
Abstract
In this study, we tested our hypothesis regarding mechanistic cross-talk between gastrointestinal inflammation and memory loss in a mouse model. Intrarectal injection of the colitis inducer 2,4,6-trinitrobenzenesulfonic acid (TNBS) in mice caused colitis via activation of nuclear factor (NF)-κB and increase in membrane permeability. TNBS treatment increased fecal and blood levels of lipopolysaccharide (LPS) and the number of Enterobacteriaceae, particularly Escherichia coli (EC), in the gut microbiota composition, but significantly reduced the number of Lactobacillus johnsonii (LJ). Indeed, we observed that the mice treated with TNBS displayed impaired memory, as assessed using the Y-maze and passive avoidance tasks. Furthermore, treatment with EC, which was isolated from the feces of mice with TNBS-induced colitis, caused memory impairment and colitis, and increased the absorption of orally administered LPS into the blood. Treatment with TNBS or EC induced NF-κB activation and tumor necrosis factor-α expression in the hippocampus of mice, as well as suppressed brain-derived neurotrophic factor expression. However, treatment with LJ restored the disturbed gut microbiota composition, lowered gut microbiota, and blood LPS levels, and attenuated both TNBS- and EC-induced memory impairment and colitis. These results suggest that the gut microbiota disturbance by extrinsic stresses can cause gastrointestinal inflammation, resulting in memory impairment.
Similar content being viewed by others


Introduction
The intensive and extensive bidirectional networks between gut microbiota and the central nervous system (CNS) are maintained through endocrine, neural, and immune pathways.1, 2 Exposure to stresses, such as pathogens, stimulates the brain to secrete hormones such as corticotrophin-releasing factor, via the hypothalamus–pituitary–adrenal axis, which disturb gut microbiota, permeability, and barrier function and increase production of endotoxins such as lipopolysaccharides (LPSs).3, 4 Moreover, these endotoxins, which are parts of bacterial chemical components, stimulate gut immune responses and constrain the secretion of the neurotransmitters serotonin and catecholamines.5, 6, 7 These neurotransmitters, cytokines, and hormones promote communication between the gut immune system (related to gut microbiota) and the CNS, and maintain homeostasis.5, 6 Dysregulation of these signals results in colitis, autism, and obesity.8, 9
Gut microbiota of healthy humans and animals reside in the ileum and colon, and consist of >1011 bacteria per gram of gut contents.10, 11 They produce toxic compounds, such as LPS. LPS is detected by receptors on macrophages, dendritic cells, and endothelial cells that are involved in the innate immune system, and LPS signals activate the biosynthesis of inflammation mediators (e.g., tumor necrosis factor (TNF) via a Toll-like receptor 4-linked nuclear factor (NF)-κB signaling pathway) to cause inflammation.12, 13 The overexpression of LPS in gut microbiota increases blood LPS levels through gut inflammation.14 Peripherally administered LPS causes systemic inflammation, as well as memory impairment by disrupting specific neural circuits within the hippocampus.15 Furthermore, Lukiw16 suggested that LPS released from the abundant Bacteroides fragilis in the gut might represent a major factor that contributes to systemic inflammation and diseases such as including Alzheimer’s disease (AD). In addition, colitis inducers, such as 2,4,6-trinitrobenzenesulfonic acid (TNBS), dextran sulfate sodium, and high-fat diet, cause colitis in vivo and increase gastrointestinal permeability.17, 18 These colitis inducers disturb gut microbiota composition and increase gut microbiota LPS levels.19, 20 In particular, TNBS increases Enterobacteriaceae population and decreases the number of Bifidobacteria and Lactobacilli.19, 20 Some antibiotics have been shown to suppress cognitive performance.21
In this study, as stresses can induce LPS levels through colitis and as LPS can affect CNS functions, we tested our hypothesis that colitis inducers could cause gastric inflammation and memory impairment by disturbing gut microbiota composition, and investigated whether fluctuations in gut microbiota composition by the colitis inducer could potentiate colitis and memory impairment in mice.
Results
TNBS caused colitis and memory impairment in mice
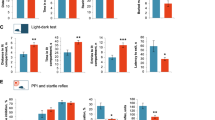
In the present study, intrarectal administration of TNBS in mice caused colon shortening, increased myeloperoxidase (MPO) activity, induced the expression of inflammatory markers, including inducible nitric oxide synthase and cyclooxygenase-2, and increased NF-κB activation in the colon (Figure 1a–c and Supplementary Figure S1a online). TNBS suppressed the expression of tight junction proteins ZO-1, claudin-1, and occludin in the colon. TNBS increased the number of Enterobacteriaceae, in particular Escherichia coli (EC, >75% of Enterobacteriaceae), but reduced the number of Bifidobacteria and Lactobacilli, including Lactobacillus johnsonii (LJ) (Figure 1d,e). TNBS treatment increased the production of gut microbiota LPS (Figure 1f). Furthermore, TNBS treatment increased blood LPS and TNF-α levels, and hippocampal TNF-α levels (Figure 1g–i). Treatment with TNBS also significantly reduced learning and memory behaviors in the Y-maze and passive avoidance tasks (Figure 1j,k). TNBS also induced NF-κB activation in the hippocampus and suppressed the expression of brain-derived neurotrophic factor (BDNF) and phosphorylation of cAMP response element-binding protein (CREB) (Figure 1l and Supplementary Figure S1b).

