
Abstract
Despite the recent description of the mucosal vaccine-induced reduction of Helicobacter pylori natural infection in a phase 3 clinical trial, the absence of immune correlates of protection slows the final development of the vaccine. In this study, we evaluated the role of interleukin (IL)-22 in mucosal vaccine-induced protection. Gastric IL-22 levels were increased in mice intranasally immunized with urease+cholera toxin and challenged with H. felis, as compared with controls. Flow cytometry analysis showed that a peak of CD4+IL-22+IL-17+ T cells infiltrating the gastric mucosa occurred in immunized mice in contrast to control mice. The inhibition of the IL-22 biological activity prevented the vaccine-induced reduction of H. pylori infection. Remarkably, anti-microbial peptides (AMPs) extracted from the stomachs of vaccinated mice, but not from the stomachs of non-immunized or immunized mice, injected with anti-IL-22 antibodies efficiently killed H. pylori in vitro. Finally, H. pylori infection in vaccinated RegIIIβ-deficient mice was not reduced as efficiently as in wild-type mice. These results demonstrate that IL-22 has a critical role in vaccine-induced protection, by promoting the expression of AMPs, such as RegIIIβ, capable of killing Helicobacter. Therefore, it can be concluded that urease-specific memory Th17/Th22 cells could constitute immune correlates of vaccine protection in humans.
Similar content being viewed by others


Introduction
The Gram-negative bacterium Helicobacter pylori infects half of the world’s population.1 Although most infected individuals develop asymptomatic chronic active gastritis, H. pylori infection may also cause complications such as gastroduodenal ulcers and increases the risk of developing gastric malignancies.1 Vaccine is an attractive approach to controlling H. pylori infections worldwide. Recently, Zeng and colleagues2 provided evidence that oral administration of H. pylori urease B subunit fused to Escherichia coli heat-labile toxin B subunit significantly reduced the acquisition of natural H. pylori infection in children. Although this study provides the proof of concept for vaccine-induced mucosal protection against H. pylori infection, much work remains to be done to increase the level of protection and to clearly define immunological correlates of protection. Indeed, whereas the vaccine protected more than 70% of recipients from infections in the first year of vaccination, the levels of protection were less than 56% in the second and third years after vaccination.2, 3 Moreover, although several preclinical studies clearly demonstrated that the Helicobacter-specific CD4+ T-cell response is key in vaccine protection,4, 5 no CD4+ T-cell responses were evaluated in this clinical trial to clearly identify immune correlates of protection.2 Running new clinical trials without defined immunological correlates of protection is extremely risky. Our aim in this study was to gain a better understanding of the urease-specific CD4+ T-cell response leading to vaccine-induced protection in mice to identify potential immune correlates of vaccine protection.
Recently, the production of interleukin (IL)-17 by Th17 cells has been identified as a player in vaccine-induced H. pylori clearance in some circumstances6, 7 but not in others.8 IL-17 is known to be linked to neutrophil recruitment and activation through the induction of granulocyte colony-stimulating factor and IL-87 mediating resistance against extracellular microbial infections.6, 7 In mice, Th17 cells are a major source of IL-22.9 By eliciting various innate defensive mechanisms from epithelial cells, IL-22 is essential for host defense against extracellular pathogens, such as bacteria and yeast, at mucosal surfaces.9 The functions of IL-22 during host defense against extracellular pathogens can be categorized within three major areas. First, by promoting epithelial proliferation, IL-22 helps to maintain and restore the integrity of the epithelial barrier function during the invasion of pathogens.9 Second, IL-22—in part synergistically with other cytokines, such as IL-17 or tumor necrosis factor-α—induces the expression of antimicrobial peptides (AMPs) involved in host defense in the skin, the airways, and the intestine.9 For example, IL-22 induces the expression of S100A7, S100A8, S100A9, β-defensin-2, and β-defensin-3 in the skin.9 IL-22 also promotes the release of regenerating islet-derived protein IIIβ (RegIIIβ) and RegIIIγ from intestinal epithelial cells during Citrobacter rodentium infection in the colon.10 Lastly, IL-22 promotes the production of inflammatory mediators, such as IL-6, granulocyte colony-stimulating factor, IL-1β, serum amyloid A, and lipopolysaccharide-binding protein.9 For instance, IL-22 is essential for the release of chemokines such as CXCL1, CXCL5, and CXCL9, as well as IL-6 and granulocyte colony-stimulating factor from airway epithelial cells during Klebsiella pneumoniae infection.11 Considering the functions of IL-22 in the control of mucosal infections and the role of Th17 cells, a major source of IL-22, in the vaccine-induced reduction of Helicobacter infection, we set out to investigate the role of IL-22 in the reduction of Helicobacter infection following immunization.
Results
IL-22 secreting CD4+ T cells infiltrate the gastric mucosa during vaccine-induced protection
In order to determine whether IL-22 has a role in vaccine-induced protection, we first evaluated whether the expression of IL-22 is increased in the stomach mucosa of immunized mice on days 4 and 5 post infection. Indeed, on days 4 and 5 post infection, vaccinated mice demonstrated a clear decrease of H. felis colonization in their stomach mucosa (Figure 1a).12 As compared with control mice, high IL-22 mRNA expression was measured in the stomach mucosa of immunized mice on days 4 and 5 post infection (Figure 1b, left panel). On day 5 post infection, IL-22 protein levels increased significantly in the stomach mucosa of immunized mice (Figure 1b, right panel).

