
Abstract
The inflammasome is a cytosolic multi-protein innate immune rheostat, sensing a variety of endogenous and environmental stimuli, and regulating homeostasis or damage control. In the gastrointestinal tract, inflammasomes orchestrate immune tolerance to microbial and potentially food-related signals or drive the initiation of inflammatory responses to invading pathogens. When inadequately regulated, intestinal inflammasome activation leads to a perpetuated inflammatory response leading to immune pathology and tissue damage. In this review, we present the main features of the predominant types of inflammasomes participating in intestinal homeostasis and inflammation. We then discuss current controversies and open questions related to their functions and implications in disease, highlighting how pathological inflammasome over-activation or impaired function impact gut homeostasis, the microbiome ecosystem, and the propensity to develop gut-associated diseases. Collectively, understanding of the molecular basis of intestinal inflammasome signaling may be translated into clinical manipulation of this fundamental pathway as a potential immune modulatory therapeutic intervention.
Similar content being viewed by others


Introduction
The intestinal microenvironment represents a potentially hostile milieu in which a multitude of microorganisms, food-, and microbial-derived antigens are separated from the sterile host by only a single layer of intestinal epithelium. In order to master the tightrope walk between tolerance to dietary and microbial antigens, and the need for aggressive immunity against invasive pathogens, the host relies on three fundamental principles of host–microbial interactions in the intestine: first, the prokaryotic community is physically separated from the host by the induction of a mucus layer that is only sparsely colonized and provides a “neutral zone” of 50–100 μm between the epithelial layer and the majority of microorganisms. Second, the host immune system is able, through a multitude of innate and adaptive mechanisms, to acquire tolerance to intestinal antigens, thereby preventing overt inflammation against constant antigenic stimulation. Third, the anti-microbial repertoire of the host comprises both constantly expressed and rapidly inducible components, including anti-microbial peptides, IgA antibodies that are secreted into the intestinal lumen, and bactericidal mechanisms of immune responses in the intestinal lamina propria, which become active upon breach of the intestinal barrier and invasion of microorganisms into the otherwise sterile tissue.
As such, the host depends on a myriad of protective mechanisms and innate immune sensing platforms, whose integrated signaling is critically important in maintaining homeostasis, while preserving the host’s ability to efficiently react through an explosive yet self-contained protective inflammatory counteraction at times when a potentially life-risking infection is encountered.1 This important “friend vs. foe” distinction is achieved through an array of germ-line-encoded pattern recognition receptors (PRRs) that recognize conserved molecular patterns, including microbial and host-derived molecules, to initiate tolerogenic vs. defense programs. The first discovered family of PRRs is the Toll-like receptors (TLRs), transmembrane receptors that can recognize multiple extracellular and intracellular signals.2, 3 Several additional classes of PRRs, classified by their ligands and downstream effector function, include C-type lectins, nucleotide-binding domain, leucine-rich repeat-containing-like receptors (NLRs), RIG-I-like receptors, and absent in melanoma (AIM2)-like receptors (ALRs).
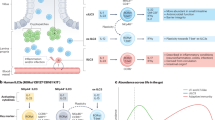
The NLR family of innate immune receptors, consisting of over 20 members discovered to date, stands out with its wide range of endogenous and exogenous triggers, and downstream functions. However, many of the ligands and functions of most NLR family members still remain largely unknown. NLRs represent cytosolic PRRs that contain three distinct domains: a central nucleotide-binding and oligomerization domain (NACHT domain), an N-terminal domain that can be either a CARD (caspase-recruitment domain), a pyrin domain or a baculovirus inhibitor of apoptosis repeat domain effector domain, and a variable number of C-terminal leucine-rich repeats (Figure 1). It is believed that the leucine-rich repeat domains interact with putative ligands, whereas the NACHT domain can bind ribonucleotides, which regulates self-oligomerization.4 Some NLRs and ALRs can form signaling platforms, called inflammasomes, which are cytoplasmic high-molecular protein complexes that are formed in response to diverse microbial or host-derived stimuli. Inflammasomes are named after their NLR or ALR protein scaffold.5

Structure and activating ligands of murine inflammasomes participating in intestinal inflammation. The nucleotide-binding domain, leucine-rich repeat-containing-like receptors (NLRs) NLRP3, NLRC4, NLRP6, NLRP1b, and the absent in melanoma (AIM2)-like receptors (ALR) member AIM2 are cytosolic sensors of a variety of endogenous and exogenous ligands. By forming inflammasomes, they prompt the cleavage of caspase-1, which subsequently results in the priming of the zymogens pro-interleukin (IL)-1β and pro-IL-18 into IL-1β and IL-18, respectively. Caspase-11, which senses cytosolic lipopolysaccharide (LPS) takes part in non-canonical inflammasome activation.
ALR proteins such as AIM2 can form an inflammasome through direct association with caspase-1 in CARD–CARD interactions. In contrast to ALRs, NLRs that contain an N-terminal pyrin domain have been shown to associate with the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), in order to recruit pro-caspase-1 to the inflammasome.6 The NACHT domain mediates inflammasome assembly, whereas the N-terminal domain mediates protein–protein interactions with downstream signaling molecules, among them caspase-1.4 Inflammasome activation triggers proteolytic cleavage and activation of caspase-1, resulting in processing and release of the proinflammatory cytokines interleukin-(IL)-1β and IL-18, and in some cases pyroptotic cell death.7 In contrast to canonical inflammasomes that activate caspase-1, non-canonical inflammasomes involve caspase-11 (or caspase-4/5 in humans). Although both caspase-1 and caspase-11 induce pyroptosis, the secretion of IL-18 and IL-1β is solely dependent on caspase-1 activity.7
Inflammasome activation is unique in its induction by a variety of endogenous and exogenous signals. In addition to responding to insults and pathogens, inflammasomes can also be activated by signals stemming from the commensal microbiota. Molecular mechanisms involving inflammasome activation have an important role in mediating an innate immune response and its associated “sterile inflammation”, which is triggered either by microbial molecules or host molecules indicative of tissue damage. Furthermore, sustained inflammatory processes predisposes to neoplasia. The role of inflammasomes in intestinal inflammation, infection, and carcinogenesis will be reviewed herein (summarized in Table 1). Non-inflammasome roles of NLRs include negative regulation of common intracellular signaling pathways, such as nuclear factor-κB (NF-κB) and mitogen-activated protein kinase pathways, regulation of anti-viral immunity, and antigen presentation on major histocomptibility complex class I.8 These are extensively reviewed elsewhere.9, 10
Canonical Inflammasomes in Gastrointestinal Physiology
NLRP3 inflammasome
The NLRP3 inflammasome (Figures 1 and 2) is expressed in both gut immune and epithelial cells, and promotes inflammation through caspase-1-dependent activation by cleavage of the pro-inflammatory cytokines IL-1β and IL-18, as well as by induction of pyroptotic cell death. It comprises the NLR family member NLRP3, the adaptor protein ASC (PYCARD), and the cysteine protease caspase-1. In quiescent states NLRP3 is expressed in low levels. Upon activation, a two-step process occurs, initiated by a priming signal that results in a NF-κB-dependent transcription of NLRP3 and pro-IL-1β (“signal I”), and ensued by a second signal that promotes inflammasome oligomerization and activation (“signal II”) culminating in caspase-1 activation, enabling cleavage of pro-IL-1β and pro-IL-18 into their biologically active forms IL-1β and IL-18.11 The ligands sensed by the NLRP3 inflammasome (“signal II”) and trigger its activation are diverse, ranging from microbial pathogen-associated molecular patterns, such as lipopolysaccharide (LPS), muramyl dipeptide, hemolysins, pore-forming toxins, nigericin, and bacterial and viral nucleic acids;12 fungal wall components zymosan and mannan; damage-associated molecular patterns such as high concentrations of ATP,13 mitochondria-derived components (mtDNA, reactive oxygen species (ROS), and phospholipid cardiolipin), and high-mobility group box 1 protein; endogenous stress such as endoplasmic reticulum stress,14 alterations in cell volume,15 or intracellular calcium influx; and exogenous stimuli or environmental irritants such as uric acid crystals, silica, asbestos, calcium phosphate, aluminum hydroxide, serum amyloid A, and cholesterol crystals.16 The diversity of inciting stimuli led to the assumption that there is a common downstream cellular event or perturbation accountable for NLRP3 activation, with potassium efflux potentially serving as a reasonable candidate and common denominator NLRP3 inflammasome-activating stimulus.17 As such, it was suggested that a member of the family of mammalian NIMA-related kinases (NEK7) responded to potassium efflux to create an NLRP3–NEK7 complex and regulate NLRP3 oligomerization and activation.18 Other suggested activation mechanisms include lysosomal destabilization by phagocytized material and generation of ROS. Activation of the NLRP3 inflammasome is further regulated by posttranscriptional mechanisms, such as ubiquitination and de-ubiquitination of NLRP3, and its downstream effector molecules.19, 20, 21, 22, 23

Mechanisms of NLRP3 inflammasome activation. NLRP3 inflammasome activation is triggered by various exogenous and endogenous danger signals, such as viral and bacterial infections (e.g., Candida albicans, cholera toxin), metabolic dysregulation and cellular damage. Some of these signals activate the NLRP3 inflammasome through known sensors, such as DHXN33 (DEAH-Box Helicase 33), which detect viral and bacterial RNA. The priming and activation of the NLRP3 inflammasome involves a two-step process, in which “signal I” initiates the nuclear factor-κB (NF-κB)-mediated transcription of NLRP3 and pro-interleukin (IL)-1β, and “signal II” involves the inflammasome assembly and caspase-1 cleavage. Although “signal I” can be triggered by a large variety of signals, “signal II” was suggested to result from several distinct cellular perturbations, namely potassium efflux, cytosolic release of lysosomal cathepsins or cytosolic release of mitochondria-derived factors such as reactive oxygen species (ROS). More specifically, pore-forming toxins activate the NLRP3 inflammasome through potassium efflux. Similarly, extracellular ATP stimulates the purinergic receptor P2X ligand-gated ion channel (P2X7) to open and leads to potassium efflux and the formation of pannexin 1 hemichannel, allowing for the entry of pathogen-associated molecular patterns and damage-associated molecular patterns into the cell and triggering NLRP3 inflammasome activation. Another cation channel, TRPM2 (transient receptor potential melastatin 2) mediates crystal- and liposome-induced mitochondrial ROS production through calcium influx, which in turn activates the NLRP3 inflammasome. Other cellular events can also trigger oxidative stress, such as endoplasmic reticulum stress, which causes the dissociation of TXNIP (thioredoxin-interacting protein) from thioredoxin (TRX), and its subsequent binding to the NLRP3 inflammasome and its activation. The NLRP3 inflammasome activity can be further modulated by extracellular cues, such as diet, and by ubiquitination and de-ubiquitination of its components by proteins, such as IAP and BRCC3 (BRCA1–BRCA2-containing complex). NLRP3 inflammasome activity has downstream effects on the gut microbial composition.