2,4,6-Trinitrobenzenesulfonic acid (TNBS) caused colitis and memory impairment in mice. Colitis markers including colon length (a), myeloperoxidase (MPO) activity (b), nuclear factor (NF)-κB activation, and inducible nitric oxide synthase (iNOS), cyclooxygenase (COX)-2, and tight-junction protein expression (c) were measured in the colon. The numbers of Enterobacteriaceae (d) and Bifidobacteria+Lactobacilli (e), and the level of fecal lipopolysaccharide (LPS) were measured in the colon fluid. White, gray, and black bars in d. indicate the composition of E. coli (EC), Klebsiella pneumoniae, and Proteus mirabilis, respectively. Fecal (f) and blood LPS (g) levels were measured using the Limulus assay. Blood (h) and hippocampal tumor necrosis factor (TNF)-α (i) levels were measured using the enzyme-linked immunosorbent assay (ELISA). Learning and memory behaviors were evaluated using the Y-maze (j) and passive-avoidance tasks (k). Brain-derived neurotrophic factor (BDNF) expression and NF-κB activation were measured in the hippocampus using immunoblotting (l). Values indicate mean±s.d. (n=10). *P<0.05.
EC impaired learning and memory impairment in mice
Treatment win TNBS in mice increased the number of Enterobacteriaceae, in particular EC. Therefore, we investigated whether oral gavage with EC could cause the colitis and memory impairment in mice. Oral gavage with EC significantly induced colon shortening and increased MPO activity in the colon of mice (Figure 2a,b). Furthermore, EC treatment increased NF-κB activation, inducible nitric oxide synthase and cyclooxygenase-2 expression, and suppressed the expression of tight-junction proteins (Figure 2c,d, and Supplementary Figure S2a,b). EC increased the number of Enterobacteriaceae including EC (> 85% of Enterobacteriaceae) and LPS production in gut microbiota but reduced the number of Bifidobacteria and Lactobacilli (Figure 2e,f). As was observed with TNBS, EC treatment also increased blood LPS and TNF-α, and hippocampal TNF-α levels (Figure 2g–i). Treatment with EC caused impairment of learning and memory in the Y-maze and passive-avoidance tasks (Figure 2j,l). EC treatment also reduced BDNF expression and CREB phosphorylation, and increased NF-κB activation in the hippocampus (Figure 2l and Supplementary Figure S2c).

E. coli (EC) caused colitis and memory impairment in mice. Colitis markers including colon length (a), myeloperoxidase (MPO) activity (b), nuclear factor (NF)-κB activation, and inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 expression (c), and tight-junction protein expression (d) were measured in the colon. The numbers of Enterobacteriaceae (e) and Bifidobacteria+Lactobacilli (f) were measured in the colon fluid. White, gray, and black bars in e. indicate the composition of EC, K. pneumoniae, and P. mirabilis, respectively. Blood lipopolysaccharide (LPS) levels were measured by Limulus assay (g). Blood (h) and hippocampal tumor necrosis factor (TNF)-α (i) levels were measured by enzyme-linked immunosorbent assay (ELISA). Learning and memory behaviors were evaluated in Y-maze (j) and passive-avoidance tasks (k). Brain-derived neurotrophic factor (BDNF) expression and NF-κB activation was measured in the hippocampus by immunoblotting (l). Values indicate mean± s.d. (n=10). *P<0.05.
LJ attenuated TNBS- or EC-induced colitis and memory impairment in mice
Treatment with TNBS reduced the number of Bifidobacteria and Lactobacilli, in particular LJ, in gut microbiota and impaired learning and memory in mice. Therefore, we isolated LJ, a type of gut bacteria that is present in at significantly low numbers in mice with TNBS-induced memory impairment, using the culture system in the selective media. Thereafter, we investigated whether administration of LJ simultaneously ameliorate colitis and memory impairment in mice with EC- or TNBS-induced colitis. Oral administration of LJ also ameliorated TNBS-induced impairment of learning and memory in Y-maze task (Figure 3a). LJ treatment increased the TNBS-suppressed expression of BDNF and phosphorylation of CREB, and suppressed the TNBS-induced activation of hippocampal NF-κB (Figure 3b and Supplementary Figure S3a). Treatment with LJ lowered TNBS-induced blood TNF-α and LPS levels, and hippocampal TNF-α levels (Figure 3c–e). Administration of LJ to TNBS-treated mice attenuated TNBS-induced colon shortening and MPO activity (Figure 3f). In addition, it reduced the activation of NF-κB and the expression of inducible nitric oxide synthase and cyclooxygenase-2 in the colon (Figure 3g,h and Supplementary Figure S3b).

L. johnsonii (LJ) attenuated 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis and memory impairment. Learning and memory behaviors were evaluated in the Y-maze task (a). Brain-derived neurotrophic factor (BDNF) expression and nuclear factor (NF)-κB activation were measured in the hippocampus using immunoblotting (b). Blood lipopolysaccharide (LPS) levels were measured using the Limulus assay (c). Blood (d) and hippocampal tumor necrosis factor (TNF)-α (e) levels were measured using enzyme-linked immunosorbent assay (ELISA). Colitis markers, including colon shortening (f), colonic myeloperoxidase (MPO) activity (g), and NF-κB activation, and inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 expression (h) and tight junction protein expression (i) were measured in the colon. Values indicate mean± s.d. (n=10). *P<0.05.
Thereafter, we examined the effect of administering LJ on EC-induced memory impairment in mice. Treatment with EC impaired the performance of mice in the Y-maze and passive avoidance tasks (Figure 4a,b). In contrast, oral administration of LJ ameliorated EC-induced learning and memory impairment. Furthermore, administration of LJ restored EC-suppressed expression of BDNF and phosphorylation of CREB in the hippocampus and suppressed EC-induced activation of NF-κB (Figure 4c and Supplementary Figure S4a). LJ administration reduced blood LPS and TNF-α levels, and hippocampal TNF-α levels (Figure 4d–f). LJ suppressed EC-induced colon shortening, colonic MPO activity, NF-κB activation, and inducible nitric oxide synthase and cyclooxygenase-2 expression (Figure 4g–i and Supplementary Figure S4b,c). In addition, administration of LJ attenuated EC-induced infiltration of macrophages and dendritic cells and increased EC-suppressed expression of colonic tight-junction proteins in mice (Figure 4j and Supplementary Figure S4d). EC treatment increased the absorption of orally administered Alexa Fluor 488-conjugated LPS (Sigma) into the blood 2 h after LPS treatment in EC-treated mice compared with that in normal control mice (Figure 4k). LJ treatment, however, inhibited the absorption of LPS into the blood. Therefore, we investigated whether LJ could increase LPS-suppressed expression of tight junction proteins in Caco-2 cells. LJ treatment increased the expression of tight-junction proteins and inhibited the activation of NF-κB in fecal LPS-stimulated Caco-2 cells, whereas fecal LPS lysate (isolated from the feces of TNBS-treated mice) significantly inhibited the expression of tight-junction proteins (Figure 4l).