Vaccine-induced reduction of H. felis infection is associated with high levels of IL-22 in the gastric mucosa. BALB/c WT mice were either vaccinated with urease and CT (vaccinated) or administered CT alone as control (control). Two weeks after vaccination, mice were challenged with H. felis. On days 4 and 5 post infection, vaccinated mice demonstrated a clear decrease of H. felis colonization in their stomach mucosa (a). On day 4 post bacterial challenge, a large increase of IL-22 mRNA expression was observed in the gastric mucosa of immunized mice as compared with controls (b, left panel). This was corroborated on day 5 post bacterial challenge by increased levels of IL-22 protein in the gastric samples of vaccinated mice, as compared with controls (b, right panel). On day 5 post bacterial challenge, increase of IL-22-dependent genes (CXCL2, S100A9, and RegIIIβ) mRNA expression was observed in the gastric mucosa of vaccinated mice as compared with controls (c). Detection of RegIIIβ by immunohistochemical staining on day 5 post bacterial challenge. Stomach tissue sections of vaccinated and infected mice were first stained with sheep anti-RegIIIβ or an irrelevant sheep antibody (sheep IgG) followed by biotinylated rabbit anti-sheep and avidin HRP (d). Pictures are representative of data obtained from five individual vaccinated and infected mice. Bars: 50 μm. The increase of RegIIIβ mRNA expression correlated with the decrease of bacterial load on day 5 post bacterial challenge (e). Lymphoid cells infiltrating the stomach mucosa were isolated for detection of IL-22-producing CD4+ T cells (upper panels) or IL-17- and IL-22-producing CD4+ T cells (lower panels) in CD45+CD3+ cells by flow cytometry analysis (f). On day 5 post bacterial challenge, the percentages and numbers of IL-22+CD4+ T cells and IL-17+IL-22+CD4+ T cells (g) infiltrating the stomach of vaccinated mice were higher in comparison with control mice. Isolated stomach mononuclear cells were stimulated with urease for 5 days in vitro. IL-22 protein concentration was measured in the cell supernatant by ELISA. We observed that mononuclear cells from vaccinated mice stimulated with urease produced higher IL-22 levels than mononuclear cells from control mice (h). Plots are representative of at least two independent experiments (n=6–8 mice per group), each dot represents a mouse. *P<0.05, **P<0.01. CT, cholera toxin; ELISA, enzyme-linked immunosorbent assay; HRP, horseradish peroxidase; IL, interleukin; WT, wild type.
In order to determine whether the upregulation of IL-22 expression levels modulates the antimicrobial defenses of the stomach mucosa, we measured the mRNA expression levels of several IL-22-induced genes. We observed that, on day 5 post infection, CXCL2, S100A9, and RegIIIβ mRNA expressions were upregulated in the stomach mucosa of immunized mice as compared with control mice (Figure 1c). The immunohistochemical staining of RegIIIβ in the stomach mucosa confirmed that gastric epithelial cells of immunized mice upregulated AMPs as compared with control mice on day 5 post infection (Figure 1d). Remarkably, the increase of RegIIIβ mRNA expression correlated with the decrease of bacterial load (Figure 1e). Unfortunately, we did not obtain the same correlation with immunohistochemical staining. Indeed, although we could detect H. felis and RegIIIβ on the same tissue section (data not shown), it was not possible to correlate the intensity of RegIIIβ staining with the absence or presence of H. felis within each gland of every mouse. This absence of clear correlation between RegIIIβ staining and H. felis infection is probably linked to the limits of immunohistochemical techniques and/or to the kinetic/sensitivity of RegIIIβ-induced bacterial killing and/or to the biology of H. felis infection. Collectively, these results show that during the vaccine-induced protection, IL-22 is produced and associated with the upregulation of AMP expression by the stomach epithelial cells.
We sought to determine whether the CD4+ T-cell population infiltrating the gastric mucosa during vaccine-induced reduction of H. pylori infection6, 12 was the source of IL-22. To this end, we isolated gastric lymphoid cells and evaluated the production of IL-22 in the CD4+ T-cell population by flow cytometry. Figure 1f (upper panels) and Figure 1g show that on day 5 post bacterial challenge, there was an increased proportion of gastric CD4+ T cells in immunized animals that produced IL-22 as compared with controls (2.06±0.60% and 0.79±0.26%, P<0.01, respectively). Time course analysis revealed that gastric CD4+IL-22+ T cells could be detected in immunized mice on day 3, peaking at day 5 (Supplementary Figure S1A online). On day 10, the CD4+IL-22+ T cells were still present in high numbers in immunized mice, whereas in control mice, the CD4+IL-22+ T cells were at the limits of detection (Supplementary Figure S1).
IL-22 is produced by a wide variety of cells belonging to the adaptive or the innate immune system.9 As we previously demonstrated that during vaccine-induced reduction of H. pylori infection, Th17 cells infiltrate the stomach mucosa and have a role in the reduction of H. pylori infection,6, 7 we first checked whether IL-22 is produced by Th17. We observed that on day 5 post bacterial challenge, a large proportion of CD4+IL-22+ T cells produced IL-17 (Figure 1f (lower panels) and Figure 1g). It has to be noted that a small but detectable number of CD4+IL-22+ T cells do not produce IL-17, these cells might be either Th22, Th1, and/or immature and/or exhausted Th17 (Figure 1f (lower panels)). Time course studies revealed that gastric CD4+IL-17+IL-22+ T cells could already be detected in immunized mice on day 3 (Supplementary Figure S1B).
In addition, we did not detect any significant IL-22 production by gastric CD45+CD3−CD4−CD5− or CD45+CD3−CD4+CD5− cells in immunized or control mice on day 5 post infection (Supplementary Figure S1C). These results suggest that cells of the innate immune system do not significantly contribute to the IL-22 production during the vaccine-induced protection. Lastly, in order to confirm that the IL-22 is produced by Helicobacter-specific CD4+ T cells, we isolated stomach lymphoid cells on day 5 post infection and stimulated these cells with urease (the vaccine antigen). Figure 1h shows that only immune cells isolated from the stomach mucosa of immunized and infected mice stimulated with urease produced IL-22.