NLRP3 inflammasome in intestinal inflammation The role of the NLRP3 inflammasome in intestinal inflammation remains unsettled. Upon induction of experimental colitis by oral administration of dextran sodium sulfate (DSS) or rectal instillation of the hapten 2,4,6-trinitrobenzenesulfonic acid, some studies suggested that mice deficient in NLRP3 inflammasome components exhibit exacerbated colitis,24, 25, 26 manifested by increased mortality, breached epithelial integrity, and enhanced translocation of commensal bacteria from the gut to the systemic circulation, as well as reduced production of cytokines, including IL-1β. In these studies, NLRP3 inflammasome-deficient mice showed enhanced intestinal leukocyte infiltration, whereas macrophages isolated from knocked-out mice failed to mount an immune response to bacterial muramyl dipeptide and neutrophils showed impaired chemotaxis and exacerbated apoptosis. Furthermore, NLRP3-deficient mice exhibited a microbiome configuration different than their wild-type (WT) counterparts.24 Bone marrow (BM) chimeric experiments suggested that NLRP3 expression in non-hematopoietic cells, i.e., epithelial cells, may regulate intestinal inflammation.25 Similarly, NLRP3 was proven protective in oxazolone-induced colitis, as NLRP3-deficient mice were more susceptible to oxazolone hapten-mediated colitis as compared with their WT counterparts. The disease severity in this model was ameliorated by exogenous administration of IL-1β or IL-18.27
However, other studies have shown an opposite trend in disease severity in mice deficient in NLRP3 inflammasome components28, 29, 30 or related NLRP3-activated pro-inflammatory cytokines.31 These discrepancies may be explained by differences in the microbiome composition among animal facilities, as co-housing of NLRP3 deficient with WT mice could reverse the NLRP3-deficient colitogenic phenotype, whereas antibiotic treatment attenuated it.29 Moreover, it has been suggested that even slight differences between bacterial strains differentially activated inflammasomes, further supporting microbial composition as an important factor determining disease severity.32 Other explanations for the observed discrepancies include differences in mouse genetic background among studies, modified experimental colitis protocols, and differences in NLRP3 ablation methods, with some studies utilizing gene-deleted mice, whereas others implementing biochemical or immunological caspase-1 and inflammasome inhibitory approaches.24, 33, 34, 35, 36, 37 In addition, context-specific posttranscriptional mechanisms were suggested to be involved in inflammasome activation, further accounting for potential inter-study differences. The later mechanisms mediate degradation of NLRP3 and pro-IL-1β proteins (as well as tumor ecrosis factor (TNF)-α and IL-6) by the ubiquitin-proteasomal pathway, while being abrogated during experimental colitis.38
In another colitis model utilizing non-steroidal anti-inflammatory drugs, IL-1β, and NLRP3 expression, as well as IL-1β and caspase-1 protein levels, were found to be elevated through an NLRP3 inflammasome-mediated pathway. Concordantly, NLRP3 and caspase-1-deficient mice were rescued from non-steroidal anti-inflammatory drug-induced enteropathy. This phenotype was exacerbated upon the administration of TLR4 agonists and attenuated in TLR4-deficient mice and in mice treated with apyrase, an ATP scavenger, or with Brilliant Blue G, a purinergic P2X7 receptor antagonist, highlighting their potential significance in this inflammatory pathway.39
NLRP3 inflammasome in infectious enteritis and colitis The NLRP3 inflammasome also has an important role in protection from intestinal pathogens. Apart from inducing the secretion of pro-inflammatory cytokines, it controls a preponderance of immune processes necessary to combat infections, such as acidification of the phagosome.40
Intestinal pathogens are capable of activating the NLRP3 inflammasome through various molecules, such as by HpmA hemolysin produced by Proteus mirabilis,41 TcdA and TcdB toxins produced by Clostridium difficile,42 Yersinia enterocolitica adhesin invasin, which interacts with β1 integrin to provide “signal I” for NLRP3 inflammasome activation, and the bacterial type three secretion system (T3SS) translocon constituting “signal II”, thereby triggering IL-18 maturation and secretion.43 Intriguingly, the Yersinia pseudotuberculosis T3SS effector protein YopK and the combination of Yersinia enterocolica YopE and YopH impaired this machinery to evade inflammasome activation and reduce IL-18 secretion, thus hampering the host response to the pathogen.44 Citrobacter rodentium, a naturally occurring pathogen and the mouse correlate of enteropathogenic Escherichia coli in humans, is used as a murine experimental colitis model.45, 46, 47 In several studies, C. rodentium infection in NLRP3-deficient mice resulted in exacerbated weight loss, augmented inflammation and increased bacterial colonization as compared to WT mice, an effect mediated by non-hematopoietic NLRP3 deficiency.48
Contrary to the protective role of NLRP3 in several pathogen infections described above, a recent study, which implemented a model of polymicrobial sepsis through cecal ligation and puncture, showed that mice deficient in NLRP3 or mice receiving a pharmacological inhibitor of NLRP3 exhibited increased survival and augmented bacterial clearance compared with WT or untreated mice. The underlying mechanism suggested to confer this protective effect in NLRP3-deficient mice was decreased autophagy and increased phagocytosis by neutrophils.49
The role of NLRP3 in Salmonella typhimurium infection remains elusive,50 with one study suggesting of a muti-protein complex formed in macrophages in the presence of S. typhimurium infection, which is composed of NLRP3 and NLRC4, both being required for IL-1β processing.51 Helicobacter pylori features two contradicting traits—whereas cagA-positive strains were shown to activate the inflammasome to exacerbate DSS-induced colitis and to promote carcinogenesis,52 other studies demonstrated that H. pylori infection conferred protection from DSS-induced colitis, both clinically and histopathologically, by the induction of the Muc2 gene and production of mucus. This effect was abrogated in NLRP3, IL-18, IL-18 receptor, and MyD88-deficient mice, alluding to an NLRP3 inflammasome-dependent mechanism.53 Similarly, H. pylori infection in MUC1-deficient mice resulted in severe and lethal gastritis, and elevated IL-1β levels compared with WT mice, whereas mice deficient in MUC1 and either NLRP3 or caspase-1 did not develop severe gastritis.54
NLRP3 has also been implicated in protozoan infections, as it was found pivotal in sensing of invading Entamoeba histolytica and in mounting a robust inflammatory response against it.55, 56
NLRP3 inflammasome in intestinal tumorigenesis Chronically persistent inflammation is considered a major risk factor for cancer development; hence, it is not surprising that NLRP3 was implicated in tumorigenesis in various solid and hematopoietic malignancies.57 In humans, NLRP3 polymorphism was associated with gastric cancer in the chinese population58 and associated with an unfavorable outcome of patients suffering of colorectal tumors in the swedish population.59 Similarly, NLRP3 expression was diminished in colorectal cancer samples compared with healthy controls.60
NLRP3-deficient mice featured increased propensity to develop colitis-associated tumors in the azoxymethane (AOM)-DSS model, mediated by NLRP3 deficiency in hematopoietic cells.26, 61 NLRP3 has been suggested to confer a protective role from colorectal cancer development in an IL-18-dependent manner, as recombinant IL-18 exogenous administration alleviated this tumorigenic phenotype in NLRP3-deficient mice.25 This NLRP3 protective mechanism may stem from NLRP3 inflammasome-induced epithelial repair62 or from its initiation of programmed cell death. Interestingly, it has been recently demonstrated that dietary constituents may modulate tumor promotion. For example, high-cholesterol diet exacerbated inflammatory responses and tumor burden in the AOM-DSS model in mice through NLRP3 inflammasome activation.63 Conversely, other studies suggested that the inhibition of the NLRP3 inflammasome may actually confer protection against colorectal tumorigenesis in the AOM-DSS model.64
Other potential neoplastic-related NLRP3 effects include regulation of metastases formation, as mice deficient in NLRP3 inflammasome components featured increased liver colorectal cancer metastatic growth, a process shown to be elicited by impaired IL-18 signaling, which hampered natural killer cell-mediated immunosurveillance and tumoricidal activity.65 In addition, the NLRP3 inflammasome was suggested to participate in immune-mediated responses to the chemotherapeutic agents gemcitabine and 5-fluorouracil.66
Nutrition as a modulator of NLRP3 inflammasome activation Dietary constituents, such as saturated fatty acids, can function as stimuli for inflammasome activation to elicit endoplasmic reticulum stress responses, including IRE1α activation.67 Titanium dioxide, used as a food additive and in pharmaceutical formulations, aggravates DSS-induced colitis by activating the NLRP3 inflammasome, as well as increasing ROS production and gut permeability.68 Cholesterol consumption can trigger inflammasome-driven intestinal inflammation, which is dependent on the cholesterol-binding protein Niemann-Pick C1-like 1 and NF-κB activation by commensal microbiota.69 Other metabolites can suppress the NLRP3 inflammasome, for instance soy protein70 or the ketone body β-hydroxybutyrate, which characterizes caloric restrictive or ketogenic diets.71 In addition, metabolic enzymes, such as hexokinase, can serve as PRRs and activate the NLRP3 inflammasome upon metabolic signals pertaining to glycolysis or the tricarboxylic acid pathway.72 Moreover, nutrition may shape the commensal intestinal microbiome, which tightly regulates inflammasome activation; for instance, a study has revealed that bile acids metabolized by the microbiome bind to the bile acid receptor TGR5 to induce protein kinase A (PKA), which in turn leads to the ubiquitination and inhibition of NLRP3, thereby protecting against insulin resistance.73 However, other studies argue that bile acids can potentially serve as an endogenous danger signal and activate the NLRP3 inflammasome.74 Finally, it has been shown that diet rich in short-chain fatty acids protects mice against colitis by binding to GPR43 located on colonic epithelial cells, which triggers potassium efflux and hyperpolarization, leading to NLRP3 inflammasome activation.75
NLRC4 inflammasome
The NLRC4 inflammasome is activated by intracellular flagellin and components of the T3SS of pathogenic Gram-negative bacteria76, 77, 78 (Figure 1). Similar to other inflammasomes, NLRC4 inflammasome formation leads to secretion of IL-1β and IL-18, but can also induce pyroptosis.76, 79 In addition, the NLRC4 inflammasome takes part in apoptosis pathways downstream to p53.80, 81
NLRC4 inflammasome in intestinal inflammation. Data regarding the role of NLRC4 in immune-mediated colitis are conflicting. Although NLRC4 deficiency was reported not to affect the severity of DSS-induced colitis in some studies,82 others showed that NLRC4-deficient mice exhibited a more severe phenotype than WT mice in response to 2% DSS administration; however, this difference was lost when higher DSS concentrations were applied.83 Severe colitis in NLRC4-deficient mice was associated with reduced colonic IL-18 secretion, suggesting that this cytokine might protect against DSS-induced colitis. However, NLRC4 deficiency did not exacerbate colitis upon IL-10R neutralization, another model for immune-mediated colitis.83 In humans, a severe neonatal-onset enterocolitis was described in a family harboring a de novo mutation in the HD1 domain of the NLRC4 gene.84 This mutation was shown to result in spontaneous cleavage of procaspase-1 and ASC multimerization in HEK293 cells. In addition, mutated macrophages showed increased production of IL-1β and IL-18, and increased cell death. Collectively, some mouse models of colitis suggest that NLRC4 confers protection against colitis, whereas human subjects harboring a gain-of-function mutation in the NLRC4 gene exhibit increased pro-inflammatory risk. This finding merits further investigation, as NLRC4 may conduce to varying effects among different mammalian species and genetic makeups.