L. johnsonii (LJ) attenuated E. coli (EC)-induced colitis and memory impairment. Learning and memory behaviors were evaluated in Y-maze (a) and passive avoidance tasks (b). Brain-derived neurotrophic factor (BDNF) expression and nuclear factor (NF)-κB activation was measured in the hippocampus using immunoblotting (c). Hippocampal (d) and blood tumor necrosis factor (TNF)-α (e) levels were measured using enzyme-linked immunosorbent assay (ELISA). Blood lipopolysaccharide (LPS) levels were measured using the Limulus assay (f). Colitis markers, including colon shortening (g), colonic myeloperoxidase (MPO) activity (h), NF-κB activation, inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 expression (i), and tight junction protein expression (j) were measured in the colon. The absorption of Alexa Fluor 488-conjugated LPS into the blood was measured 2 h after its oral administration (k). Effect of the fecal LPS fraction on the expression of tight-junction proteins and the activation of NF-κB was measured in Caco-2 cells (l). The fecal LPS was prepared from the colon fluid of mice with 2,4,6-trinitrobenzenesulfonic acid (TNBS)-treated colitis. Values indicate mean± s.d. (n=10). *P<0.05.
TNBS and EC disturbed gut microbiota composition
To understand the role of gut microbiota in learning and memory impairment induced by TNBS or EC, we treated mice with TNBS or EC and analyzed gut microbiota composition by pyrosequencing. There were no significant differences in bacterial richness and diversity between the fecal samples of the mice, as demonstrated by the number of sequences analyzed, estimated operational taxonomic unit richness, and coverage (Supplementary Table S1). When comparing the results of taxonomy-based analysis between the gut microbiota of mice treated with or without TNBS, TNBS treatment was found to significantly modulate the microbiota population (Supplementary Figure S5 and Supplementary Table S2,3). At the phylum level, TNBS treatment resulted in a significant increase in Proteobacteria (a major group of Gram-negative bacteria that include Enterobacteriaceae) and Bacteroidetes, and a decrease in Firmicutes (Figure 5a). At the family level, TNBS treatment suppressed the population of Lachnospiraceae and Rikenellaceae, and increased the population of Bacteroidales including EF602759_f and EU845084_f (Figure 5b). As a result, TNBS treatment increased the ratio of Proteobacteria to Firmicutes, Proteobacteria to Bacteroidetes, and Bacteroidetes to Firmicutes (Figure 5c–e). Treatment with EC also increased the Proteobacteria and Firmicutes populations in gut microbiota compared with those of normal control mice (Figure 5a). EC treatment led to a significant increase in the ratios of Proteobacteria to Firmicutes and Bacteroides to Firmicutes. At the family level, EC treatment suppressed the population of Lachnospiraceae and Rikenellaceae, and increased the population of Bacteroidales including EF602759_f and EU845084_f (Figure 5b).

Effects of 2,4,6-trinitrobenzenesulfonic acid (TNBS), E. coli (EC), and L. johnsonii (LJ) on the gut microbiota composition in mice. Effect on the phylum (a) and family levels (b). The ratios of Proteobacteria to Firmicutes (P/F) (c), Bacteroidetes to Firmicutes (B/F) (d), and Proteobacteria to Bacteroidetes (P/B) (e), were analyzed by pyrosequencing of the bacterial 16S rRNA fragments (d). (f) Principal coordinate analysis (PCoA) plot. The plot shows the clustering pattern among mice treated with vehicle alone (NOR), TNBS alone, EC alone, LJ in the presence of EC (EC+LJ) based on weighted pairwise Fast UniFrac analysis. Effect on the number of Enterobacteriaceae (g) and Bifidobacteria+Lactobacilli (h) was measured by using the culture of selective media. Black, gray, and white bars in (g) indicate the composition of EC, K. pneumoniae, and P. mirabilis, respectively. Values indicate mean± s.d. (n=5). *P<0.05.
In addition, we examined the effect of LJ administration on EC-disturbed gut microbiota composition in mice with EC-induced memory impairment (Figure 5a,b). Oral administration of LJ restored the population of Firmicutes, which was decreased by EC, and Proteobacteria, which was increased by EC. Furthermore, LJ administration restored EC-disturbed gut microbiota composition to the levels of normal control mice, as evaluated using PCoA analysis (Figure 5f). LJ treatment decreased the EC-induced Enterobacteriaceae population in gut microbiota using the culture in the selective media but increased that of Bifidobacteria and Lactobacilli population, which was decreased by EC (Figure 5g,h).
LPS isolated from EC accelerated memory impairment in mice
Treatment with TNBS or EC in mice significantly increased fecal and blood LPS levels, hippocampal TNF-α level, and NF-κB activation. Moreover, fecal LPS significantly induced the activation of NF-κB in Caco-2 cells. Therefore, we isolated LPS from EC, intraperitoneally injected LPS in mice, and investigated learning and memory behaviors using the Y-maze and passive avoidance tasks (Figure 6a,b). Treatment with LPS significantly impaired learning and memory, increased the activation of NF-κB, and suppressed BDNF expression and CREB phosphorylation in the hippocampus (Figure 6c). Furthermore, LPS treatment increased blood LPS levels (Figure 6d) and hippocampal LPS and TNF-α levels (Figure 6e,f).

