. 32, 129–139 (1991).
van Tol, E.A., Verspaget, H.W. & Lamers, C.B. Effects of CCK-8 and CCK-33 on human natural killer cell activity: studies on intestinal lamina propria and peripheral blood mononuclear cells. Immunopharmacology 25, 11–18 (1993).
Reardon, C. et al. Lymphocyte-derived ACh regulates local innate but not adaptive immunity. Proc. Natl Acad. Sci. USA 110, 1410–1415 (2013).
Zhang, J.G., Cong, B., Li, Q.X., Chen, H.Y., Qin, J. & Fu, L.H. Cholecystokinin octapeptide regulates lipopolysaccharide-activated B cells co-stimulatory molecule expression and cytokines production in vitro. Immunopharmacol. Immunotoxicol. 33, 157–163 (2011).
Liu, L. et al. Somatostatin improved B cells mature in macaques during intestinal ischemia-reperfusion. PLoS ONE 10, e0133692 (2015).
Sung, E.Z., Da Silva, N.F., Goodyear, S.J., McTernan, P.G., Arasaradnam, R.P. & Nwokolo, C.U. Ghrelin promotes nuclear factor kappa-B activation in a human B-lymphocyte cell line. Mol. Biol. Rep. 38, 4833–4838 (2011).
Saada, S. et al. Differential expression of neurotensin and specific receptors, NTSR1 and NTSR2, in normal and malignant human B lymphocytes. J. Immunol. 189, 5293–5303 (2012).
Chen, J., Dong, J.T., Li, X.J., Gu, Y., Cheng, Z.J. & Cai, Y.K. Glucagon-like peptide-2 protects impaired intestinal mucosal barriers in obstructive jaundice rats. World J. Gastroenterol. 21, 484–490 (2015).
Hanna, M.K. et al. Individual neuropeptides regulate gut-associated lymphoid tissue integrity, intestinal immunoglobulin A levels, and respiratory antibacterial immunity. J. Parent. Enteral Nutr. 24, 261–268 (2000).
Jia, X. et al. CCK8 negatively regulates the TLR9-induced activation of human peripheral blood pDCs by targeting TRAF6 signaling. Eur. J. Immunol. 44, 489–499 (2014).
Kao, J.Y., Pierzchala, A., Rathinavelu, S., Zavros, Y., Tessier, A. & Merchant, J.L. Somatostatin inhibits dendritic cell responsiveness to Helicobacter pylori. Regul. Pept. 134, 23–29 (2006).
da Silva, L., Neves, B.M., Moura, L., Cruz, M.T. & Carvalho, E. Neurotensin downregulates the pro-inflammatory properties of skin dendritic cells and increases epidermal growth factor expression. Biochim. Biophys. Acta 1813, 1863–1871 (2011).
Li, Q. et al. Cholecystokinin octapeptide significantly suppresses collagen-induced arthritis in mice by inhibiting Th17 polarization primed by dendritic cells. Cell Immunol. 272, 53–60 (2011).
Dunzendorfer, S., Kaser, A., Meierhofer, C., Tilg, H. & Wiedermann, C.J. Cutting edge: peripheral neuropeptides attract immature and arrest mature blood-derived dendritic cells. J. Immunol. 166, 2167–2172 (2001).
Miyamoto, S. et al. Cholecystokinin plays a novel protective role in diabetic kidney through anti-inflammatory actions on macrophage: anti-inflammatory effect of cholecystokinin. Diabetes 61, 897–907 (2012).
Li, S. et al. CCK-8 inhibits LPS-induced IL-1beta production in pulmonary interstitial macrophages by modulating PKA, p38, and NF-kappaB pathway. Shock 27, 678–686 (2007).
De la Fuente, M., Campos, M., Del Rio, M. & Hernanz, A. Inhibition of murine peritoneal macrophage functions by sulfated cholecystokinin octapeptide. Regul. Pept. 55, 47–56 (1995).