NLRC4 inflammasome in infectious enteritis and colitis. The NLRC4 inflammasome has an important role in the maintenance of balance between tolerance towards intestinal commensals and inflammatory responses to pathogens. Murine intestinal mononuclear phagocytes produce IL-1β through the activation of the NLRC4 inflammasome during S. typhimurium and Pseudomonas aeruginosa infections, but not upon exposure to commensal bacteria.85 Accordingly, NLRC4-deficient mice infected with Salmonella showed exacerbated colitis (on the BALB/c genetic background)85 and increased mortality due to uncontrolled bacterial replication and systemic dissemination (on the C57BL/6 genetic background).83 This protective property of NLRC4 was dependent on the mode of infection, as it occurred when the pathogen was administered orally, whereas an intraperitoneal injection of S. typhimurium resulted in a similar susceptibility to infection compared with WT controls.85 Furthermore, it was also dependent on flagellar recognition by NLRC4, as infection of NLRC4-deficient mice with an aflagellate mutant of S. typhimurium featured a comparable mortality rate to that of WT mice.83 The increased burden of S. typhimurium in epithelial cells in NLRC4-deficient mice was suggested to arise from impaired expulsion of infected enterocytes.86 A genetic approach and BM transplantation assays implicated the epithelium as the predominant source of NLRC4 in this setting.
C. rodentium colonization and pathology has similarly been shown to be dependent on the expression of a T3SS,87 which triggers NLRC4 inflammasome activation.77 Following oral administration of C. rodentium, NLRC4-deficient mice exhibited enhanced infection, manifesting in significant weight loss, increased bacterial load and exacerbated histological features of hyperplasia, leukocyte infiltration, and edema compared with their WT counterparts.88, 89 Furthermore, NLRC4-deficient mice exhibited elevated secretion of both interferon-γ and IL-17A, suggestive of a hyperactivation of the adaptive immune system. Similar to S. typhimurium infection, NLRC4 expression in non-hematopoietic cells was shown to confer protection during C. rodentium infection, suggesting that the protective role of NLRC4 originates from colonic epithelial cells, rather than from BM-derived immune cells.89 Notably, infected caspase-1-deficient mice showed a more severe phenotype compared with NLRP3- and NLRC4-deficient mice, although the in vitro induction of caspase-1 was not dependent on NLRC4 inflammasome activation.
NLRC4 inflammasome in intestinal tumorigenesis. Data on the roles of NLRC4 in tumorigenesis remain sparse. In one study, NLRC4-deficient mice exposed to AOM-DSS featured a significant increase in tumor load and aggressiveness compared with WT mice. Consequently, tumor development was suggested to be caspase-1-dependent and influenced by activation of the NLRC4 inflammasome.82 However, a different study utilizing the same model has not replicated this observation.26
NLRP6 inflammasome
NLRP6, originally termed PYPAF5, is highly expressed in the kidney, liver, and lungs, as well as in the large and small intestine (Figures 1 and 3). Studies over the past years have implicated NLRP6 in the regulation of host defense against microorganisms and intestinal homeostasis through diverse mechanisms, including inflammasome formation, NF-κB, and mitogen-activated protein kinase signaling regulation, as well as anti-microbial and anti-viral immunity. In both mice and humans, NLRP6 is exceedingly abundant in the intestinal epithelium, in particular in enterocytes and mucus-secreting goblet cells,90, 91, 92, 93, 94 where it is upregulated in the late-gestation fetal intestine.95 In addition, NLRP6 was also suggested to be expressed in myofibroblasts.91 In humans, NLRP6 transcript and protein are primarily detected in intestinal epithelial cells of the duodenum, jejunum, and ileum,92 whereas in mice NLRP6 is expressed in both the small and the large intestines.90

Mechanisms of NLRP6 inflammasome activation. NLRP6 senses viral and bacterial compounds and metabolites and its activation sustains homeostasis by mucus production, anti-microbial peptides (AMPs) secretion and the maintenance of a healthy microbial composition.
NLRP6 has been implicated in the response to viral pathogens and bacterial components of the commensal microbiome. NLRP6, through the helicase DHX15, binds to viral RNA and regulates the expression of interferon-stimulated genes that are central for anti-viral immunity, through the mitochondrial adapter protein MAVS.96 Interestingly, following encephalomyocarditis virus infection, NLRP6 expression was upregulated and NLRP6-deficient mice exhibited enhanced viral load96 if infection occurred by the gastrointestinal, but not by the systemic route. This suggests that NLRP6 functions as a viral sensor to control infection with enteric viruses.
The intestinal expression of NLRP6 at the site of a rich microbial community suggests a possible role for the microbiome in the regulation of NLRP6. Indeed, an unbiased metabolomics screen identified potential microbiome-derived modulators of NLRP6 activity, among them the bile acid derivative taurine as a positive regulator of NLRP6 signaling and IL-18 production, and the polyamine spermine and amino acid histamine as negative regulators of NLRP6 activity.97 In contrast to viral recognition, bacterial metabolite modulation of NLRP6 is associated with inflammasome activation and IL-18 production. Consequently, in germ-free or antibiotics-treated mice, NLRP6 activation and downstream IL-18 production is abrogated.
A recent study suggested that protozoan members of the microbiota, such as the commensal protist Tritrichomonas musculis, similarly contribute to the modulation of epithelial inflammasome activity and IL-18 induction. Given the centrality of NLRP6 in determining intestinal IL-18 levels, this finding may suggest an additional commensal activator for this NLR; however, the precise identity of the specific epithelial inflammasome responsible for the described induction of IL-18 has not been addressed to date.98 Together, these studies suggest that a wide array of microbial factors regulate the expression and activation of NLRP6. In addition, NLRP6 expression might be regulated by non-microbial factors, notably stress induced by water deprivation could downregulate NLRP6 expression through corticotrophin-releasing hormone.99
Evidence for transcriptional regulation of NLRP6 came from transcription factor-binding analysis, which identified binding sites for both retinoid X receptor and peroxisome proliferator-activated receptor-γ at the NLRP6 promoter region.95 However, although in vitro expression of NLRP6 was induced by peroxisome proliferator-activated receptor-γ agonist, the in vivo effect of peroxisome proliferator-activated receptor-γ on NLRP6 requires further study.99, 95
Intestinal activation of NLRP6 has several downstream implications, among them a contribution to the formation of a mucus layer that protects the epithelium against invasion of pathogens and physical damage. Consequently, NLRP6-deficient mice showed dysfunctional mucus granule exocytosis, possibly through autophagy regulation.93 The role of NLRP6 in mucus secretion was further described in a recent study identifying “sentinel” goblet cells that are localized at the upper part of the crypt and respond to TLR ligands such as LPS and flagellin. Upon endocytotic uptake of TLR ligands, sentinel goblet cells are expelled from the epithelium, triggering MUC2 secretion by adjacent goblet cells in the colonic crypt.94 Importantly, sentinel goblet cells might mediate mucus response to bacterial invasion into the inner mucus layer in an NLRP6-dependent manner, but are not necessary for the formation of a normal mucus layer under homeostatic conditions, as NLRP6-deficient mice featured an intact inner mucus layer in this study.94 Of note, although ASC and caspase-1 are likewise involved in the generation of a protective mucus layer, IL-18 and IL-1β were dispensable for this process, suggesting an inflammasome-independent function of NLRP6 in goblet cells.93 As a consequence of abrogated mucus production, NLRP6-deficient mice were unable to clear enteric pathogens from the intestinal mucosa and were highly susceptible to persistent colonization.93
In addition to its role in maintaining a protective mucus layer, NLRP6 also participates in the secretion of antimicrobial peptides from epithelial cells,97, 100 including angiogenin-4, intelectin-1, resistin-like-β, and others.97 In contrast to mucus secretion, this arm of the epithelial functions of NLRP6 was dependent on IL-18, indicating differential regulation of goblet cell mucus secretion vs. paracrine amplification of anti-microbial peptide production. In the absence of a functional NLRP6-associated inflammasome, a distorted antimicrobial peptide configuration develops, which can partially be restored by IL-18 supplementation.97
NLRP6-deficient mice also feature alterations in bacterial composition and function (dysbiosis), which are dominantly transmissible to WT mice, at least in some animal facilities.90, 101 Upon microbial transfer, the dysbiotic microbiota, through combinatorial levels of the metabolites taurine, histamine, and spermine, are able to suppress inflammasome signaling in the new host.97 Therefore, the dysbiotic microbiota dampens IL-18 levels and the downstream antimicrobial peptides, and modifies the intestinal niche to support microbial persistence.97 Conventionalization of germ-free NLRP6-deficient mice results in the de novo development of dysbiosis, suggestive of an NLRP6 causative role in regulating microbiome community structure.97 With that said, the relative contribution of NLRP6 signaling in impacting microbiome composition and function, in comparison with husbandry and vivarium-mediated effects, merit further studies. As such, NLRP6 deficiency-mediated dysbiosis may be greatly influenced in its severity and composition by local vivarium microbiome configuration and its diversity.
The dysbiotic microbiota that was observed under NLRP6 deficiency in some vivaria supports the development of pathologies, including intestinal inflammation, metabolic disease, and cancer,90, 91, 102, 103 and enhances the development of disease in cohoused WT mice. Similarly, mice deficient in the inflammasome adaptor ASC, caspase-1, and IL-18 featured, in some facilities, dysbiotic microbiota that was transmissible to WT mice.90 The intestinal inflammatory state of NLRP6-deficient mice and WT recipients of the dysbiotic microbiota was shown to be enhanced through CCL5, induced by the microbiota, and inflammatory cells were recruited to supports colitis.90 Inflammation in NLRP6-deficient mice was also associated with enhanced intestinal tumorigenesis driven by IL-6 signaling.91, 101, 103 Furthermore, the dysbiotic microbiota facilitated exacerbated translocation of microbial products into the portal vein, activating TLRs to produce TNF-α in the liver, and promoted the progression of non-alcoholic fatty liver to non-alcoholic steatohepatitis.102
Data concerning the role of NLRP6 in human gastrointestinal diseases remain scarce. In patients with non-alcoholic steatohepatitis and portal fibrosis, adipose tissue NLRP6 and circulating levels of IL-18 were significantly upregulated.104 Notably, although in mice NLRP6 was suggested to have a role in protection from colorectal cancer, in human colorectal cancer samples NLRP6 expression showed a minor difference compared with controls.60 Thus, the functional role of NLRP6 in humans and its role as an inflammasome regulator awaits further studies.