Intraperitoneal injection of lipopolysaccharide (LPS) isolated from E. coli (EC) impaired learning and memory in mice. Learning and memory behaviors were evaluated in Y-maze (a) and passive-avoidance tasks (b). Brain-derived neurotrophic factor (BDNF) expression and nuclear factor (NF)-κB activation was measured in the hippocampus by immunoblotting (c). Hippocampal (d) and blood tumor necrosis factor (TNF)-α (e) levels were measured by enzyme-linked immunosorbent assay (ELISA). Blood LPS levels were measured by Limulus assay (f). Values indicate mean± s.d. (n=10). *P<0.05.
Discussion
Various physiological factors, such as a high-fat diet, physiological stresses, and pathogenic infections, elevate the level of blood LPS.22, 23 LPS concentration is the highest in the gastrointestinal tract including the colon, where trillions of commensal bacteria reside.24, 25 Therefore, a great portion of LPS in the blood may originate from Gram-negative bacteria in the gut microbiota. Normally, LPS in the gastrointestinal tract cannot easily penetrate across the healthy intestinal epithelium.26 However, gut microbiota LPS is well-known to be involved in the initiation and propagation of intestinal inflammation.27, 28 Gut permeability disorders caused by the suppression of tight junction protein expression through gut inflammation increase the paracellular flux of bacteria and their LPS.29 Therefore, the blood LPS levels are markedly elevated in gut permeability disorders such as inflammatory bowel disease, leading to endotoxemia and systemic inflammation.
In the present study, we found that treatment with TNBS caused colitis, significantly disturbed the gut microbiota composition, in particular by increasing the Proteobacteria to Bacteroidetes ratio and EC, and increased gut microbiota LPS production. Oral gavage of EC in mice caused colitis, suppressed the expression of tight junction proteins, increased the absorption of orally administered LPS into the blood, and, as a result, increased intestinal permeability. Consistent with our finding, it was previously reported that patients with inflammatory bowel disease exhibit high LPS levels in the blood.27 This increase in LPS levels may cause systemic inflammation and accelerate the occurrence of degenerative diseases including AD. We also found that treatment with TNBS or EC increased LPS levels in gut microbiota and blood, and enhanced memory impairment. These treatments increased NF-κB activation and TNF-α expression in the hippocampus, both of which have important roles in the consolidation of memory information and spatial memory that enables navigation.30 In addition, the treatments reduced BDNF expression and CREB phosphorylation. Furthermore, an intraperitoneal injection of LPS isolated from EC, a member of gut microbiota, in mice caused memory impairment, induced NF-κB activation, and reduced BDNF expression in the hippocampus. Czerniawski and Guzowski15 reported that peripheral injection of LPS hippocampus-dependently impaired context discrimination memory. We also found that repetitive peritoneal injection of LPS caused memory impairment and induced NF-κB activation and TNF-α expression in the hippocampus. However, little LPS (∼0.025% of an intravenously administered dose) crossed the blood–brain barrier.31, 32 Hasegawa-Ishii et al.33 reported that one peritoneal injection of LPS activated hippocampal astrocytes through interaction between the cells of the brain–immune interface and cytokine signals, and, when repetitively injected, LPS activated microglia. Buttini et al.34 reported that a peritoneal injection of LPS activated microglia, which produced proinflammatory cytokines. These reports suggested that chronic endotoxemia may impair memory by activating microglia in the hypothalamus, although whether LPS directly or indirectly activated microglia in the brain remains distinctly unproven.
Importantly, glial cells, especially microglia and astrocytes, are involved in the pathogenesis of various CNS diseases, such as AD.35, 36 Microglia, which are the brain-resident macrophages, have an important role in the immune surveillance of the CNS against injury or pathogen infection. The exposure of microglia to pathogens and their antigens (e.g., LPS) lead to the secretion of various inflammatory mediators, such as TNF-α, which are associated with a number of neurodegenerative diseases.37 Astrocytes, such as microglia, readily become activated in response to injury and regulate neuroinflammatory events, such as microglia.36 Overactivation of glial cells can cause neuronal damage, which may result in neuropathological changes in a number of CNS disorders, such as memory impairment. Therefore, suppression of NF-κB activation and TNF-α expression in glia is regarded as an important therapeutic strategy for neuroinflammation-mediated diseases. TNBS treatment reduced the beneficial gut bacteria (Bifidobacteria and Lactobacilli) including LJ and altered the neurotrophic factor BDNF. Moreover, the change in the BDNF signaling pathway is relevant to a range of human neuronal and psychiatric disorders, such as AD. Our study demonstrated that treatment with LJ ameliorated TNBS- or EC-induced memory impairment in mice, increased BDNF expression, and inhibited NF-κB activation. These results supported the idea that the disturbance of gut microbiota, such as the suppression of the beneficial bacteria, may cause memory impairment and eventually AD. Mechanistically, we found that LJ treatment increased TNBS- or EC-induced suppression of tight junction proteins and inhibited TNBS- or EC-induced activation of NF-κB and expression of TNF-α, resulting in the attenuation of colitis. Furthermore, treatment with LJ restored EC-disturbed gut microbiota composition and suppressed TNBS- or EC-induced gut microbiota LPS level. Basically, these results supported the idea that LJ can ameliorate colitis and memory impairment by restoring gut microbiota composition and suppressing gut microbiota LPS production.