Collectively, these results show that a gastric infiltration of Helicobacter-specific CD4+IL-22+ T cells (mainly of Th17 origin) develops as a result of immunization and challenge. The temporal association of the infiltration of the gastric mucosa by IL-22+CD4+ T cells with the reduction of H. felis infection implicates IL-22 in the elimination of the bacteria from the stomachs of immunized mice.
IL-22 deficiency precludes vaccine-induced protection
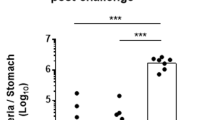
In order to determine whether IL-22 has a role in the reduction of bacterial load, we blocked the biological activity of IL-22 in vaccinated mice. In comparison with vaccinated mice injected with control antibodies, vaccinated mice injected with anti-IL-22 monoclonal antibodies13 still had high levels of H. felis infection on day 5 post infection (Figure 2a). Similarly, vaccinated mice injected with anti-IL-22 antibodies still had high levels of H. pylori infection on days 7 (Figure 2b, left and middle panels showing two independent experiments) and 10 post infection (Figure 2c). Of note, the injection of anti-IL-22 antibodies to control mice did not increase H. felis or H. pylori infection (Figure 2a, b (left and middle panels) and Figure 2c). In order to validate this experiment, we vaccinated and infected IL-22-deficient mice. In a first step, we measured serum antibody responses in wild-type (WT) and IL-22-deficient mice and found that both groups of mice were able to mount an antibody response against urease (Supplementary Figure S2A). In addition, splenocytes of both groups of mice proliferated at similar levels when exposed to urease (Supplementary Figure S2B). Taken together, these results suggest that the immunization protocol was efficient at promoting urease-specific CD4+ T-cell responses in both strains of mice. However, following immunization, although WT mice reduced H. felis and H. pylori infections, IL-22-deficient mice still had high levels of H. felis and H. pylori infections at the time they were killed (Figure 2a, b (right panel)).

IL-22 deficiency precludes vaccine-induced reduction of H. pylori infection. BALB/c WT or IL-22-deficient mice were either vaccinated with urease and CT (Vaccinated) or administered CT alone (Control). Two weeks after vaccination, mice were challenged with H. felis (a) or H. pylori (b and c). Vaccinated mice were intraperitoneally injected with 200 μg of anti-IL-22-neutralizing monoclonal antibody (IgG2a) or with purified non-specific rat IgG2a monoclonal antibody at days 1 and 3 after H. felis challenge or at days 1, 3, 5, 7, and 9 after H. pylori challenge. Mice were killed on days 5, 7, or 10 post infection and the stomachs were recovered for detection of Helicobacter infection either by qPCR (a) or the numeration of CFU (b and c). IL-22Fc therapy decreases H. felis and H. pylori infection. Three weeks after infection with H. felis or H. pylori, 50 μg of IL-22Fc fusion protein (provided by Genentech) were intraperitoneally injected on days 0 and 3 and killed on day 5. As control, infected mice received vehicle, a PBS solution. At the time the mice were killed, the stomachs were recovered for detection of Helicobacter infection either by qPCR (d) or the numeration of CFU (e). Each dot represents a mouse. *P<0.05, **P<0.01. CFU, colony-forming unit; CT, cholera toxin; IL, interleukin; n.s., not significant; PBS, phosphate-buffered saline; WT, wild type.
Collectively, these data demonstrate that IL-22 has a role in vaccine-induced protection.
IL-22 therapy decreases Helicobacter infection
As IL-22 deficiency precludes vaccine-induced reduction of H. pylori infection, we tested whether IL-22 injection can promote the reduction of Helicobacter infection in non-immunized mice. To this end, we injected the IL-22Fc fusion protein to chronically infected mice. The fusion of IL-22 to the Fc fragment of immunoglobulins can dramatically increase the half life of the molecule in vivo and therefore potentialize the therapeutic effect of IL-22.14 Moreover, the injection of IL-22Fc fusion protein has already been shown to confer protection to mice inoculated with the gut pathogen C. rodentium.14 Three weeks after infection with H. felis or H. pylori, 50 μg of IL-22Fc fusion protein was injected on days 0 and 3 and the mice were killed on day 5. As a control, infected mice received vehicle (phosphate-buffered saline). Remarkably, mice injected with IL-22Fc fusion protein were less infected than control mice (Figure 2d, e).
The reduction of Helicobacter infection is not dependent on the pro-inflammatory properties of IL-22
Because IL-22 is associated with chronic inflammatory diseases like psoriasis,9 we evaluated whether the pro-inflammatory properties of IL-22 have a role in the vaccine or IL-22Fc-induced reduction of H. felis infection. Figure 3a shows that the injection of anti-IL-22 antibodies did not decrease the levels of inflammatory cytokines/chemokines produced within the stomach mucosa of vaccinated mice on day 5 post bacterial infection. In addition, the injection of anti-IL-22 antibodies did not decrease infiltration of gastric inflammatory cells in vaccinated WT mice on day 5 post H. felis infection (Figure 3b, c). Moreover, vaccinated IL-22-deficient mice on day 5 post H. felis infection demonstrated gastric inflammatory cells infiltration similar to that of their WT counterparts (Figure 3c and Supplementary Figure S2C (upper panels)). In line with these observations, the injection of IL-22Fc fusion protein to chronically H. felis-infected mice did not promote the infiltration of inflammatory cells or increase inflammatory markers within the stomach mucosa (Figure 3c, Supplementary Figure S2C (lower panels) and Figure 3d). Collectively, these data show that the pro-inflammatory properties of IL-22 are dispensable for the reduction of H. felis infection.