In addition to the expression and function in intestinal epithelial cells, a recent study suggested that NLRP6 expression was apparent in intestinal inflammatory monocytes that were recruited into the intestines upon induction of chemical injury.105 Adoptive transfer of these myeloid cells from WT mice into NLRP6-deficient mice protected them from colitis and associated mortality. Monocytes from NLRP6-deficient mice featured reduced production of IL-18-dependent TNF-α, suggesting that NLRP6 enhances TNF-α production through the regulation of IL-18.105 In contrast, in an earlier study, NLRP6 was shown to suppress TNF-α in BM-derived macrophages through negative regulation of canonical NF-κBand mitogen-activated protein kinase pathways in myeloid cells.106 Consequently, in response to systemic infection with the intracellular pathogens Listeria monocytogenes and S. typhimurium, NLRP6-deficient mice presented enhanced inflammatory responses and myeloid cell recruitment, which facilitated accelerated clearance of the infectious agents.106
Although NLRP6 has emerged in recent years as a central regulator of host–microbial interactions, controversies and open questions regarding the mechanisms of action and cell type-specific activity remain to be answered. Further studies are thus required to determine the function of NLRP6 in a tissue- and cell type-specific context, and to generate a model of action for this NLR in disease regulation.
NLRP1 inflammasome
NLRP1 forms an inflammasome complex with caspase-1 upon activation by muramyl dipeptide in humans and by Bacillus anthracis toxin in mice107 (Figure 1), whereas ASC is not required for in vitro NLRP1-mediated caspase-1 activation.108 Upon DSS administration, mice deficient in NLRP1b, one of the three tandem paralogs of the human NLRP1 gene, developed exacerbated colitis compared with controls, characterized by acute distal colon inflammation and impaired epithelial cell barrier function. Treatment with antibiotics rescued the mice from disease109 and the colitogenic phenotype of DSS-treated mice was transferrable to WT mice upon co-housing with NLRP1b-deficient mice. These experiments suggest that the microbiome has a role in the pathogenesis of experimental colitis in NLRP1b-deficient mice. IL-1β and IL-18 were reduced in colon explants of NLRP1b-deficient mice compared with controls. Complementation of these cytokines during DSS treatment attenuated disease severity, hinting at their role as mediators of NLRP1b-induced protection in this model.109
NLRP1 expression was examined in human patients with colorectal cancer, and was found to be reduced in tumors as compared with adjacent healthy tissue, whereas expression levels of NLRP3 and NLRC4 were upregulated or remained unchanged, respectively.109 To clarify the role of NLRP1 in a model of inflammation-driven colon tumorigenesis, NLRP1b-deficient mice were treated with AOM-DSS. A significantly increased number of polyps and enhanced inflammation, hyperplasia, and dysplasia were observed in colons of NLRP1b-deficient mice compared with WT controls109 and the disease in these mice extended to the entire colon. As in the DSS-induced colitis model, production of IL-1β and IL-18 by colonic explants of NLRP1b-deficient mice was reduced. In addition, elevated levels of IL-6 and the anti-inflammatory cytokines IL-10 were detected. BM chimera experiments showed NLRP1b-deficient recipient mice to feature a more severe phenotype regardless of a WT or NLRP1b donor genotypes, indicating that NLRP1 acts through the non-hematopoietic compartment, probably intestinal epithelial cells, to protect the host from AOM-DSS inflicted carcinogenesis.
AIM2 inflammasome
AIM2 is an interferon-inducible cytoplasmic DNA sensor that belongs to the HIN-200 family110, 111, 112 (Figure 1). There is contradictory evidence regarding the role of AIM2 in DSS-induced colitis. Although AIM2 deficiency did not affect disease severity according to some observations,113 others proposed a protective role114, 115 with DSS treatment in AIM2-deficient mice inducing enhanced weight loss, slower recovery rate, and exacerbated histologic features compared with WT controls. This protective role of AIM2 was found to emerge from the non-hematopoietic compartment.
Pre-treatment of AIM2-deficient mice with antibiotics attenuated disease phenotype,114 suggesting that severe disease in AIM2-deficient mice may involve gut bacterial dysbiosis. Indeed, E. coli and Enterobacteriaceae were reported to be 1,000-fold higher in abundance in feces collected from AIM2-deficient mice compared with WT mice according to one study,114 whereas another study suggested that Prevotella, Bacteroides, and intestinal Bacteriodes were enriched, and the TM7 phylum was depleted in AIM2-deficient mice compared with WT mice.115 Intriguingly, co-housing of AIM2-deficient mice with WT mice induced milder colitis in the former114 and transfer of microbiome from AIM2-deficient mice into germ-free mice reproduced the sensitivity to DSS-induced colitis,114 highlighting a potential role of dysbiosis in the pathogenesis of the disease. These alterations in the microbiome assembly could be triggered by impairment in AIM2-dependent homeostatic mechanisms, such as diminished IL-18, IL-1β, and anti-microbial peptide levels, increased expression of several defensins and IL-22BP, and reduced caspase-1 activation and aberrant intestinal tissue repair seen in AIM2-deficient mice.115 Accordingly, in vivo infusion of IL-18 led to a reduction of colonic E. coli growth in mutated mice and protected them against DSS-induced colitis by generation of anti-microbial peptides.114 Moreover, intestinal epithelial cells treated with recombinant IL-18 reduced the expression of IL-22BP115
AIM2 has been also recently implicated in radiation-induced enteritis. Mice lacking AIM2 (or caspase-1) were protected from radiation-induced intestinal damage due to abrogation of caspase-1-mediated pyroptosis of epithelial cells in the intestinal crypts.116 In addition, AIM2 was shown to be critical in the maintenance of the intestinal epithelial barrier integrity against invading pathogens, as AIM2-deficient mice showed increased susceptibility to mucosal infection by S. typhimurium, but not to systemic infection by the same pathogen.117 Mutated mice exhibited exacerbated weight loss, intestinal inflammation, and increased epithelial permeability due to reduced expression of some tight-junction proteins mediated by Akt phosphorylation.117
AIM2 has been implicated in human colorectal carcinogenesis, as inactivating germ-line mutations in the AIM2 gene were characterized in tumors sampled from patients with hereditary non-polyposis colorectal cancer.118, 119 Moreover, colorectal tumor tissue was found to show reduced levels of AIM2 compared with adjacent normal tissue120 and its levels correlated with poor prognosis and tumor progression.60, 120 In mice, several studies suggested a protective role of non-hematopoietic cell-derived AIM2 in colitis-associated cancer113, 121 via mechanisms distinct from inflammasome regulation, such as excessive Akt activation,113 or aberrant Wnt signaling triggering uncontrolled proliferation of intestinal stem cells.121 This trait was exacerbated in conditions of gut dysbiosis and rescued upon microbiota transplantation from WT mice,121 showcasing the synergy between host and microbiome factors in mediating this phenotype.
NLR family apoptosis inhibitory proteins
NLR family apoptosis inhibitory proteins (NAIPs) are cytosolic receptors important for antibacterial immunity, which belong to the IAP family and are characterized by three N-terminal baculovirus inhibitor of apoptosis repeat domains (Figure 1). NAIPs recognize Gram-negative bacteria, including components of T3SS and bacterial flagella.122, 123 NAIPs and NLRC4 are homologous, and upon activation may assemble into an active inflammasome to induce caspase-1-mediated cleavage of IL-1β and IL-18, as well as pyroptosis (reviewed in Vance124). However, their role in regulating apoptosis remains unclear to date.20, 125, 126 The mouse genome contains six paralogs of the human NAIP (Naip1–6), which were shown to be mostly expressed in the colon and innate immune cells.127 Mice lacking all six NAIPs exhibited delayed tissue pathology compared with their WT counterparts when exposed to DSS.127 The former showed reduced weight loss, disease activity index, colon shortening, delayed immune cell infiltration and were protected from mucosal damage and loss of mucous-producing goblet cells.
NAIPs were found to be crucial in pathogen restriction, as NAIP1-6-deficient mice infected with S. typhimurium displayed an elevated bacterial load in their cecal mucosa compared with heterozygous littermates or WT mice. This phenotype was attributed to their impaired expulsion of infected enterocytes from the mucosa.86
Implementation of the AOM-DSS model in NAIP1-6-deficient mice showed heavier tumor burden and increased tumor size compared with WT mice, which was driven by increased activation of signal transducer and activator of transcription 3 (STAT3).127 In addition, tumor tissue isolated from NAIP1-6-deficient mice expressed pro-inflammatory cytokines, matrix metalloproteinases (MMPs), and Timp1, pertaining to tumor growth and invasion. However, no differences in IL-18 and IL-1β levels were detected both during DSS-induced colitis and in the AOM-DSS model. It was therefore postulated that NAIPs regulate STAT3 phosphorylation independent of the inflammasome axis.127 As NAIPs are expressed both in epithelial and myeloid cells, NAIPs-specific deletions were utilized to establish the niche responsible for their cancer protective properties. While deletion of NAIPs in epithelial cells (villin+) showed more severe tumor burden similar to the whole-body deficiency, deletion in the myeloid lineage (lysm+) showed a phenotype indistinguishable from WT mice, indicating that NAIPs expression in epithelial cells is responsible to its protection in the AOM-DSS model. Increased tumor burden was additionally shown in AOM without DSS, suggesting lack of correlation between tumorigenesis and inflammation in NAIP1-6 deficiency. Concordantly in humans, NAIP expression was reduced in colon adenocarcinoma compared with healthy tissue.128
NLRP12
NLRP12 possesses both inflammasome-dependent and inflammasome-independent properties, and is predominantly expressed by myeloid cells, where it has a pivotal role in gut homeostasis. Although one report showed that co-expression of NLRP12 and ASC resulted in the activation of both NF-κB and caspase-1,129 the majority of studies suggested that NLRP12 was a negative regulator of inflammation via suppression of NF-κB and ERK activation in macrophages, and did not function through inflammasome formation.
Upon DSS challenge, NLRP12 is downregulated to allow the production of the pro-inflammatory cytokines IL-1β and TNF-α, a process mediated by TLR4 triggering PR domain zinc finger protein 1 (Blimp-1) upregulation.130 NLRP12-deficient mice were found susceptible to colitis and tumorigenesis, a phenotype associated with increased production of pro-inflammatory cytokines, chemokines, and tumorigenic factors.131, 132 Although inflammation was mediated by both the hematopoietic and the non-hematopoietic compartments, tumorigenesis was chiefly mediated by the non-hematopoietic compartment.133 Interestingly, NLRP12 can be hijacked by intestinal pathogens. S. typhimurium resisted innate host defenses by activating NLRP12 and thereby suppressing NF-κB and mitogen-activated protein kinase signaling pathways, and reducing pro-inflammatory and anti-microbial molecule release, allowing the bacteria to persist in macrophages. Accordingly, NLRP12-deficient mice showed reduced bacterial burdens in response to orally administered S. typhimurium infection.134
Another prominent NLR, which negatively regulates innate immune responses, is NLRC3. Albeit not implicated in inflammasome formation, it has been recently shown to associate with phophoinositide 3-kinase and subsequently inhibit downstream mammalian target of rapamycin pathways.135 Mice lacking NLRC3 treated by AOM-DSS featured more severe colitis and aggressive tumorigenicity compared with their WT counterparts, an effect mediated primarily by epithelial cells, but also by hematopoietic cells.