Finally, the brain affects the composition of gut microbiota and regulates immune responses via the hypothalamus–pituitary–adrenal axis and the disturbance of gut microbiota has been shown to affect anxiety-like and dementia-like behaviors.4, 38 The gut-to-brain axis is regulated through the central and enteric nervous responses and the neural, endocrine, and immune responses.1, 39 Therefore, the gut–brain axis is important for maintaining homeostasis. Interestingly, gut microbiota produces neuroactive molecules, such as acetylcholine and serotonin.1, 40 These findings suggest that the changes in gut microbiota composition by diet, drugs, disease, and probiotics correlate with changes in levels of circulating cytokines, some of which can affect brain function. However, gut microbiota produces endotoxins including LPS, which can cause inflammatory diseases (e.g., colitis). Gastrointestinal inflammation may increase the absorption of LPS into the blood and cause systemic inflammatory diseases such as cognitive failure, hepatitis, and rheumatoid arthritis. As such, EC caused colitis, memory impairment, and recessive BDNF expression. However, LJ, which increased membrane tight junction protein expression and inhibited NF-κB activation, ameliorated EC-induced colitis and memory impairment. Thus, the induction of gut microbiota LPS by diets and diseases may increase the occurrence of systemic inflammatory diseases such as AD, which may then change the secretion of neuroactive molecules in brains. In addition, the disturbance of gut microbiota composition directly affects the production of neuroactive molecules. In conclusion, our study supports that gut inflammation induced by the disturbance of gut microbiota may cause memory impairment and maintaining beneficial gut microbiota composition and inhibiting LPS production by beneficial gut bacteria, which suppress endotoxin-producing bacteria, such as EC, is important to suppress and prevent the occurrence of inflammatory degenerative diseases, including memory impairment.
Methods
Culture of gut bacteria. LJ and EC isolated from mouse gut microbiota were cultured in GAM broth (BD, Radnor, PA) and selective media MRS (BD), BL, and DHL (Nissui Pharm, Tokyo, Japan). Briefly, LJ and EC were cultured in 0.5 l of GAM broth at 37 °C (an optical density at 600 nm, 1.0–1.2), collected by centrifugation (5,000 g for 20 min), and washed with saline twice. The collected cells (5 × 109 CFU ml−1) were suspended in Saline (for cell experiments, heated at 72 °C for 30 min) or 1% glucose (for oral administration to mice).
For the analysis of gut microbiota by selective media, fresh colon content (∼0.1 g) from each group was collected in sterilized plastic cups, carefully suspended in 9 volumes of dilution media, diluted 10-fold in a stepwise manner, and inoculated directly onto agar plates with blood liver medium (BL, Bifidobacteria/Lactobacilli-selective medium, Nissui Pharm) and hydrogen sulfate lactose medium (DHL, Enterobacteriaceae-selective medium, Eiken Chem, Tokyo, Japan).41 DHL agar plates were aerobically cultured for 1 day at 37 °C and BL agar plates were anaerobically cultured for 3 days at 37 °C.
LJ and EC were selected of the colonies substantially grown in BL and DHL, respectively, and identified by Gram staining, a sugar utilization test (API 50 CHL or API 20 E Kit, bioMerieux, Seoul, Korea), and 16S rRNA sequencing (ABI 3730XL DNA analysis). These were deposited in the National Center for Biotechnology Information (NCBI)’s short read archive under accession number KY751910 and KY751911.
Isolation of LPS from EC. LPS was extracted as described previously with some modifications.42 Briefly, EC was cultured in tryptic soy broth (500 ml) for 24 h at 37 °C and collected by centrifugation at 10 000 g for 5 min. The pellets were washed twice in 0.15 M phosphate-buffered saline (PBS, pH 7.2) containing 0.15 mM CaCl2 and 0.5 mM MgCl2, suspended in 50 ml PBS, and sonicated for 30 min on ice. The sonicate was incubated with proteinase K (100 μg ml−1, Sigma, St Louis, MO) at 65 °C for 2 h and subsequently treated with RNase (40 μg ml−1, Sigma) and DNase (20 μg ml−1, Sigma) in the presence of 1 μl/ml of 20% MgSO4 and 4 μl ml−1 of chloroform at 37 °C overnight. The reaction solution was extracted with the same volume of 90% phenol with vigorous shaking at 65–70 °C for 15 min, transferred to polypropylene tubes, and centrifuged at 8,500 g for 15 min. The supernatants were treated with 10 volumes of 95% cold ethanol in the presence of 0.5 M sodium acetate at −20 °C overnight and centrifuged at 2,000 g at 4 °C for 10 min. The resulting pellet was suspended in distilled water, dialyzed twice against double distilled water at 4 °C, then lyophilized, and used in the present experiment as LPS.
Culture of Caco-2 cells. Caco-2 cells were purchased from Korea Cell Line Bank (Seoul, Korea) and cultured at 37 °C in a 5% CO2–95% air atmosphere in DMEM (Sigma) containing 10% fetal bovine serum and 1% antibiotic–antimycotic. To measure the effect of LJ on the expression of tight junction proteins in vitro, cells were treated with 100 ng ml−1 of the fecal LPS fraction in the presence or absence of LJ for 24 h and the expression levels of tight junction proteins in their lysate were measured by immunoblotting.
Animals. Male ICR mice (25–27 g, 5 weeks old) were supplied from RaonBio. (Gyeonggi-do, Korea). All mice were housed in wire cages at 20–22 °C and 50±10% humidity, fed standard laboratory chow, and water ad libitum. Mice were used in the experiments after the acclimation for more than 1 week. Each group in all experiments consisted of 10 mice.
All animal experiments were approved by The Committee for the Care and Use of Laboratory Animals in Kyung Hee University and performed in accordance with The Kyung Hee University Guidelines for Laboratory Animals Care and Usage (IRB Number KHUASP(SE)-15-092).
Preparation of experimental colitic mice. For the preparation of mice with TNBS-induced colitis, 2.5% (w/v) TNBS solution (100 μl, dissolved in 50% ethanol) was intrarectally injected into the colon of mice anesthetized with ether43 (Supplementary Figure S6a). To distribute TNBS entirely within the colon, mice were held in a vertical position for 30 s after treatment with TNBS. Normal control group was treated with saline instead of TNBS. Mice were killed 24 h, 8th day, and 15th day after TNBS treatment.
For the preparation of EC-induced colitis, mice were orally administered EC suspension (1 × 109 CFU, suspended in 100 μl of 1% glucose) once a day for 5 days (Supplementary Figure S6b). Normal control group was treated with 1% glucose instead of EC. Mice were killed 24 h, 8th day, and 15th day after EC treatment.