Saia, R.S., Mestriner, F.L., Bertozi, G., Cunha, F.Q. & Carnio, E.C. Cholecystokinin inhibits inducible nitric oxide synthase expression by lipopolysaccharide-stimulated peritoneal macrophages. Mediators Inflamm. 2014, 896029 (2014).
Cunningham, M.E. et al. Cholecystokinin-stimulated monocytes produce inflammatory cytokines and eicosanoids. Am. J Gastroenterol. 90, 621–626 (1995).
Liang, C.P., Han, S., Li, G., Tabas, I. & Tall, A.R. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes 61, 2609–2620 (2012).
Hogan, A.E. et al. Glucagon-like peptide 1 analogue therapy directly modulates innate immune-mediated inflammation in individuals with type 2 diabetes mellitus. Diabetologia 57, 781–784 (2014).
Xie, S. et al. GLP-2 suppresses LPS-induced inflammation in macrophages by inhibiting ERK phosphorylation and NF-kappaB activation. Cell Physiol. Biochem. 34, 590–602 (2014).
Komorowski, J. & Stepien, H. Somatostatin (SRIF) stimulates the release of interleukin-6 (IL-6) from human peripheral blood monocytes (PBM) in vitro. Neuropeptides 29, 77–81 (1995).
Peluso, G., Petillo, O., Melone, M.A., Mazzarella, G., Ranieri, M. & Tajana, G.F. Modulation of cytokine production in activated human monocytes by somatostatin. Neuropeptides 30, 443–451 (1996).
Arakawa, M. et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 59, 1030–1037 (2010).
Dixit, V.D. et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Invest. 114, 57–66 (2004).
Kellokoski, E., Kunnari, A., Jokela, M., Makela, S., Kesaniemi, Y.A. & Horkko, S. Ghrelin and obestatin modulate early atherogenic processes on cells: enhancement of monocyte adhesion and oxidized low-density lipoprotein binding. Metabolism 58, 1572–1580 (2009).
Tanaka, M. et al. Differential effects of GLP-1 receptor agonist on foam cell formation in monocytes between non-obese and obese subjects. Metabolism 65, 1–11 (2016).
Mitchell, P.D. et al. Glucagon-like peptide-1 receptor expression on human eosinophils and its regulation of eosinophil activation. Clin. Exp. Allergy. 47, 331–338 (2017).
De la Fuente, M., Carrasco, M. & Hernanz, A. Modulation of human neutrophil function in vitro by gastrin. J. Endocrinol. 153, 475–483 (1997).
Goldman, R., Bar-Shavit, Z. & Romeo, D. Neurotensin modulates human neutrophil locomotion and phagocytic capability. FEBS Lett. 159, 63–67 (1983).
Carrasco, M., Del Rio, M., Hernanz, A. & De la Fuente, M. Inhibition of human neutrophil functions by sulfated and nonsulfated cholecystokinin octapeptides. Peptides 18, 415–422 (1997).
Adeyemi, E.O., Savage, A.P., Bloom, S.R. & Hodgson, H.J. Somatostatin inhibits neutrophil elastase release in vitro. Peptides 11, 869–871 (1990).
Robbins, R.A., Nelson, K.J., Gossman, G.L. & Rubinstein, I. Neurotensin stimulates neutrophil adherence to bronchial epithelial cells in vitro. Life Sci. 56, 1353–1359 (1995).
Varol, C. et al. Long-acting glucose-dependent insulinotropic polypeptide ameliorates obesity-induced adipose tissue inflammation. J. Immunol. 193, 4002–4009 (2014).
Fukamachi, S. et al. Topical cholecystokinin depresses itch-associated scratching behavior in mice. J. Invest. Dermatol. 131, 956–961 (2011).
Vergara, P., Saavedra, Y. & Juanola, C. Neuroendocrine control of intestinal mucosal mast cells under physiological conditions. Neurogastroenterol. Motility 14, 35–42 (2002).
Saavedra, Y. & Vergara, P. Somatostatin inhibits intestinal mucosal mast cell degranulation in normal conditions and during mast cell hyperplasia. Regul. Pept. 111, 67–75 (2003).