Non-Canonical Inflammasome Signaling in Gastrointestinal Physiology
The non-canonical inflammasome pathway entails caspase-11 activation in mice or caspase-4 (and possibly caspase-5) activation in humans.136, 137, 138, 139 Caspase-11 senses LPS, a cell wall component of Gram-negative bacteria,139 and is activated by its lipid-A moiety138, 140 (Figure 1). Although cytosolic bacteria, such as Burkholderia thailandensis, Burkholderia pseudomallei, and cytosolic mutants of S. typhimurium and Legionella pneumophilia, could directly activate caspase-11, bacteria residing in vacuoles, such as WT Salmonella and Legionella, were none-inflammasome activating141 and required interferon-induced IRGB10 and guanylate-binding proteins for the vacuole lysis and activation of a caspase-11 inflammasome.142, 143 LPS binding to caspase-11 subsequently leads to oligomerization and activation of other caspases,137 which in turn cleave Gasdermin D to induce pyroptosis144 and activate the NLRP3 inflammasome, possibly by processing pannexin 1 and triggering potassium efflux.145 This observation was replicated in humans, as a human monocytic cell line THP1 showed LPS-induced caspase-4-mediated pyroptosis and IL-1β maturation, which was independent of TLR4. Although caspase-4 was sufficient to induce pyroptosis, IL-1β maturation relied on downstream NLRP3 inflammasome activation.146
Non-canonical inflammasome activation in intestinal inflammation
Caspase-11 was found to be protective in the DSS experimental colitis model, although the underlying mechanism remains elusive. Some studies propose that caspase-11-deficient mice were susceptible to severe colitis compared with their WT counterparts due to reduced production of IL-18 and IL-1β,147, 148 by both hematopoietic and non-hematopoietic cells.147 This impaired cytokine production led in turn to epithelial barrier dysfunction with decreased epithelial proliferation and increased cell death. Exogenous administration of recombinant IL-18 rescued the disease phenotype, highlighting its role in the maintenance of intestinal barrier integrity.148 However, other studies could not replicate the same reduction in IL-18 levels in caspase-11 deficiency.149 They rather showed that caspase-11-deficient mice feature intestinal dysbiosis, manifested by depletion in Prevotella species and enrichment in Bacteroidetes. Co-housing of mutated mice with WT mice did not alter their increased susceptibility to DSS-induced colitis, despite normalizing their microbiome assembly.149
Non-canonical inflammasome activation in infectious enteritis and colitis
Caspase-11 was shown to exert a pro-inflammatory role and mediate host defense in the gut against a large variety of pathogens.141, 150, 151, 152, 153, 154 It was found to be essential for caspase-1 activation and cytokine processing, such as IL-18 and IL-1β, in macrophages infected with E. coli, C. rodentium and Vibrio cholerae. Intriguingly, loss of caspase-11 protected mice from a lethal dose of LPS.139, 155
Mice deficient in caspase-11 showed enhanced colonization upon infection with the intracellular enteric pathogen S. typhimurium concurrent with reduced histopathological features of cecal inflammation. Similarly, knockdown of caspase-4 in human epithelial cell lines resulted in attenuated mature IL-18 secretion upon Salmonella infection and also upon extracellular pathogen infection with enteropathogenic E. coli. One proposed mechanism was diminished shedding of infected epithelial cells due to reduced pyroptosis.156 Likewise, murine macrophages manipulated to enhance ROS production through nucleotide-binding and oligomerization-domain containing 2 (NOD2) or RIP2 deficiency showed increased caspase-11 expression and activation upon C. rodentium infection, and subsequent non-canonical NLRP3 inflammasome activation. Clinically, these mice were protected from C. rodentium infection.157
Intriguingly, it should be noted that caspase-11 possesses two contradicting roles. It confers protection in chemically induced colitis, whereas, conversely, excessive activation by LPS during overwhelming infections result in septic shock.138, 140 Future studies are needed to define how environmental cues may control the pro- vs. anti-inflammatory roles of caspase-11.
Integrative Inflammasome Signaling in Gastrointestinal Physiology
As previously alluded in this review, inflammasomes often tend to act in concert to form intertwining interactions among themselves and with other PRRs of the innate immune system. The orchestration of this elaborate network of multiple activating and suppressive signals is essential to successfully produce a single net inflammatory effect during encounter of risk to the host, while avoiding activation of inflammatory arms and ensuing tissue damage by spurious signals. This integrative signaling activity is characterized by a few basic principles.
First, inflammasomes sense a wide range of signals, encompassing ubiquitous microbial motifs (e.g., muramyl dipeptide sensed by NLRP1 and double-stranded DNA sensed by AIM2) and virulence factors (e.g., T3SS sensed by NAIPs), as well as direct and indirect markers of cellular stress, such as ROS and lysosomal destabilization. This vast repertoire of ligands sensed by inflammasomes ensures efficient and sensitive surveillance to the multitude of antigens present in the intestine upon reaching the host.
Second, inflammasome activation exhibits a great deal of redundancy, as a single insult by a pathogen, or by tissue damage can activate more than one inflammasome, and often other PRRs. For example, L. monocytogenes is capable of engaging several inflammasomes (NLRP3, NLRC4, or AIM2)158 and S. typhimurium can trigger both NLRP3 and NLRC4 activation.51 This strategy not only provides backup in case one mechanism is dysfunctional or bypassed, but also fine-tunes the inflammatory response based on tissue- or cell type-specific expression of inflammasome sensors and the spatial and temporal localization of the stimulus. For instance, S. typhimurium flagellin is sensed by TLR5 when present in the extracellular compartment and by NLRC4 when intracellular,85 and NLRP3 inflammasome activation can be potentiated by simultaneous activation of TLRs by virulent bacteria, producing a rapid IRAK-1-dependent inflammatory response and pyroptosis.159
Third, inflammasomes interact with other PRRs either positively (“cross-activation”), thereby leading to signal amplification and the initiation of inflammatory responses, or negatively (“cross-inhibition”) to terminate these responses or as means of prioritizing between inflammatory signals. Cross-activation occurs, for instance, in the case of diverse ligands that constitute “signal I” for the NLRP3 inflammasome or the co-assembly of NAIP and NLRC4 to create an inflammasome complex, whereas cross-inhibition can be exemplified in the case of NLRP6 interfering with signaling pathways downstream of TLRs.160
Fourth, albeit superfluous and diverse, stimuli that trigger inflammasome activation eventually lead to common signaling transduction pathways and converge into distinct effector mechanisms, i.e., the priming of IL-1β and IL-18, and pyroptosis.
These integration principles are of utmost importance to intestinal homoeostasis, as harmful stimuli should be promptly distinguished from incidental signals coming from the commensal microbiota, thereby inciting a robust, yet contained inflammatory response in the former context, while maintaining tolerance in the later.
Inflammasome in Human Gastrointestinal Disease
Inflammasomes have a pivotal role in human immune homeostasis and mutations in inflammasome components emerged as the underlying etiologies in various mono- and multi-genic inflammatory disorders, including congenital auto-inflammatory syndromes, autoimmune and auto-inflammatory diseases, metabolic diseases, and infectious disorders. Hereditary gain-of-function mutations in the pyrin gene are associated with familial Mediterranean fever161 and gain-of-function mutations in the NLRP3 gene have been categorized under the title cryopyrin-associated periodic syndromes. These include three conditions: Muckle-Wells syndrome, familial cold autoinflammatory syndrome, and neonatal-onset multisystem inflammatory disease (NOMID) also known as chronic infantile neurologic cutaneous articular (CINCA).162 In addition, a gain-of-function mutation in NLRC4 has been linked to macrophage activation syndrome163 and, as previously discussed, neonatal-onset enterocolitis.84 Ligands sensed by the inflammasome have been suggested to invoke, contribute, or exacerbate chronic inflammatory conditions, such as gout, atherosclerosis, obesity, diabetes, and neurodegenerative diseases.102, 164, 165, 166 Furthermore, inflammasomes were suggested to participate in complex human physiological processes, such as aging167, 168 and cardiac remodeling after myocardial infection,169 as well as contributing to iatrogenic complications, such as foreign body response to implanted biomaterials.170
Inflammatory bowel disease (IBD), comprising Crohn’s disease (CD), ulcerative colitis (UC), and indeterminate colitis, is a multifactorial condition that has been suggested to be linked to a variety of nod-like receptor and inflammasome perturbations. Pioneering works showed increased IL-18 expression in epithelial and lamina propria monomuclear immune cells isolated from patients with CD, and increased production of IL-1β in colonic mucosal tissue or isolated mononuclear immune cells obtained from patients with IBD.171, 172, 173 These cytokine levels or degree of imbalance were correlated with the disease severity171, 174 and recombinant IL-18 stimulation triggered a proliferative response in intestinal mucosal lymphocytes obtained from patients with CD, but not in those extracted from healthy controls.175
Single-nucleotide polymorphisms in inflammasome-related genes have been detected in patients with IBD: NOD2, NLRP1, NLRP3, IL-1Ra,176, 177, 178, 179, 180, 181, 182 IL-18, and IL-18RAP.183 One of the highly studied polymorphisms that could potentially increase susceptibility to intestinal inflammation, IBD, and colorectal cancer involves the NOD2 gene.184, 185 The cytosolic PRR NOD2 is activated by peptidoglycan to generate a proinflammatory immune response that includes secretion of cytokines, induction of autophagy, and the production of antimicrobial peptides that contribute to the maintenance of microbiota composition.186, 187, 188 NOD2-deficient mice exhibited increased loads of commensals and altered ability to control colonizing bacteria in their terminal ilea.189, 190, 191 Moreover, mutations in the ATG16L gene, such as T300A, which are associated with elevated risk for CD, have been functionally linked to impaired autophagy, a process known to upregulate inflammasome activation.192, 193, 194 Macrophages deficient in ATG16L failed to undergo autophagy, similar to mononuclear cells derived from CD patients, and thus exhibited constitutive activation of caspase-1, increased IL-1β production, and increased response to TLR stimulation.194, 195
Some of the findings highlighted throughout this review in murine models of colitis coincide with observations in IBD patients. For instance, NLRP3-deficient mice feature reduced anti-microbial activity and altered expression of colonic defensins, which somewhat resembles similar features in CD patients.24, 196, 197 Moreover, serum samples of patients with IBD exhibited increased levels of free IL-22, a cytokine found to be associated with NLRP3 and NLRP6 inflammasomes activation during intestinal tissue damage, and postulated to mediate tumorigenesis in the recovery phase.198 Colonic biopsies obtained from patients with active UC were significantly enriched in NLRP1 compared with biopsies from healthy subjects;109 colonic mucosa samples from patients with CD showed increased expression of NLRP3 and ASC compared with healthy controls37 and samples obtained from UC patients showed a correlation between NLRP3 expression levels and disease severity.27 In addition, biopsies collected from terminal ileum of pediatric patients with IBD showed altered epithelial NLRP6 expression.199 Furthermore, ileal biopsies from patients with CD and UC revealed increased prevalence of intracellular adherent-invasive E. coli that were capable of inducing IL-1β through an NLRP3-dependent mechanism in macrophages.200 AIM2 was shown to be upregulated in epithelial cells obtained from colon biopsies of IBD patients and downregulated by anti-TNF-α therapy.201 Mutations and epigenetic silencing of the AIM2 promoter were detected in colorectal tumors119 and complete lack of AIM2 expression was associated with an up to threefold increase in mortality compared with AIM2-positive tumor samples.120 Non-canonical inflammasome activation was also implicated in human IBD. Caspase-4 and caspase-5 were upregulated in colon biopsies from patients with UC suffering from active disease compared with patients in remission; caspase-4 expression was upregulated in patients with IBD, who did not respond to the anti-TNF-α agent infliximab;147 Finally, caspase-4 and caspase-5 were upregulated in biopsies obtained from patients with colitis-associated tumors. The precise integrated roles of inflammasomes in human intestinal inflammation and tumorigenesis merit further studies.