For the preparation of LPS-induced memory impairment, mice were intraperitoneally administered LPS solution (8 μg kg−1, dissolved in saline) once a day for 10 days (Supplementary Figure S6c). The LPS was isolated from EC. Normal control group was treated with 1% glucose instead of EC. Mice were killed 48 h after the final administration of LPS treatment.
To measure the anti-colitic and memory impairment-ameliorating effect of LJ, mice were randomly divided into four groups: normal control, LJ-treated group in normal control, colitis control group induced by treatment with TNBS or EC, and LJ-treated group in mice with colitis (Supplementary Figure S6d,e). Colitis was induced by the intrarectal injection of 2.5% (w/v) TNBS solution (100 μl, dissolved in 50% ethanol) into the colon of mice anesthetized with ether or the oral administration of EC (1 × 109 CFU, suspended in 100 μl of 1% glucose) once a day for 5 days. Normal group was treated with vehicle instead of TNBS or EC. LJ (1 × 109 CFU per mouse, suspended in 0.1 ml of 1% glucose) were orally administered once a day for 5 days from 72 h after the final treatment with TNBS or EC. Normal group was treated with 1% glucose (vehicle) alone instead of LJ. Memory behaviors were measured 2 h after the final administration of LJ in Y-maze and passive avoidance task. Mice were killed 2 h after the measurement of memory behaviors. The hippocampus and colon were removed. The colons were opened longitudinally and gently washed with ice-cold PBS. The specimens were stored at −80 °C until used in the experiment for the assays of enzyme activity, enzyme-linked immunosorbent assay, and immunoblotting.
Passive avoidance task. Passive avoidance task was performed in a two-compartment acrylic box connected a lighted compartment (20 × 20 × 20 cm) to a dark compartment (20 × 20 × 20 cm) by an entrance hole (5 × 5 cm) according to the method of Jung et al.44 Briefly, in the acquisition trial a mouse was placed in the lighted compartment 1st, 8th, and 15th day after treatment with TNBS, EC, or its LPS and, when the mouse entered the dark chamber, a 0.3 mA electrical shock for 2 s was given through floor grids. A retention trial was performed 24 h after the acquisition trial and a latency time to re-enter the dark chamber was measured.
Y-maze task. Y-maze was performed in a three-arm horizontal maze (40 cm-long and 3 cm-wide with 12 cm-high walls) in which the arms are symmetrically disposed at 120° angles from each other according to the method of Jung et al.44 The maze floor and walls were made from dark opaque polyvinyl plastic. A mouse was initially placed within one arm 1st, 8th, and 15th day after treatment with TNBS, EC, or its LPS, and the sequence (i.e., ACABC, etc.) and number of arm entries were recorded manually for each mouse for 8 min. An actual alternation was defined as entries into all three arms on consecutive choices (i.e., ABC, CAB, or BAC but not ABA). Maze arms were thoroughly cleaned between tasks to remove residual odors. Alternation (%) was indicated as follows: alternation (%)=[(number of alternations)/(number of total arm entries−2)] × 100. The number of arm entries served as an indicator of locomotor activity.
Assay of MPO activity. A mouse colon was homogenized in 10 mM potassium phosphate buffer (pH 7.0) containing 0.5% hexadecyl trimethyl ammonium bromide and centrifuged for 10 min at 20 000 g at 4 °C.45 The supernatant (50 μl) was added to the reaction mixture (0.1 mM H2O2 and 1.6 mM tetramethyl benzidine), incubated at 37 °C for 2 min, and then periodically monitored the absorbance at 650 nm for 5 min. MPO activity was calculated as the quantity of enzyme degrading 1 μmol ml−1 of peroxide, and expressed in unit per mg protein.
Enzyme-linked immunosorbent assay and immunoblotting. Mouse colon or hippocampus was homogenized in the RIPA lysis buffer containing 1% phosphatase inhibitor cocktail and 1% protease inhibitor cocktail on ice and centrifuged at 15 000 g at 4 °C for 15 min. Cultured Caco-2 cells were homogenized in RIPA lysis buffer containing 1% phosphatase inhibitor cocktail and 1% protease inhibitor cocktail on ice. The cytokine levels in the supernatants were measured using enzyme-linked immunosorbent assay kit (Ebioscience, Atlanta, GA).
For the immunoblotting, the supernatants of the colon and cultured cell homogenates were subjected to SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane.45 Proteins were probed with antibodies, detected with horseradish peroxidase-conjugated secondary antibodies, and visualized with ECL detection kit.
Limulus amoebocyte lysate assay. Blood and fecal endotoxin contents were determined using the diazo-coupled limulus amoebocyte lysate assays (Cape Cod, E. Falmouth, MA) according to the method of Kim et al.41 Briefly, for the determination of blood endotoxin concentration, plasma was diluted in pyrogen-free water 10-fold, inactivated at 70 °C for 10 min, and then incubated with limulus amoebocyte lysate for 30 min at 37 °C. Addition of reagents led to formation of a magenta derivative that absorbs light at 545 nm.
For the determination of fecal endotoxin concentration, 20 mg of feces from colons was placed in 50 ml of PBS in a pyrogen-free tube and sonicated for 1 h on ice.41 After centrifugation at 400 g for 15 min, the upper 30 ml was collected, sterilized by filtration through a 0.45 μm filter followed by re-filtration through a 0.22 μm filter, and inactivated for 10 min at 70 °C. The filtered sonicate was used for the determination of endotoxin.