The reduction of Helicobacter infection is not dependent on pro-inflammatory properties of IL-22. BALB/c WT mice were either vaccinated with urease and CT (Vaccinated) or administered CT alone (Control). Two weeks after vaccination, mice were injected with 200 μg of anti-IL-22-neutralizing monoclonal antibody (rat IgG2a) or with purified non-specific rat IgG2a monoclonal antibody at days 1 and 3 after H. felis infection and killed on day 5 post infection. At the time the mice were killed, stomach tissues were recovered to perform qPCR to assess mRNA expression of inflammatory markers (IL-1β, IL-17, IL-23p19, and CXCL2; a) and histological analysis was conducted to evaluate tissue damage (b, c). Pictures are representative of score tissue damage of 0 (control rat IgG; anti-IL-22) and 4 (Vaccinated, rat IgG, anti-IL-22). H. felis-infected mice were injected with 50 μg of IL-22Fc fusion protein or PBS on days 0 and 3 and killed on day 5. At the time the mice were killed, stomach tissues were recovered to perform histological analysis (c) and qPCR was conducted to assess mRNA expression of inflammatory markers (d). Each dot represents a mouse. *P=0.05. CT, cholera toxin; IL, interleukin; n.s., not significant; PBS, phosphate-buffered saline; WT, wild type.
IL-22-induced antimicrobial peptides have a critical role in the reduction of Helicobacter infection
It has been shown that IL-22 induces the expression of AMPs involved in host defense in the skin, the airways, and the intestine.9 Similarly, we observed an upregulation of AMPs in the stomach mucosa of vaccinated mice associated with reduced H. felis infection (Figure 1c–e and Supplementary Figure S3A). We detected an increase in the expression of RegIIIβ, S100A9, and RegIIIγ, whose expressions have been reported to be dependent on the biological activity of IL-22.10 Indeed, vaccinated and infected WT mice injected with neutralizing anti-IL-22 antibodies or IL-22-deficient mice demonstrated a clear decrease of RegIIIβ, S100A9, and RegIIIγ expression in the stomach mucosa as compared with vaccinated WT mice (Figure 4a, b (left pictures)). In addition, the injection of IL-22Fc fusion protein promoted the expression of RegIIIβ, S100A9, and RegIIIγ in the stomach mucosa as compared with vehicle-injected mice (Figure 4a, b (middle and right pictures)).

IL-22-induced antimicrobial peptides have a critical role in the reduction of Helicobacter infection. The stomach mucosa of WT or IL-22-deficient vaccinated, control mice or chronically infected WT mice injected with rat IgG, anti-IL-22 antibodies, or recombinant IL-22Fc fusion protein were recovered to perform qPCR to quantify mRNA expression levels of RegIIIβ, S100A9, and RegIIIγ (a). Stomach tissue sections of vaccinated and infected mice injected with anti-IL-22 antibodies or mice chronically infected with H. felis were stained to detect RegIIIβ (b). Pictures are representative of data obtained from five or six individual mice. Bars: 50 μm. Three weeks after infection with H. felis, 50 μg of IL-22Fc fusion protein or PBS was injected on days 0 and 3 and the mice were killed on day 5. At the time the mice were killed, AMPs were extracted from the stomach and RegIIIβ expression detected by western blot (c). This western blot is representative of data obtained from six individual mice. The stomach mucosa of vaccinated or control WT mice were recovered at day 5 after H. felis infection to extract AMPs. H. felis or H. pylori were incubated with AMPs or hBD3, an AMP known to efficiently kill Helicobacter in vitro and stained with DiBAC4(3) to discriminate intact non-fluorescent and killed fluorescent Helicobacter (d). Pictures are representative of AMP-mediated Helicobacter killing; pictures are representative of data obtained from five or six individual mice in at least two independent experiments (d). Three weeks after infection with H. felis, WT or RegIIIβ−/− C57BL/6 mice were injected with 50 μg of IL-22Fc fusion protein or PBS on days 0 and 3 and killed on day 5 (e). At the time the mice were killed, the stomachs were recovered for detection of H. felis infection by qPCR. Two weeks after vaccination, WT and RegIIIβ-deficient mice were challenged with H. pylori and killed on day 10 post infection. The stomachs were recovered for detection of H. pylori infection by the numeration of CFU (f). Each dot represents a mouse. *P<0.05, **P<0.01, ***P<0.001. AMP, anti-microbial peptide; CT, cholera toxin; CFU, colony forming unit; DiBAC4(3), bis-(1,3-dibutylbarbituric acid) trimethine oxonol; hBD3, human β-defensin 3; IL, interleukin; n.s., not significant; PBS, phosphate-buffered saline; WT, wild type.
In order to evaluate whether the upregulation of AMPs by the stomach mucosa is responsible for the reduction of Helicobacter infection, we extracted AMPs from the stomach mucosa of vaccinated and infected mice to evaluate their potential Helicobacter killing activities. As expected, RegIIIβ was detectable by western blot of AMPs from mice with an increased level of IL-22 (Figure 4c). Dead bacteria were visualized by staining with DIBAC4(3) fluorescent dye.15 AMPs extracted from the stomach of vaccinated mice killed H. pylori and H. felis as efficiently as the recombinant human β-defensin 3 (Figure 4d and Table 1). Human β-defensin 3 is an AMP known to efficiently kill H. pylori in vitro.16 In contrast, AMPs extracted from control mice did not kill H. pylori or H. felis (Figure 4d and Table 1). Notably, AMPs extracted from the stomachs of vaccinated and infected WT mice injected with anti-IL-22 antibodies or from IL-22-deficient mice also did not kill H. pylori or H. felis (Table 1). Remarkably, AMPs extracted from the stomach mucosa of chronically infected mice injected with IL-22Fc killed H. pylori and H. felis (Table 1).