Hirayama, T. et al. Ghrelin and obestatin promote the allergic action in rat peritoneal mast cells as basic secretagogues. Peptides 31, 2109–2113 (2010).
Grundemar, L. & Hakanson, R. Neuropeptide Y, peptide YY and C-terminal fragments release histamine from rat peritoneal mast cells. Br. J. Pharmacol. 104, 776–778 (1991).
Li, E., Zhao, A., Shea-Donohue, T. & Singer, S.M. Mast cell-mediated changes in smooth muscle contractility during mouse giardiasis. Infect. Immun. 75, 4514–4518 (2007).
Jenny, M. et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J. 21, 6338–6347 (2002).
Li, H.J. et al. Distinct cellular origins for serotonin-expressing and enterochromaffin-like cells in the gastric corpus. Gastroenterology 146 3, e753 (2014).
Lieb, K., Fiebich, B.L., Busse-Grawitz, M., Hull, M., Berger, M. & Bauer, J. Effects of substance P and selected other neuropeptides on the synthesis of interleukin-1 beta and interleukin-6 in human monocytes: a re-examination. J. Neuroimmunol. 67, 77–81 (1996).
Goverse, G., Stakenborg, M. & Matteoli, G. The intestinal cholinergic anti-inflammatory pathway. J. Physiol. 594, 5771–5780 (2016).
Bohorquez, D.V., Samsa, L.A., Roholt, A., Medicetty, S., Chandra, R. & Liddle, R.A. An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PLoS ONE 9, e89881 (2014).
Bohorquez, D.V. et al. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Invest. 125, 782–786 (2015).
Davies, G.A., Bryant, A.R., Reynolds, J.D., Jirik, F.R. & Sharkey, K.A. Prion diseases and the gastrointestinal tract. Canadian J. Gastroenterol. 20, 18–24 (2006).
Drummond, C.G., Bolock, A.M., Ma, C., Luke, C.J., Good, M. & Coyne, C.B. Enteroviruses infect human enteroids and induce antiviral signaling in a cell lineage-specific manner. Proc. Natl Acad. Sci. USA 114, 1672–1677 (2017).
Luyer, M.D., Greve, J.W.M., Hadfoune, M., Jacobs, J.A., Dejong, C.H. & Buurman, W.A. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J. Exp. Med. 202, 1023–1029 (2005).
Glatzle, J. et al. Chylomicron components activate duodenal vagal afferents via a cholecystokinin A receptor-mediated pathway to inhibit gastric motor function in the rat. J. Physiol. 550 (Pt 2), 657–664 (2003).
Lubbers, T. et al. Chylomicron formation and glucagon-like peptide 1 receptor are involved in activation of the nutritional anti-inflammatory pathway. J. Nutr. Biochem. 22, 1105–1111 (2011).
Wu, R. et al. Orexigenic hormone ghrelin attenuates local and remote organ injury after intestinal ischemia-reperfusion. PLoS ONE 3, e2026 (2008).
Sigalet, D.L. et al. Enteric neural pathways mediate the anti-inflammatory actions of glucagon-like peptide 2. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G211–G221 (2007).
Lubbers, T., Luyer, M.D., de Haan, J.J., Hadfoune, M., Buurman, W.A. & Greve, J.W. Lipid-rich enteral nutrition reduces postoperative ileus in rats via activation of cholecystokinin-receptors. Ann. Surg. 249, 481–487 (2009).
Eisner, F., Martin, E.M., Kuper, M.A., Raybould, H.E. & Glatzle, J. CCK1-receptor stimulation protects against gut mediator-induced lung damage during endotoxemia. Cell. Phys. Biochem. 32, 1878–1890 (2013).
Ghia, J.E., Blennerhassett, P. & Collins, S.M. Vagus nerve integrity and experimental colitis. Am. J. Physiol.-Gastrointest. Liver Physiol. 293, G560–G567 (2007).
Matteoli, G. et al. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut 63, 938–948 (2014).
de Haan, J.J. et al. Lipid-rich enteral nutrition regulates mucosal mast cell activation via the vagal anti-inflammatory reflex. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G383–G391 (2013).