The emerging comprehension of inflammasome-related mechanisms in the pathogenesis of intestinal inflammation may be harnessed to devise inflammasome-targeted human therapeutics. Anakinra is an IL-1 receptor antagonist widely used for the treatment of rheumatoid arthritis. However, several reports indicated that this approach was unsuccessful in IBD patients.202 IL-18 inhibition by recombinant IL-18 binding protein, on the other hand, has been proven successful in a case report of NLRC4-associated hyperinflammation.203 Other points of intervention along inflammasome activation pathways include fatty acid oxidation by NOX4, which can be inhibited by GKT137831 or VAS-2870;204 caspase-1 inhibition by pralnacasan;28 NF-κB inhibition by A20;205 and NLRP3 inhibition by glyburide.37 Treatment with small molecular compounds shown to inhibit the NLRP3 inflammasome in DSS-induced colitis models may pose candidate therapeutic agents,e.g.: rhapontin,206 MCC950,207 1-ethyl-5-methyl-2-phenyl-1H-benzo[d]imidazole (Fc11a-2),208 andrographolide,64 3-(2-Oxo-2-phenylethylidene)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (compound 1),209 asiatic acid,210 cannabinoid receptor 2 agonists,211 and celastrol.212 Similarly, colchicine demonstrated NLRP3 inhibitory properties and disease attenuation in non-steroidal anti-inflammatory drug-induced intestinal injury.213 Alternatively, targeting inflammasome ubiquitination may effectively modulate its activation status.23 Prebiotics, such as yeast cell wall mannooligosaccharide,214 or dietary modifications with omega-3 fatty acid supplementation,215 alpinetin,216, 217 and bile-acid derived compounds73, 74 may alter the activation status of the NLRP3 inflammasome. Microbiome-modulated metabolites (“postbiotics”), such as Fumigaclavine C, a fungal metabolite, may achieve NLRP3 inflammasome inhibition and attenuate colitis.218 Another potentially promising future mode of intervention includes modulating of the gut microbiome towards an immunomodulatory configuration. However, current knowledge is yet insufficiently present to reproducibly implement this promising avenue. Future approaches may include administration of probiotic agents to manipulate inflammasomes. For instance Kaposi’s sarcoma-associated herpesvirus Orf63 is a viral homolog of the human NLRP1, which can block NLRP1-dependent innate immune responses, such as caspase-1 activation and IL-1β and IL-18 processing, and thereby may serve as a future inflammasome modulatory agent.219
Conclusions and Prospects
Growing evidence implicates inflammasomes and their integrated signaling cascades as fundamentally important components contributing to both intestinal homeostasis and gut-associated physiological inflammatory responses. As such, inflammasomes, situated at a strategic interface between the sterile host and a potentially harmful microenvironment, constitute important platforms enabling the host to sense and make sense of numerous endogenous and exogenous stimuli. Using inflammasome activation signals and their downstream intercalated effector arms, the host is able to integrate complex sets of environmental stimuli into a comprehensive signal, thereby facilitating its ability to decide between tolerating a signal or initiating a potent inflammatory response against it.
Many gaps in our understanding of inflammasome signaling in the gastrointestinal tract and its physiological responses will constitute areas of exciting research in years to come. These include the elucidation of roles of multiple currently unknown nod-like receptors and their putative roles in gastrointestinal physiology and immunopathology. New inflammasome activators, including potential eukaryotic ligands, will constitute exciting new areas of research. Likewise, even in extensively studied inflammasomes such as the NLRP3 inflammasome, surprisingly little insight has been gathered on the actual molecular interactions and activation cascade that link its many modulatory ligands and the actual formation of an active multi-protein complex. Equally interesting will be the study of inflammasome disassembly and how it may be regulated by the host in order to timely terminate inflammatory responses. Similarly, growing evidence implicates posttranslational modifications in contributing to inflammasome signaling. Understanding these upstream and downstream inflammasome modulatory responses may enhance our understanding in realizing the “full picture” by which the host utilizes multiple redundant innate immune platforms to ensure a timely, self-sustained, and optimally targeted inflammatory response, while avoiding chronic inflammation, immune pathology, and their devastating complications. Similarly, the role of inflammasomes in modulating the adaptive immune response and metabolic homeostasis is only beginning to be deciphered. Studies occasionally yield contradicting results, which can derive from different experimental conditions and variations in intestinal microbiome compositions in different vivaria. Many of these seemingly contradicting responses may represent an opportunity to explore local environmental variations contributing to inflammasome-associated responses.
Moreover, in transitioning from animal modeling to human-based studies, individualized human complexity in host genetics,220, 221, 222, 223, 224, 225 the gut microbiome,226 dieting,227 and immune responses228 should be taken into account in assessing the net contributions of environmental and endogenous factors to gastrointestinal inflammasome activation in humans. Notwithstanding these challenges and limitations regarding gut-associated inflammasome signaling, its regulatory roles in controlling dietary–microbiome–host immune interactions seem to constitute an important and previously under-appreciated component in a variety of immune and immune-modulated disorders, ranging from auto-inflammatory diseases to cancer, the metabolic syndrome, and neurodegenerative disorders. Especially promising is the recent identification of inflammasome-modulating small molecules, putatively representing exciting opportunity for clinical interventions in tackling these common and currently cureless disorders.
References
Thaiss, C.A., Zmora, N., Levy, M. & Elinav, E. The microbiome and innate immunity. Nature 535, 65–74 (2016).
Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1, 135–145 (2001).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 (2004).
Duncan, J.A. et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl Acad. Sci. USA 104, 8041–8046 (2007).
Lamkanfi, M. & Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 (2012).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Lamkanfi, M. & Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Lupfer, C. & Kanneganti, T.-D. The expanding role of NLRs in antiviral immunity. Immunol. Rev. 255, 13–24 (2013).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Schroder, K. et al. The inflammasomes. Cell 140, 821–832 (2010).
Zaki, M.H., Lamkanfi, M. & Kanneganti, T.-D. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32, 171–179 (2011).
Mitoma, H. et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39, 123–135 (2013).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Murakami, T. et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl Acad. Sci. USA 109, 11282–11287 (2012).
Compan, V. et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500 (2012).
Zambetti, L.P., Laudisi, F., Licandro, G., Ricciardi-Castagnoli, P. & Mortellaro, A. The rhapsody of NLRPs: master players of inflammation...and a lot more. Immunol. Res. 53, 78–90 (2012).
Muñoz-Planillo, R. et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
He, Y., Zeng, M.Y., Yang, D., Motro, B. & Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016).
Song, H. et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 7, 13727 (2016).
Roy, N., Deveraux, Q.L., Takahashi, R., Salvesen, G.S. & Reed, J.C. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 16, 6914–6925 (1997).
Rodgers, M.A. et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 211, 1333–1347 (2014).
Lopez-Castejon, G. et al. Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J. Biol. Chem. 288, 2721–2733 (2013).
Py, B.F., Kim, M.-S., Vakifahmetoglu-Norberg, H. & Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013).
Hirota, S.A. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372 (2011).
Zaki, M.H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Allen, I.C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Itani, S. et al. NLRP3 inflammasome has a protective effect against oxazolone-induced colitis: a possible role in ulcerative colitis. Sci. Rep. 6, 39075 (2016).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Bauer, C., Duewell, P., Lehr, H.-A., Endres, S. & Schnurr, M. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig. Dis. 30 (Suppl 1), 82–90 (2012).
Siegmund, B., Lehr, H.A., Fantuzzi, G. & Dinarello, C.A. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl Acad. Sci. USA 98, 13249–13254 (2001).
Ishikura, T. et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J. Gastroenterol. Hepatol. 18, 960–969 (2003).
Becker, H.M., Apladas, A., Scharl, M., Fried, M. & Rogler, G. Probiotic Escherichia coli Nissle 1917 and commensal E. coli K12 differentially affect the inflammasome in intestinal epithelial cells. Digestion 89, 110–118 (2014).
Loher, F. et al. The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1T-cell activation. J. Pharmacol. Exp. Ther. 308, 583–590 (2004).
Bauer, C. et al. The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-α mRNA expression. Dig. Dis. Sci. 52, 1642–1652 (2007).
Siegmund, B. et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-γ and TNF-α production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R1264–R1273 (2001).
Sivakumar, P.V. et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 50, 812–820 (2002).
Liu, L. et al. The pathogenic role of NLRP3 inflammasome activation in inflammatory bowel diseases of both mice and humans. J. Crohn’s Colitis jjw219 (2016) doi:10.1093/ecco-jcc/jjw219.
Filardy, A.A., He, J., Bennink, J., Yewdell, J. & Kelsall, B.L. Posttranscriptional control of NLRP3 inflammasome activation in colonic macrophages. Mucosal Immunol. 9, 850–858 (2016).
Higashimori, A. et al. Mechanisms of NLRP3 inflammasome activation and its role in NSAID-induced enteropathy. Mucosal Immunol 9, 659–668 (2016).
Sokolovska, A. et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat. Immunol. 14, 543–553 (2013).
Seo, S.-U. et al. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 42, 744–755 (2015).
Ng, J. et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139, 542–552 552–3 (2010).
Thinwa, J., Segovia, J.A., Bose, S. & Dube, P.H. Integrin-mediated first signal for inflammasome activation in intestinal epithelial cells. J. Immunol. 193, 1373–1382 (2014).
Brodsky, I.E. et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387 (2010).
Borenshtein, D., McBee, M.E. & Schauer, D.B. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 24, 32–37 (2008).
Eckmann, L. Animal models of inflammatory bowel disease: lessons from enteric infections. Ann. N. Y. Acad. Sci. 1072, 28–38 (2006).
Mundy, R., MacDonald, T.T., Dougan, G., Frankel, G. & Wiles, S. Citrobacter rodentium of mice and man. Cell. Microbiol. 7, 1697–1706 (2005).
Song-Zhao, G.X. et al. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 7, 763–774 (2013).
Jin, L., Batra, S. & Jeyaseelan, S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J. Immunol. 198, 1253–1262 (2016).