In order to investigate whether RegIIIβ has a role in the IL-22-induced reduction of H. felis infection in vivo, we injected IL-22Fc into chronically infected RegIIIβ-deficient mice. Remarkably, RegIIIβ-deficient mice did not reduce H. felis infection as efficiently as WT mice (Figure 4e). Lastly, in order to investigate whether RegIIIβ has a role in vaccine-induced protection, we vaccinated and challenged RegIIIβ-deficient mice. In a first step, we measured serum antibody responses in WT and RegIIIβ-deficient mice and found that both groups of mice were able to mount an antibody response against urease (Supplementary Figure S2D). In addition, splenocytes of both groups of mice proliferated at similar levels when exposed to urease (Supplementary Figure S2E). Taken together, these results suggest that the immunization protocol was efficient at promoting urease-specific CD4+ T-cell responses in both strains of mice. However, following immunization, although WT mice exhibited reduced H. pylori infection, RegIIIβ-deficient mice did not (Figure 4f).
Collectively, these data demonstrate that IL-22-induced AMPs, among them RegIIIβ, produced by the stomach mucosa kill H. pylori during a period of vaccine-induced protection.
Discussion
This study highlights the importance of IL-22 in the generation of vaccine-induced protective immunity directed toward a gut pathogen. In this study, we provided evidence that the CD4+ T cells infiltrating the gastric mucosa of H. felis-challenged vaccinated mice produce IL-22 (Figure 1f, g). These CD4+IL-22+ T cells mainly belong to the Th17 subset because they also produced IL-17 (Figure 1d, g). Neutralization of the IL-22 biological activity by injecting anti-IL-22 antibodies or by using IL-22-deficient mice decreased the efficacy of the vaccine to reduce H. felis and H. pylori infections (Figure 2). In addition, injection of IL-22Fc fusion protein into mice chronically infected with H. felis or H. pylori significantly decreased bacterial loads (Figure 2d, e). We next provided evidence that H. felis or H. pylori were killed by AMPs extracted from the stomach of vaccinated mice but not from control mice. Remarkably, the H. felis or H. pylori killing activities of stomach AMPs were dependent on IL-22. Indeed, AMPs extracted from the stomach of vaccinated mice injected with anti-IL-22 antibodies or vaccinated IL-22-deficient mice lacked anti-H. felis or H. pylori killing activity (Table 1). Moreover, AMPs extracted from the stomach of chronically Helicobacter-infected mice injected with IL-22Fc fusion protein efficiently killed H. felis or H. pylori (Table 1). In addition, we observed that H. pylori infection in vaccinated RegIIIβ-deficient mice was not reduced as efficiently as in WT mice (Figure 4f). Collectively, these results show an essential role for IL-22-dependent AMPs, such as RegIIIβ, in vaccine-induced protection.
Stomach epithelial cells express IL-22 receptor,17, 18 and the increase of STAT3 phosphorylation (Supplementary Figure S3B) detected within the stomach tissue during vaccine-induced reduction of Helicobacter infection suggests that gastric epithelial cells are stimulated by IL-22 in vivo. The upregulation of the IL-22-dependent RegIIIβ by gastric epithelial cells supports this interpretation (Figure 1d). By promoting the expression of AMPs such as RegIIIβ, RegIIIγ, S1008A, and S100A9, IL-22, either produced by ILC319 or Th1720, has been shown to have a key role in the stomach immune defenses directed against Candida albicans. Similarly, in our study, we observed that IL-22-dependent AMPs have critical roles in protecting the stomach from H. pylori infection. In our experimental conditions, we did not observe any production of IL-22 by ILC3 in vaccinated mice on day 5 post bacterial infection (Supplementary Figure S1C). Similarly, recent studies by Zhuang et al. and Shiu et al.17, 21 did not find any production of IL-22 by innate immune cells in the stomach of mice on day 35 post H. pylori or H. felis infection. Given that H. pylori is a noninvasive bacterium that subverts the detection mechanisms of the innate immune system,22 it can be postulated that IL-23-driven activation of IL-22 production by ILC323 is minimal in the stomach mucosa of H. pylori-infected non-vaccinated hosts. In vaccinated and infected mice, although IL-23 is produced at high levels (Figure 3a), it might be expected that because ILC3 are rare cells, ILC3 constitute a very minor source of IL-22 compared with infiltrating urease-specific CD4+ Th17 cells (Figure 1f, g).
Paradoxically, AMPs are already known to be induced in gastric epithelial cells of patients chronically infected by H. pylori.24, 25 A recent paper from the group of Wehkamp16 showed that AMPs, such as human beta defensin (hBD1) 1 and 2 (hBD2) and elafin, are highly induced in the stomach mucosa of H. pylori-infected individuals. However, hBD1, hBD2, and elafin cannot kill H. pylori.16 By comparison, other AMPs, such as human β-defensin 3 and LL37, showed strong bactericidal activities against H. pylori16, 26 but are negligibly expressed in the gastric mucosa of infected individuals. Our study demonstrates that vaccination or injection of IL-22Fc fusion protein alone modulates the AMPs produced by gastric epithelial cells, leading to the exclusion of H. pylori from its gastric mucosal niche. We identified RegIIIβ as one of IL-22-dependent AMPs responsible for H. pylori killing in vivo (Figure 4e, f). However, other AMPs like S100A9 (Figures 1c and 4a) or other factors such as IL-22-dependent mucins27 might also participate in the vaccine-induced reduction of H. pylori infection in vivo. In addition, IL-22-independent mechanisms, such as IL-17-dependent6, 7 or -independent8 neutrophil infiltration, participate in the protective immune response leading to the reduction of Helicobacter infection.5 These IL-22-independent mechanisms are probably at least in part responsible for the reduction of the bacterial load observed in vaccinated mice injected with anti-IL-22 antibodies (Figure 2b).