Dalli, J., Colas, R.A., Arnardottir, H. & Serhan, C.N. Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity 46, 92–105 (2017).
Matteoli, G. & Boeckxstaens, G.E. The vagal innervation of the gut and immune homeostasis. Gut 62, 1214–1222 (2013).
Murray, M.J. & Murray, A.B. Anorexia of infection as a mechanism of host defense. Am. J. Clin. Nutr. 32, 593–596 (1979).
Wing, E.J. & Young, J.B. Acute starvation protects mice against Listeria monocytogenes. Infect. Immun. 28, 771–776 (1980).
Feingold, K.R. et al. FGF21 is increased by inflammatory stimuli and protects leptin-deficient ob/ob mice from the toxicity of sepsis. Endocrinology 153, 2689–2700 (2012).
Ayres, J.S. & Schneider, D.S. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS Biol. 7, e1000150 (2009).
Castex, N., Fioramonti, J., Ducos de Lahitte, J., Luffau, G., More, J. & Bueno, L. Brain Fos expression and intestinal motor alterations during nematode-induced inflammation in the rat. Am. J. Physiol. 274 (1 Pt 1), G210–G216 (1998).
Gay, J., More, J., Bueno, L. & Fioramonti, J. CCK-induced Fos expression in brain stem is enhanced after intestinal nematode infection in rats. Brain Res. 942, 124–127 (2002).
Procaccini, C., Jirillo, E. & Matarese, G. Leptin as an immunomodulator. Mol. Aspects Med. 33, 35–45 (2012).
Lord, G.M., Matarese, G., Howard, L.K., Baker, R.J., Bloom, S.R. & Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394, 897–901 (1998).
Batra, A. et al. Leptin: a critical regulator of CD4(+) T-cell polarization in vitro and in vivo. Endocrinology 151, 56–62 (2009).
Tu, T., Koski, K.G. & Scott, M.E. Mechanisms underlying reduced expulsion of a murine nematode infection during protein deficiency. Parasitology 135, 81–93 (2008).
McDermott, J.R., Bartram, R.E., Knight, P.A., Miller, H.R.P., Garrod, D.R. & Grencis, R.K. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc. Natl Acad. Sci. USA 100, 7761–7766 (2003).
Acknowledgements
F.R. and F.M.G. are supported by grants from European Foundation for the Study of Diabetes and Boehringer Ingelheim Basic Research Programme; the Wellcome Trust (grants 106262/Z/14/Z, 106263/Z/14/Z and 100574/Z/12/Z); the Medical Research Council Metabolic Diseases Unit (grants MRC_MC_UU_12012/3 and MRC_MC_UU_12012/5); and the Novo Nordisk Foundation. F.M.G. and F.R. have research collaborations with AstraZeneca/MedImmune. F.M.G. has received honoraria for speaking at symposia organized by Novo Nordisk and is a member of the external scientific advisory board of BioKier. F.R. has received honoraria for speaking at symposia organized by MSD. J.J.W. is supported by the Royal Society (RG160039).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Author contributions
J.J.W., F.R., and F.M.G. wrote the article.
Rights and permissions
About this article

Cite this article
Worthington, J., Reimann, F. & Gribble, F. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol 11, 3–20 (2018). https://doi.org/10.1038/mi.2017.73
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.73
This article is cited by
-
Establishment of a chicken intestinal organoid culture system to assess deoxynivalenol-induced damage of the intestinal barrier function
Journal of Animal Science and Biotechnology (2024)
-
Robust differentiation of human enteroendocrine cells from intestinal stem cells
Nature Communications (2022)
-
“Every cell is an immune cell; contributions of non-hematopoietic cells to anti-helminth immunity”
Mucosal Immunology (2022)
-
The gut hormone Allatostatin C/Somatostatin regulates food intake and metabolic homeostasis under nutrient stress
Nature Communications (2022)
-
Intestinal Models for Personalized Medicine: from Conventional Models to Microfluidic Primary Intestine-on-a-chip
Stem Cell Reviews and Reports (2022)