Jong, H. K. De et al. Limited role for ASC and NLRP3 during in vivo Salmonella typhimurium infection. BMC Immunol. 15, 30 (2014).
Man, S.M. et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl Acad. Sci. USA 111, 7403–7408 (2014).
Suzuki, N. et al. Mutual reinforcement of inflammation and carcinogenesis by the Helicobacter pylori CagA oncoprotein. Sci. Rep. 5, 10024 (2015).
Engler, D.B. et al. Helicobacter pylori–specific protection against inflammatory bowel disease requires the NLRP3 inflammasome and IL-18. Inflamm. Bowel Dis. 21, 854–861 (2015).
Ng, G.Z. et al. The MUC1 mucin protects against Helicobacter pylori pathogenesis in mice by regulation of the NLRP3 inflammasome. Gut 65, 1087–1099 (2016).
Mortimer, L., Moreau, F., Cornick, S. & Chadee, K. The NLRP3 inflammasome is a pathogen sensor for invasive Entamoeba histolytica via activation of α5β1 integrin at the macrophage-amebae intercellular junction. PLoS Pathog. 11, e1004887 (2015).
Mortimer, L., Moreau, F., Cornick, S. & Chadee, K. Gal-lectin-dependent contact activates the inflammasome by invasive Entamoeba histolytica. Mucosal Immunol. 7, 829–841 (2014).
Janowski, A.M., Kolb, R., Zhang, W. & Sutterwala, F.S. Beneficial and detrimental roles of NLRs in carcinogenesis. Front. Immunol 4, 370 (2013).
Castaño-Rodríguez, N., Kaakoush, N.O., Goh, K.-L., Fock, K.M. & Mitchell, H.M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS ONE 9, e98899 (2014).
Ungerbäck, J. et al. Genetic variation and alterations of genes involved in NFκB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 33, 2126–2134 (2012).
Liu, R. et al. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget 6, 33456–33469 (2015).
Zaki, M.H., Vogel, P., Body-Malapel, M., Lamkanfi, M. & Kanneganti, T.-D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 185, 4912–4920 (2010).
Radtke, F. & Clevers, H. Self-renewal and cancer of the gut: two sides of a coin. Science 307, 1904–1909 (2005).
Du, Q. et al. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem. Pharmacol. 105, 42–54 (2016).
Guo, W. et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 10, 972–985 (2014).
Dupaul-Chicoine, J. et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43, 751–763 (2015).
Bruchard, M. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64 (2013).
Robblee, M.M. et al. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 14, 2611–2623 (2016).
Ruiz, P.A. et al. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut gutjnl-2015-310297 (2016) doi:10.1136/gutjnl-2015-310297.
Progatzky, F. et al. Dietary cholesterol directly induces acute inflammasome-dependent intestinal inflammation. Nat. Commun. 5, 5864 (2014).
Bitzer, Z.T. et al. Soy protein concentrate mitigates markers of colonic inflammation and loss of gut barrier function in vitro and in vivo. J. Nutr. Biochem. 40, 201–208 (2017).
Youm, Y.-H. et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 21, 263–269 (2015).
Wolf, A.J. et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166, 624–636 (2016).
Guo, C. et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 944 (2016).
Zhao, S. et al. Deoxycholic acid triggers NLRP3 inflammasome activation and aggravates dss-induced colitis in mice. Front. Immunol. 7, 536 (2016).
Macia, L. et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 6, 6734 (2015).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Miao, E.A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl Acad. Sci. USA 107, 3076–3080 (2010).
Miao, E.A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575 (2006).
Sutterwala, F.S. & Flavell, R.A. NLRC4/IPAF: a CARD carrying member of the NLR family. Clin. Immunol. 130, 2–6 (2009).
Sadasivam, S. et al. Caspase-1 activator Ipaf is a p53-inducible gene involved in apoptosis. Oncogene 24, 627–636 (2005).
Thalappilly, S., Sadasivam, S., Radha, V. & Swarup, G. Involvement of caspase 1 and its activator Ipaf upstream of mitochondrial events in apoptosis. FEBS J. 273, 2766–2778 (2006).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl Acad. Sci. USA 107, 21635–21640 (2010).
Carvalho, F.A. et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 5, 288–298 (2012).
Romberg, N. et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 46, 1135–1139 (2014).
Franchi, L. et al. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 13, 449–456 (2012).
Sellin, M.E. et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict salmonella replication in the intestinal mucosa. Cell Host Microbe 16, 237–248 (2014).
Deng, W. et al. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl Acad. Sci. USA 101, 3597–3602 (2004).
Liu, Z. et al. Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 287, 16955–16964 (2012).
Nordlander, S., Pott, J. & Maloy, K.J. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 7, 775–785 (2013).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Normand, S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl Acad. Sci. USA 108, 9601–9606 (2011).
Gremel, G. et al. The human gastrointestinal tract-specific transcriptome and proteome as defined by RNA sequencing and antibody-based profiling. J. Gastroenterol. 50, 46–57 (2015).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Birchenough, G.M.H., Nyström, E.E.L., Johansson, M.E.V. & Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542 (2016).
Kempster, S.L. et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G253–G263 (2011).
Wang, P. et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830 (2015).
Levy, M. et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 (2015).
Chudnovskiy, A. et al. Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 167, 444–456.e14 (2016).
Sun, Y. et al. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology 144, 1478–1487 1487–8 (2013).
Hooper, L.V., Stappenbeck, T.S., Hong, C.V. & Gordon, J.I. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 4, 269–273 (2003).
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Chen, G.Y., Liu, M., Wang, F., Bertin, J. & Núñez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 186, 7187–7194 (2011).
Mehta, R. et al. Expression of NALPs in adipose and the fibrotic progression of non-alcoholic fatty liver disease in obese subjects. BMC Gastroenterol. 14, 208 (2014).
Seregin, S.S. et al. NLRP6 function in inflammatory monocytes reduces susceptibility to chemically induced intestinal injury. Mucosal Immunol. 10, 434–445 (2016).
Anand, P.K. et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393 (2012).
Nour, A.M. et al. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect. Immun. 77, 1262–1271 (2009).
Faustin, B. et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724 (2007).
Williams, T.M. et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunol. 194, 3369–3380 (2015).
Bürckstümmer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Wilson, J.E. et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 21, 906–913 (2015).
Hu, S. et al. The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep. 13, 1922–1936 (2015).
Ratsimandresy, R.A., Indramohan, M., Dorfleutner, A. & Stehlik, C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell. Mol. Immunol. 14, 127–142 (2016).
Hu, B. et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768 (2016).
Hu, G.-Q. et al. AIM2 contributes to the maintenance of intestinal integrity via Akt and protects against Salmonella mucosal infection. Mucosal Immunol. 9, 1330–1339 (2016).
Schulmann, K. et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology 128, 590–599 (2005).
Woerner, S.M. et al. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosomes Cancer 46, 1080–1089 (2007).
Dihlmann, S. et al. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int. J. Cancer 135, 2387–2396 (2014).
Man, S.M. et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 162, 45–58 (2015).
Zhang, L. et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409 (2015).
Hu, Z. et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404 (2015).
Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 32, 84–89 (2015).
Maier, J.K.X. et al. The neuronal apoptosis inhibitory protein is a direct inhibitor of caspases 3 and 7. J. Neurosci. 22, 2035–2043 (2002).
Davoodi, J., Ghahremani, M.-H., Es-Haghi, A., Mohammad-Gholi, A. & Mackenzie, A. Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int. J. Biochem. Cell Biol. 42, 958–964 (2010).
Allam, R. et al. Epithelial NAIPs protect against colonic tumorigenesis. J. Exp. Med. 212, 369–383 (2015).
Endo, T. et al. Expression of IAP family proteins in colon cancers from patients with different age groups. Cancer Immunol. Immunother. 53, 770–776 (2004).
Wang, L. et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J. Biol. Chem. 277, 29874–29880 (2002).
Shi, F. et al. Inflammasome-independent role of NLRP12 in suppressing colonic inflammation regulated by Blimp-1. Oncotarget 7, 30575–30584 (2016).
Tuncer, S., Fiorillo, M.T. & Sorrentino, R. The multifaceted nature of NLRP12. J. Leukoc. Biol. 96, 991–1000 (2014).
Zaki, M.H. et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 (2011).
Allen, I.C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36, 742–754 (2012).
Zaki, M.H., Man, S.M., Vogel, P., Lamkanfi, M. & Kanneganti, T.-D. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc. Natl Acad. Sci. USA 111, 385–390 (2014).
Karki, R. et al. NLRC3 is an inhibitory sensor of PI3K–mTOR pathways in cancer. Nature 540, 583–587 (2016).
Vanaja, S.K., Rathinam, V.A.K. & Fitzgerald, K.A. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 25, 308–315 (2015).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Hagar, J.A., Powell, D.A., Aachoui, Y., Ernst, R.K. & Miao, E.A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Aachoui, Y. et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978 (2013).
Man, S.M. et al. IRGB10 liberates bacterial ligands for sensing by the AIM2 and Caspase-11-NLRP3 inflammasomes. Cell 167, 382–396.e17 (2016).
Meunier, E. et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509, 366–370 (2014).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Rühl, S. & Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 45, 2927–2936 (2015).
Schmid-Burgk, J.L. et al. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 45, 2911–2917 (2015).
Williams, T.M. et al. Caspase-11 attenuates gastrointestinal inflammation and experimental colitis pathogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G139–G150 (2015).
Oficjalska, K. et al. Protective role for caspase-11 during acute experimental murine colitis. J. Immunol. 194, 1252–1260 (2015).
Demon, D. et al. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 7, 1480–1491 (2014).
Case, C.L. et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl Acad. Sci. USA 110, 1851–1856 (2013).
Broz, P. et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291 (2012).
Gabrielli, E. et al. Induction of caspase-11 by aspartyl proteinases of Candida albicans and implication in promoting inflammatory response. Infect. Immun. 83, 1940–1948 (2015).
Kobayashi, T. et al. The Shigella OspC3 effector inhibits Caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13, 570–583 (2013).
Coutermarsh-Ott, S.L. et al. Caspase-11 modulates inflammation and attenuates Toxoplasma gondii pathogenesis. Mediators Inflamm. 2016, 9848263 (2016).
Pallett, M.A. et al. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol. (2016) doi:10.1038/mi.2016.77.
Knodler, L.A. et al. Noncanonical inflammasome activation of Caspase-4/Caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16, 249–256 (2014).
Lupfer, C.R. et al. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog. 10, e1004410 (2014).
Franchi, L., Muñoz-Planillo, R. & Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 (2012).
Lin, K.-M. et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl Acad. Sci. USA 111, 775–780 (2014).
Elinav, E., Henao-Mejia, J. & Flavell, R.A. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol. 6, 4–13 (2013).
Chae, J.J. et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity 34, 755–768 (2011).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Canna, S.W. et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 46, 1140–1146 (2014).
Sheedy, F.J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820 (2013).
Rainone, V. et al. Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. Int. J. Obes. (Lond) 40, 1026–1033 (2016).