Although we observed that IL-22 led to the reduction of H. pylori infection in vaccinated animals, Zhuang et al.17 showed that IL-22 is detectable in chronically infected individuals and Obonyo et al.28 showed that WT C57BL/6 mice infected with H. felis highly expressed gastric IL-22 mRNA with an apparent increase over time but without any corresponding reduction of H. felis colonization. The absence of IL-22-mediated H. pylori/H. felis clearance in chronic infection might come from the lack or presence of additional key molecules, such as IL-10, IL-17, tumor necrosis factor-α, or IL-22BP known to modulate the anti-bacterial properties of IL-22 and/or the presence of cells suppressing the overall immune responses such as T-regulatory cells and/or the myeloid-derived suppressor cells.17, 29, 30
The necessity to induce a Helicobacter-specific CD4+ T-cell response associated with an inflammatory reaction is a common theme in vaccine-induced reduction of Helicobacter infection from the gastric mucosa. Helicobacter-specific Th1, Th2, and/or Th17 cells are most likely activated by IL-23-producing protease-activated receptor 2-dependent dendritic cells31 within the Peyer’s patches32 and/or mesenteric lymph nodes and/or directly within the stomach mucosa of vaccinated and infected mice. Activated CD4+ T cells secrete a large set of cytokines such as IFNγ, IL-17, and tumor necrosis factor-α that promote in some circumstances the recruitment and activation of inflammatory cells such as neutrophils,7 inflammatory monocytes,33 or mast cells,12 which participate in the reduction of bacterial load. Conflicting results have been published on the role of the different actors of the inflammatory response such as mast cells,34 neutrophils,12, 33 and IL-178 in vaccine protection. These discordant observations probably reflect the presence of redundant or compensatory mechanisms used by the mucosal immune system to mount inflammatory responses capable of reducing Helicobacter infection in vaccinated and infected mice. In addition, it might also indicate, as illustrated in our study, that the CD4+ T-cell response via the secretion of factors such as IL-22 controls non-inflammatory mechanisms having important roles in vaccine protection.
In conclusion, our study reinforces the finding that IL-22-dependent cross-talk exists at mucosal surfaces between activated memory Th17/Th22 cells and epithelial cells to enhance their anti-bacterial defenses.9, 20, 35, 36 With respect to the clinical development of a future H. pylori mucosal vaccine, our data suggest that it might be of great interest to correlate the presence of circulation and/or stomach urease-specific memory Th17/Th22 cells with vaccine efficacy to develop immune correlates of vaccine protection. In addition, we identified IL-22 as a key molecule capable of reducing H. pylori infection in vivo without inducing inflammatory processes. It thus can be suggested that IL-22 might be of clinical value in conjunction with prophylactic/therapeutic vaccines or antibiotic therapies in order to increase H. pylori clearance efficiency in chronically infected individuals.
Methods
Mice. BALB/c or C57BL/6 mice (6–8-week-old) were purchased from Harlan, Horst, The Netherlands. Female IL-22-deficient (IL-22−/−) BALB/c mice were provided by the Ludwig Institute for Cancer Research, (Brussels Branch, Belgium). C57BL/6 RegIIIβ-deficient mice (RegIIIβ−/−) were provided by the CRCM (Marseille, France). Mice were bred under conventional, non-specific-pathogen-free conditions. This study was approved by the Canton of Vaud Veterinary Office (authorization no. 836.9).
Bacteria and Infection. H. pylori P49, kindly provided by Harry Kleanthous (Acambis, Cambridge, MA), is a human clinical isolate adapted to mice. H. pylori P49 expresses VacA but not CagA. H. pylori P49 was grown on Helicobacter pylori-selective agar plate (Oxoid, Basingstoke, UK) and brain heart infusion broth supplemented with 0.25% yeast extract and 10% fetal calf serum (PAA, Pasching, Austria) under microaerophilic conditions. The H. felis strain (ATCC 49179) was grown biphasically under microaerophilic conditions. H. pylori P49 and H. felis infections were induced by orogastric gavage as described previously.12
Immunization. Mice were immunized intranasally four times at 1-week intervals with 30 μg of recombinant H. pylori urease (kindly provided by Sanofi-Pasteur, Lyon, France) combined with 5 μg of cholera toxin (Calbiochem, Lucerne, Switzerland). Control mice were administered only cholera toxin.
Assessment of Helicobacter colonization. H. felis colonization was assessed by measuring the copy number of H. felis flaB gene by quantitative PCR as described previously.6 H. pylori colonization was assessed by plating to numerate colony-forming units as described previously.12
Quantitative PCR. RNA extraction, reverse transcription, and PCR amplification were performed as described previously.6 The primers used in this study were purchased from Qiagen (Valencia, CA): IL-22 (QT00128324); CXCL2 (QT00113253); RegIIIβ (QT00239302); RegIIIγ (QT00147455); and S100A9 (Calgranulin B; QT00105252).
Histology and immunohistochemistry. For histology, stomach tissues were fixed in neutral buffered 10% formalin and subsequently embedded in paraffin. Sections of 4 μm thickness were stained with hematoxylin and eosin. Indexes of histopathological damage were calculated by the addition of inflammatory and atrophy grades assessed in the antral and fundal mucosa as described previously.6 Immunohistochemistry was performed as described previously.37 Briefly, paraffin-embedded stomach tissue sections (4 μm) were decorated with anti-RegIIIβ sheep antibodies (R&D Systems, Minneapolis, MN) followed by biotinylated rabbit anti-sheep (Abcam, Cambridge, UK) and avidin horseradish peroxidase complex (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA). RegIIIβ+ cells were visualized using diaminobenzidine tetrahydrochloride substrate kit for peroxidase (Vector Laboratories). Mayer’s hematoxylin was used as a nuclear counterstain. Endogenous peroxidase was quenched with 1% H2O2, and endogenous biotin was blocked with an avidin biotin blocking kit (Vector Laboratories).