Gut microbiota analysis by pyrosequencing. For the analysis of gut microbiota by pyrosequencing, genomic DNA was extracted from mouse fecal samples using a commercial DNA isolation kit (QIAamp DNA Stool Mini Kit, Qiagen, Hilden, Germany) following the manufacturer’s protocol. Amplification of genomic DNA was performed using barcoded primers that targeted the V1 to V3 regions of the bacterial 16S rRNA gene. The amplification, sequencing, and basic analysis were performed according to the method of Kim et al.41 by using a 454 GS FLX Titanium Sequencing System (Roche, Branford, CT). Sequences for each sample were sorted by a unique barcode and low-quality reads (average quality score <25 or read length <300 bp) were removed. Sequence reads were identified using the EzTaxon-e database (http://eztaxon-e.ezbiocloud.net/) on the basis of 16S rRNA sequence data. Number of sequence analyzed, observed diversity richness (operational taxonomic units), estimated OTUs (ACE and Chao1), and coverage indicated in Supplementary Table S1 were calculated using the Mothur program and defined considering a cutoff value of 97% similarity with the 16S rRNA gene sequences. 454 pyrosquencing reads have been deposited in the NCBI’s short read archive under accession number PRJNA345398.
In vivo intestinal permeability assay of LPS. Mice treated with or without EC were dosed with 0.1 ml of Alexa LPS (25 μg ml−1) per 25 g body weight. Normal control mice were dosed with a corresponding dose of PBS. Exactly 2 h after the administration of LPS, the mice were killed (CO2 and decapitation) and blood was collected from the neck into 50 ml Falcon tubes containing 100 μl EDTA (0.5 M, pH 8, Thermo Fisher Scientific, Waltham, MA). Blood was centrifuged for 10 min (1,500 g and 4 °C). The resulting plasma was mixed with the same volume of PBS and the fluorescence was measured in black 96-well microtitre plates (Proxiplate-96F, Perkin Elmer, Waltham, MA) using a FLOUstar Omega (BMG, LABTECH, Ortenberg, Germany) with excitation at 495 nm and emission at 540 nm.
Statistical analysis. Data are indicated as the mean±s.d. Statistical analysis of the data was performed with analysis of variance and Duncan’s test. Differences with a P<0.05 were considered to be statistically significant.
Accession codes
References
Grenham, S., Clarke, G., Cryan, J.F. & Dinan, T.G. Brain-gut-microbe communication in health and disease. Front Physiol 2, 94 (2011).
Moloney, R.D., Desbonnet, L., Clarke, G., Dinan, T.G. & Cryan, J.F. The microbiome: stress, health and disease. Mamm. Genome 25, 49–74 (2014).
Bailey, M.T. & Coe, C.L. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev. Psychobiol. 35, 146–155 (1999).
Mayer, E.A., Tillisch, K. & Gupta, A. Gutbrain axis and the microbiota. J. Clin. Invest. 125, 926–938 (2015).
Linthorst, A.C. & Reul, J.M. Brain neurotransmission during peripheral inflammation. Ann. N. Y. Acad. Sci. 840, 139–152 (1998).
Fung, T.C., Olson, C.A. & Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 20, 145–155 (2017).
Yang, N.J. & Chiu, I.M. Bacterial signaling to the nervous system through toxins and metabolites. J. Mol. Biol. 429, 587–605 (2017).
Petrof, E.O., Claud, E.C., Gloor, G.B. & Allen-Vercoe, E. Microbial ecosystems therapeutics: a new paradigm in medicine? Benef. Microbes 4, 53–65 (2013).
Choi, H.H. & Cho, Y.S. Fecal microbiota transplantation: current applications, effectiveness, and future perspectives. Clin. Endosc. 49, 257–265 (2016).
Arboleya, S., Watkins, C., Stanton, C. & Ross, R.P. Gut bifidobacteria populations in human health and aging. Front. Microbiol. 7, 1204 (2016).
Harmsen, H.J. & de Goffau, M.C. The human gut microbiota. Adv. Exp. Med. Biol. 902, 95–108 (2016).
Zhang, G. & Ghosh, S. Molecular mechanisms of NF-kappaB activation induced by bacterial lipopolysaccharide through Toll-like receptors. J. Endotoxin Res. 6, 453–457 (2000).
Doyle, S.L. & O'Neill, L.A. Toll-like receptors: from the discovery of NF-kappaB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol 72, 1102–1113 (2006).
Jiang, T. et al. Apple-derived pectin modulates gut microbiota, improves gut barrier function, and attenuates metabolic endotoxemia in rats with diet-induced obesity. Nutrients 8, 126 (2016).
Czerniawski, J. & Guzowski, J.F. Acute neuroinflammation impairs context discrimination memory and disrupts pattern separation processes in hippocampus. J. Neurosci. 34, 12470–12480 (2014).
Lukiw, W.J. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front. Microbiol. 7, 1544 (2016).
Kyoko, O.O. et al. Expressions of tight junction proteins Occludin and Claudin-1 are under the circadian control in the mouse large intestine: implications in intestinal permeability and susceptibility to colitis. PLoS ONE 9, e98016 (2014).
Xiong, Y. et al. Citrus nobiletin ameliorates experimental colitis by reducing inflammation and restoring impaired intestinal barrier function. Mol. Nutr. Food Res. 59, 829–842 (2015).
Kim, K.A., Jang, S.E., Jeong, J.J., Yu, D.H., Han, M.J. & Kim, D.H. Doenjang, a Korean soybean paste, ameliorates TNBS-induced colitis in mice by suppressing gut microbial lipopolysaccharide production and NF-κB activation. J. Funct. Foods 11, 417–427 (2014).
Lee, I.A., Bae, E.A., Hyun, Y.J. & Kim, D.H. Dextran sulfate sodium and 2,4,6-trinitrobenzene sulfonic acid induce lipid peroxidation by the proliferation of intestinal gram-negative bacteria in mice. J. Inflamm. (Lond.) 7, 7 (2010).
Khalifa, A.E. Antiinfective agents affecting cognition: a review. J. Chemother. 19, 620–631 (2007).
Gnauck, A., Lentle, R.G. & Kruger, M.C. Chasing a ghost?—Issues with the determination of circulating levels of endotoxin in human blood. Crit. Rev. Clin. Lab. Sci. 53, 197–215 (2016).