Schroder, K., Zhou, R. & Tschopp, J. The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300 (2010).
Youm, Y.-H. et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 1, 56–68 (2012).
Youm, Y.-H. et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 18, 519–532 (2013).
Mezzaroma, E. et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl Acad. Sci. USA 108, 19725–19730 (2011).
Malik, A.F. et al. Inflammasome components Asc and caspase-1 mediate biomaterial-induced inflammation and foreign body response. Proc. Natl Acad. Sci. USA 108, 20095–20100 (2011).
Pizarro, T.T. et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J. Immunol. 162, 6829–6835 (1999).
Ligumsky, M., Simon, P.L., Karmeli, F. & Rachmilewitz, D. Role of interleukin 1 in inflammatory bowel disease—enhanced production during active disease. Gut 31, 686–689 (1990).
Mahida, Y.R., Wu, K. & Jewell, D.P. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut 30, 835–838 (1989).
Casini-Raggi, V. et al. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J. Immunol. 154, 2434–2440 (1995).
Kanai, T. et al. Interleukin 18 is a potent proliferative factor for intestinal mucosal lymphocytes in Crohn’s disease. Gastroenterology 119, 1514–1523 (2000).
Cummings, J.R.F. et al. The genetics of NOD-like receptors in Crohn’s disease. Tissue Antigens 76, 48–56 (2010).
Iudicibus, S.De. et al. Genetic predictors of glucocorticoid response in pediatric patients with inflammatory bowel diseases. J. Clin. Gastroenterol. 45, e1–e7 (2011).
Yamamoto-Furusho, J.K. et al. Interleukin 1 β (IL-1B) and IL-1 antagonist receptor (IL-1RN) gene polymorphisms are associated with the genetic susceptibility and steroid dependence in patients with ulcerative colitis. J. Clin. Gastroenterol. 45, 531–535 (2011).
Tountas, N.A. et al. Functional and ethnic association of allele 2 of the interleukin-1 receptor antagonist gene in ulcerative colitis. Gastroenterology 117, 806–813 (1999).
Villani, A.-C. et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 41, 71–76 (2009).
Schoultz, I. et al. Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn’s disease in Swedish men. Am. J. Gastroenterol. 104, 1180–1188 (2009).
Roberts, R.L. et al. Evidence of interaction of CARD8 rs2043211 with NALP3 rs35829419 in Crohn’s disease. Genes Immun. 11, 351–356 (2010).
Thompson, S.R. & Humphries, S.E. Interleukin-18 genetics and inflammatory disease susceptibility. Genes Immun. 8, 91–99 (2007).
Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 8, 458–466 (2008).
van Heel, D.A. et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet (London, England) 365, 1794–1796 (2005).
Philpott, D.J., Sorbara, M.T., Robertson, S.J., Croitoru, K. & Girardin, S.E. NOD proteins: regulators of inflammation in health and disease. Nat. Rev. Immunol. 14, 9–23 (2014).
Grimes, C.L., Ariyananda, L.D.Z., Melnyk, J.E. & O’Shea, E.K. The innate immune protein nod2 binds directly to MDP, a bacterial cell wall fragment. J. Am. Chem. Soc. 134, 13535–13537 (2012).
Vaishnava, S., Behrendt, C.L., Ismail, A.S., Eckmann, L. & Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl Acad. Sci. USA 105, 20858–20863 (2008).
Ramanan, D., Tang, M.S., Bowcutt, R., Loke, P. & Cadwell, K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 41, 311–324 (2014).
Petnicki-Ocwieja, T. et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl Acad. Sci. USA 106, 15813–15818 (2009).
Kobayashi, K.S. et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005).
Rioux, J.D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 (2007).
Hampe, J. et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211 (2007).
Lassen, K.G. et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl Acad. Sci. USA 111, 7741–7746 (2014).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008).
Simms, L.A. et al. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut 57, 903–910 (2008).
Wehkamp, J. et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 53, 1658–1664 (2004).
Huber, S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012).
Alipour, M. et al. Mucosal barrier depletion and loss of bacterial diversity are primary abnormalities in paediatric ulcerative colitis. J. Crohn’s Colitis 10, 462–471 (2016).
la Fuente, M.De. et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int. J. Med. Microbiol. 304, 384–392 (2014).
Vanhove, W. et al. Strong upregulation of AIM2 and IFI16 inflammasomes in the mucosa of patients with active inflammatory bowel disease. Inflamm. Bowel Dis. 21, 2673–2682 (2015).
Carter, J.D., Valeriano, J. & Vasey, F.B. Crohn disease worsened by anakinra administration. J. Clin. Rheumatol. 9, 276–277 (2003).
Canna, S.W. et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. pii: S0091-6749(16)31352-5 (2016) doi:10.1016/j.jaci.2016.10.022.
Moon, J.-S. et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 22, 1002–1012 (2016).
Duong, B.H. et al. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 42, 55–67 (2015).
Wei, W. et al. Rhapontin ameliorates colonic epithelial dysfunction in experimental colitis through SIRT1 signaling. Int. Immunopharmacol. 42, 185–194 (2017).
Coll, R.C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 (2015).
Liu, W. et al. A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem. Pharmacol. 85, 1504–1512 (2013).
Wang, Y. et al. 3-(2-Oxo-2-phenylethylidene)-2,3,6,7-tetrahydro-1H-pyrazino[2,1-a]isoquinolin-4(11bH)-one (compound 1), a novel potent Nrf2/ARE inducer, protects against DSS-induced colitis via inhibiting NLRP3 inflammasome. Biochem. Pharmacol. 101, 71–86 (2016).
Guo, W. et al. Asiatic acid ameliorates dextran sulfate sodium-induced murine experimental colitis via suppressing mitochondria-mediated NLRP3 inflammasome activation. Int. Immunopharmacol. 24, 232–238 (2015).
Ke, P. et al. Activation of cannabinoid receptor 2 ameliorates DSS-induced colitis through inhibiting nlrp3 inflammasome in macrophages. PLoS ONE 11, e0155076 (2016).
Shaker, M.E., Ashamallah, S.A. & Houssen, M.E. Celastrol ameliorates murine colitis via modulating oxidative stress, inflammatory cytokines and intestinal homeostasis. Chem. Biol. Interact. 210, 26–33 (2014).
Otani, K. et al. Colchicine prevents NSAID-induced small intestinal injury by inhibiting activation of the NLRP3 inflammasome. Sci. Rep. 6, 32587 (2016).
Ferenczi, S., Szegi, K., Winkler, Z., Barna, T. & Kovács, K.J. Oligomannan prebiotic attenuates immunological, clinical and behavioral symptoms in mouse model of inflammatory bowel disease. Sci. Rep. 6, 34132 (2016).
Yan, Y. et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38, 1154–1163 (2013).
He, X. et al. Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci. Rep. 6, 28370 (2016).
Márquez-Flores, Y.K., Villegas, I., Cárdeno, A., Rosillo, M.Á. & Alarcón-de-la-Lastra, C. Apigenin supplementation protects the development of dextran sulfate sodium-induced murine experimental colitis by inhibiting canonical and non-canonical inflammasome signaling pathways. J. Nutr. Biochem. 30, 143–152 (2016).
Guo, W. et al. Fumigaclavine C ameliorates dextran sulfate sodium-induced murine experimental colitis via NLRP3 inflammasome inhibition. J. Pharmacol. Sci. 129, 101–106 (2015).
Gregory, S.M. et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334 (2011).
Li, Y. et al. A functional genomics approach to understand variation in cytokine production in humans. Cell 167, 1099–1110.e14 (2016).
Lee, M.N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980–1246980 (2014).
Kumar, V., Wijmenga, C. & Xavier, R.J. Genetics of immune-mediated disorders: from genome-wide association to molecular mechanism. Curr. Opin. Immunol. 31, 51–57 (2014).
Li, Y. et al. Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat. Med. 22, 952–960 (2016).
Netea, M.G., Wijmenga, C. & O’Neill, L.A.J. Genetic variation in Toll-like receptors and disease susceptibility. Nat. Immunol. 13, 535–542 (2012).
Aguirre-Gamboa, R. et al. Differential effects of environmental and genetic factors on T and B cell immune traits. Cell Rep. 17, 2474–2487 (2016).
Ahern, P.P., Faith, J.J. & Gordon, J.I. Mining the human gut microbiota for effector strains that shape the immune system. Immunity 40, 815–823 (2014).
Zeevi, D. et al. Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1094 (2015).
Horst, R.T. et al. Host and environmental factors influencing individual human cytokine responses. Cell 167, 1111–1124.e13 (2016).
Acknowledgements
We thank the members of the Elinav lab for discussions and apologize for authors whose work was not cited because of space constraints. N.Z. is supported by the Gilead Sciences International Research Scholars Program in Liver Disease. E.E. is supported by Y. and R. Ungar, the Abisch Frenkel Foundation for the Promotion of Life Sciences, the Gurwin Family Fund for Scientific Research, the Leona M. and Harry B. Helmsley Charitable Trust, the Crown Endowment Fund for Immunological Research, the estate of J. Gitlitz, the estate of L. Hershkovich, the Benoziyo Endowment Fund for the Advancement of Science, the Adelis Foundation, J.L. and V. Schwartz, A. and G. Markovitz, A. and C. Adelson, the French National Center for Scientific Research (CNRS), D.L. Schwarz, the V.R. Schwartz Research Fellow Chair, L. Steinberg, J.N. Halpern, A. Edelheit, and by grants funded by the European Research Council, a Marie Curie Integration grant, the German-Israeli Foundation for Scientific Research and Development, the Israel Science Foundation, the Minerva Foundation, the Rising Tide Foundation, the Helmholtz Foundation, and the European Foundation for the Study of Diabetes. E.E. is the incumbent of the Rina Gudinski Career Development Chair and a senior fellow of the Canadian Institute For Advanced Research (CIFAR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Zmora, N., Levy, M., Pevsner-Fishcer, M. et al. Inflammasomes and intestinal inflammation. Mucosal Immunol 10, 865–883 (2017). https://doi.org/10.1038/mi.2017.19
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.19
This article is cited by
-
Decreased expression of the NLRP6 inflammasome is associated with increased intestinal permeability and inflammation in obesity with type 2 diabetes
Cellular and Molecular Life Sciences (2024)
-
Curcumin attenuates aflatoxin B1-induced ileum injury in ducks by inhibiting NLRP3 inflammasome and regulating TLR4/NF-κB signaling pathway
Mycotoxin Research (2024)
-
Diesel Exhaust Particle (DEP)-induced glucose intolerance is driven by an intestinal innate immune response and NLRP3 activation in mice
Particle and Fibre Toxicology (2023)
-
The deposition of lanthanum carbonate may activate macrophages to induce gastrointestinal mucosal injury in patients with chronic kidney disease: an in vitro caco-2/THP-1 macrophage coculture model study
JBIC Journal of Biological Inorganic Chemistry (2023)
-
The metabolic nature of inflammatory bowel diseases
Nature Reviews Gastroenterology & Hepatology (2022)