Preparation of gastric lymphoid cell suspension. The isolation of the gastric lymphoid cells was performed as previously described.12
Anti-urease antibody response. The determination of the anti-urease antibody titers was performed as described previously.12
Flow cytometry. Cells were resuspended in fluorescence-activated cell sorter buffer (1% bovine serum albumin, 5 mmol l−1 EDTA in phosphate-buffered saline) and incubated with conjugated monoclonal antibodies in the presence of Fc blockers (2.4G2, Becton Dickinson (BD), San Jose, CA). Dead cells were excluded using 7-AAD (Beckman Coulter, Brea, CA); all data acquisition was performed on a Cytomics FC 500 (Beckman Coulter). We used the following reagents for the staining procedures: anti-CD3-FITC, anti-CD45-Biotin, anti-CD3-PECy5, Rat IgG1-PE, Streptavidin-PECF594 (BD), anti-CD5-FITC, anti-IL-17A-FITC, anti-CD4-PECy7 (Biolegend, San Diego, CA), and anti-IL-22-PE (eBioscience, San Diego, CA). For intracellular cytokine staining, gastric lymphoid cells were first stimulated with phorbol myristate acetate and ionomycin, then surface stained with anti-mouse CD4, CD3, and CD45 antibodies, permeabilized, and fixed. Cells were then co-stained for intracellular IL-17 and IL-22 as described previously.6 For the detection of intracellular IL-22 in ILC, gastric lymphoid cells were stimulated with phorbol myristate acetate, ionomycin, and IL-23 (10 ng ml−1). Fluorescence-activated cell sorter data were analyzed using FlowJo V10 (FLOWJO LLC, Ashland, OR).
AMP extraction. Cationic AMPs were extracted as described previously.15 Briefly, stomachs were frozen immediately in liquid nitrogen and were thoroughly ground using a cooled mortar and pestle. Proteins were extracted under gentle agitation for 2 h in 5% acetic acid with addition of Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland). The acid soluble proteins in the supernatant were lyophilized and resuspended in 0.05% acetic acid. Cationic proteins were extracted overnight at 4 °C with a weak cation exchange matrix (Macro Prep CMR, Bio-Rad Laboratories, Hercules, CA). The AMP-enriched cationic proteins were eluted with 5% acetic acid, lyophilized, and resuspended in 0.01% acetic acid. Protein concentrations were determined by BCA assay.
In vitro killing assay. H. pylori P49 or H. felis culture (D0600nm=0.6) were washed twice with phosphate-buffered saline and diluted at 4 × 106 bacteria per ml in phosphate-buffered saline. In 96-well round-bottomed plates, we mixed 50 μl of bacterial suspension with 10 μg of stomach cationic peptides or 2 μg of human β-defensin 3 (Peprotech, London, UK). The mixture was incubated 55 min at 37 °C (CO2 incubator). During the last 10 min of incubation bis-(1,3-dibutylbarbituric acid) trimethine oxonol (DiBAC4(3)) (Enzo Life Sciences, Lausen, Switzerland) (0.1 μg ml−1) was added to discriminate intact non-fluorescent and depolarized fluorescent Helicobacter.
Urease-induced specific proliferation and IL-22 production. A total of 1 × 105 splenocytes labeled with carboxyfluorescein-diacetate-succinimidyl-ester38 were stimulated with purified urease (10 μg ml−1) in RPMI 10% fetal calf serum in 96-well plates. After 4 days of incubation at 37 °C, carboxyfluorescein-diacetate-succinimidyl-ester dilution was measured by fluorescence-activated cell sorter using Cytomics FC 500 (Beckman Coulter). Proliferation index was determined using proliferation with concanavalin A (1 μg ml−1) (C5275-5MG, Sigma-Aldrich, St. Louis, MO) to normalize urease-specific proliferation. Urease-induced IL-22 secretion was determined by incubating purified urease (10 μg ml−1) with 1 × 106 stomach mononuclear cells for 5 days in RPMI 10% fetal calf serum in 12-well plates. IL-22 protein concentrations were measured in the cell supernatant by enzyme-linked immunosorbent assay using Quantikinekit (R&D Systems).
Western blot. For each sample, total protein concentration was measured using BCA assay. Twenty micrograms of proteins was loaded and separated on 12% sodium dodecyl sulfate-polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane (Pall Corporation, DF Mexico, Mexico). After a 1 h blocking step at room temperature in 3.5% bovine serum albumin, the membrane was incubated overnight at 4 °C with primary antibodies: rabbit anti-Stat3-P (Tyr705) (Cell Signaling Technology, Danvers, MA), mouse anti-Actin (Sigma), or sheep anti-RegIIIβ (R&D Systems). Following washes, the membrane was incubated at room temperature with secondary biotinylated antibodies (donkey anti-rabbit IgG (GE Healthcare, Buckinghamshire, UK), sheep anti-mouse IgG (GE Healthcare), or rabbit anti-sheep IgG (Abcam)) followed by streptavidin-horseradish peroxidase incubation. Following washes, chemiluminescence revelation was carried out on a Fusion-FX (Vilber Lourmat, Eberhardzell, Germany) with WesternBright ECL mix (Advansta Corporation, Menlo Park, CA).
Statistics. Data distribution was compared by Mann–Whitney tests and two-way analysis of variance using GraphPad (GraphPad Software, San Diego, CA), with P=0.05 as the limit of significance.
References
Graham, D.Y. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology 148, 719–731 e713 (2015).
Zeng, M. et al. Efficacy, safety, and immunogenicity of an oral recombinant Helicobacter pylori vaccine in children in China: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 386, 1457–1464 (2015).
Sutton, P. At last, vaccine-induced protection against Helicobacter pylori. Lancet 386, 1424–1425 (2015).
Ermak, T.H. et al. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J. Exp. Med 188, 2277–2288 (1998).