Kim, K.A., Gu, W., Lee, I.A., Joh, E.H. & Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 7, e47713 (2011).
Metzler-Zebeli, B.U., Mann, E., Schmitz-Esser, S., Wagner, M., Ritzmann, M. & Zebeli, Q. Changing dietary calcium-phosphorus level and cereal source selectively alters abundance of bacteria and metabolites in the upper gastrointestinal tracts of weaned pigs. Appl. Environ. Microbiol. 79, 7264–7272 (2013).
Seki, E. & Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J. Physiol. 590, 447–458 (2012).
Guo, S., Al-Sadi, R., Said, H.M. & Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 182, 375–387 (2013).
Caradonna, L., Amati, L., Magrone, T., Pellegrino, N.M., Jirillo, E. & Caccavo, D. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J. Endotoxin Res. 6, 205–214 (2000).
Kim, K.A., Jeong, J.J., Yoo, S.Y. & Kim, D.H. Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC Microbiol. 16, 9 (2016).
Moreira, A.P., Texeira, T.F., Ferreira, A.B., Peluzio Mdo, C. & Alfenas Rde, C. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br. J. Nutr. 108, 801–809 (2012).
Sheridan, G.K. & Murphy, K.J. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 3, 130181 (2013).
Singh, A.K. & Jiang, Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology 201, 197–207 (2004).
Banks, W.A. & Robinson, S.M. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav. Immun. 24, 102–109 (2010).
Hasegawa-Ishii, S., Inaba, M., Umegaki, H., Unno, K., Wakabayashi, K. & Shimada, A. Endotoxemia-induced cytokine-mediated responses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci. Rep. 6, 25457 (2016).
Buttini, M., Limonta, S. & Boddeke, H.W. Peripheral administration of lipopolysaccharide induces activation of microglial cells in rat brain. Neurochem. Int. 29, 25–35 (1996).
Dzamba, D., Harantova, L., Butenko, O. & Anderova, M. Glial cells - the key elements of Alzheimer’s disease. Curr. Alzheimer Res. 13, 894–911 (2016).
Minagar, A., Shapshak, P., Fujimura, R., Ownby, R., Heyes, M. & Eisdorfer, C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J. Neurol. Sci. 202, 13–23 (2002).
Amor, S., Puentes, F., Baker, D. & van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 129, 154–169 (2010).
Mu, C., Yang, Y. & Zhu, W. Gut microbiota: the brain peacekeeper. Front. Microbiol. 7, 345 (2016).
Carabotti, M., Scirocco, A., Maselli, M.A. & Severi, C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 28, 203–209 (2015).
Wall, R., Cryan, J.F., Ross, R.P., Fitzgerald, G.F., Dinan, T.G. & Stanton, C. Bacterial neuroactive compounds produced by psychobiotics. Adv. Exp. Med. Biol. 817, 221–239 (2014).
Kim, K.A., Gu, W., Lee, I.A., Joh, E.H. & Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 7, e47713 (2012).
Simmons, D.A., Lüderitz, O. & Westphal, O. The immunochemistry of Salmonella chemotype VI O-antigens. The structure of oligosaccharides from Salmonella group G (o 13,22) lipopolysaccharides. Biochem. J. 97, 807–814 (1965).
Joh, E.H., Lee, I.A., Jung, I.H. & Kim, D.H. Ginsenoside Rb1 and its metabolite compound K inhibit IRAK-1 activation - the key step of inflammation. Biochem. Pharmacol. 82, 278–286 (2011).
Jung, I.H., Jung, M.A., Kim, E.J., Han, M.J. & Kim, D.H. Lactobacillus pentosus var. plantarum C29 protects scopolamine-induced memory deficit in mice. J. Appl. Microbiol. 113, 1498–1506 (2012).
Kim, K.A., Lee, I.A., Gu, W., Hyam, S.R. & Kim, D.H. β-Sitosterol attenuates high-fat diet-induced intestinal inflammation in mice by inhibiting the binding of lipopolysaccharide to toll-like receptor 4 in the NF-κB pathway. Mol. Nutr. Food Res. 58, 963–972 (2014).
Acknowledgements
This study was supported by grants from the Bio & Medical Technology Development Program (2013M3A9B6076413) of the National Research Foundation (NRF) funded by the Korean government (MSIP).
Author contributions
S.-E.J., M.J.H. and D.-H.K. conceived and designed the study. S.-E.J. and S.-M.L. performed most of the experiments. J.-J.J., H.-M.J. and J.-J.L. performed part of the experiments. S.-E.J., S.-M.L., M.J.H. and D.-H.K. analyzed data. S.-E.J., M.J.H. and D.-H.K. drafted the paper. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Jang, SE., Lim, SM., Jeong, JJ. et al. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol 11, 369–379 (2018). https://doi.org/10.1038/mi.2017.49
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.49
This article is cited by
-
Bifidobacterium bifidum and Lactobacillus paracasei alleviate sarcopenia and cognitive impairment in aged mice by regulating gut microbiota-mediated AKT, NF-κB, and FOXO3a signaling pathways
Immunity & Ageing (2023)
-
Inhibition of microfold cells ameliorates early pathological phenotypes by modulating microglial functions in Alzheimer’s disease mouse model
Journal of Neuroinflammation (2023)
-
ADSCs stimulated by VEGF-C alleviate intestinal inflammation via dual mechanisms of enhancing lymphatic drainage by a VEGF-C/VEGFR-3-dependent mechanism and inhibiting the NF-κB pathway by the secretome
Stem Cell Research & Therapy (2022)
-
Estrogen receptor β deficiency impairs gut microbiota: a possible mechanism of IBD-induced anxiety-like behavior
Microbiome (2022)
-
Enterococcus faecium and Pediococcus acidilactici deteriorate Enterobacteriaceae-induced depression and colitis in mice
Scientific Reports (2022)