Moyat, M. & Velin, D. Immune responses to Helicobacter pylori infection. World J Gastroenterol 20, 5583–5593 (2014).
Velin, D. et al. Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterology 136, 2237–2246 e2231 (2009).
DeLyria, E.S., Redline, R.W. & Blanchard, T.G. Vaccination of mice against H pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136, 247–256 (2009).
DeLyria, E.S. et al. Vaccine-induced immunity against Helicobacter pylori in the absence of IL-17A. Helicobacter 16, 169–178 (2011).
Rutz, S., Eidenschenk, C. & Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev 252, 116–132 (2013).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Aujla, S.J. et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med 14, 275–281 (2008).
Velin, D., Bachmann, D., Bouzourene, H. & Michetti, P. Mast cells are critical mediators of vaccine-induced Helicobacter clearance in the mouse model. Gastroenterology 129, 142–155 (2005).
Van Belle, A.B. et al. IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J. Immunol 188, 462–469 (2012).
Basu, R. et al. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37, 1061–1075 (2012).
Nuding, S., Fellermann, K., Wehkamp, J., Mueller, H.A. & Stange, E.F. A flow cytometric assay to monitor antimicrobial activity of defensins and cationic tissue extracts. J. Microbiol. Methods 65, 335–345 (2006).
Nuding, S. et al. Gastric antimicrobial peptides fail to eradicate Helicobacter pylori infection due to selective induction and resistance. PloS One 8, e73867 (2013).
Zhuang, Y. et al. A pro-inflammatory role for Th22 cells in Helicobacter pylori-associated gastritis. Gut 64, 1368–1378 (2015).
Wolk, K., Kunz, S., Witte, E., Friedrich, M., Asadullah, K. & Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 21, 241–254 (2004).
Zelante, T. et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385 (2013).
De Luca, A. et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol 3, 361–373 (2010).
Shiu, J. et al. Gastric LTi cells promote lymphoid follicle formation but are limited by IRAK-M and do not alter microbial growth. Mucosal Immunol 8, 1047–1059 (2015).
Salama, N.R., Hartung, M.L. & Muller, A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol 11, 385–399 (2013).
Walker, J.A., Barlow, J.L. & McKenzie, A.N. Innate lymphoid cells—how did we miss them? Nat Rev Immunol 13, 75–87 (2013).
Bajaj-Elliott, M. et al. Modulation of host antimicrobial peptide (beta-defensins 1 and 2) expression during gastritis. Gut 51, 356–361 (2002).
Bauer, B., Wex, T., Kuester, D., Meyer, T. & Malfertheiner, P. Differential expression of human beta defensin 2 and 3 in gastric mucosa of Helicobacter pylori-infected individuals. Helicobacter 18, 6–12 (2013).
Bauer, B., Pang, E., Holland, C., Kessler, M., Bartfeld, S. & Meyer, T.F. The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe 11, 576–586 (2012).
Kawakubo, M. et al. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science 305, 1003–1006 (2004).
Obonyo, M., Rickman, B. & Guiney, D.G. Effects of myeloid differentiation primary response gene 88 (MyD88) activation on Helicobacter infection in vivo and induction of a Th17 response. Helicobacter 16, 398–404 (2011).
Lundgren, A. et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun 73, 523–531 (2005).
Kao, J.Y. et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138, 1046–1054 (2010).
Velin, D. et al. PAR2 promotes vaccine-induced protection against Helicobacter infection in mice. Gastroenterology 141, 1273–1282 1282 e1271 (2011).
Nagai, S. et al. Role of Peyer's patches in the induction of Helicobacter pylori-induced gastritis. Proc. Natl. Acad. Sci. USA 104, 8971–8976 (2007).
Moyat, M., Mack, M., Bouzourene, H. & Velin, D. Role of inflammatory monocytes in vaccine-induced reduction of Helicobacter felis infection. Infect. Immun 83, 4217–4228 (2015).
Ding, H., Nedrud, J.G., Wershil, B., Redline, R.W., Blanchard, T.G. & Czinn, S.J. Partial protection against Helicobacter pylori in the absence of mast cells in mice. Infect. Immun 77, 5543–5550 (2009).
Ivanov II et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Liang, S.C. et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med 203, 2271–2279 (2006).
Bernasconi, E. et al. Granulocyte-macrophage colony-stimulating factor elicits bone marrow-derived cells that promote efficient colonic mucosal healing. Inflamm. Bowel Dis 16, 428–441 (2010).
Quah, B.J., Warren, H.S. & Parish, C.R. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc 2, 2049–2056 (2007).
Acknowledgements
We thank the Mouse Pathology Facility (University of Lausanne) for performing the RegIIIβ staining and A. Nahimana for access to the Cytomics FC 500. We thank B. Corthesy for advice on AMP extraction and B.J. Marsland for critical reading of the manuscript. This work was funded by Swiss National Foundation grant 310030-141145 (to D. Velin).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Moyat, M., Bouzourene, H., Ouyang, W. et al. IL-22-induced antimicrobial peptides are key determinants of mucosal vaccine-induced protection against H. pylori in mice. Mucosal Immunol 10, 271–281 (2017). https://doi.org/10.1038/mi.2016.38
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2016.38
This article is cited by
-
IL-22 hinders antiviral T cell responses and exacerbates ZIKV encephalitis in immunocompetent neonatal mice
Journal of Neuroinflammation (2020)
-
Epitope-loaded nanoemulsion delivery system with ability of extending antigen release elicits potent Th1 response for intranasal vaccine against Helicobacter pylori
Journal of Nanobiotechnology (2019)
-
Maf deficiency in T cells dysregulates Treg - TH17 balance leading to spontaneous colitis
Scientific Reports (2019)
-
Interleukin-6: designing specific therapeutics for a complex cytokine
Nature Reviews Drug Discovery (2018)
