
Abstract
A hallmark of celiac disease is autoantibodies to transglutaminase 2 (TG2). By visualizing TG2-specific antibodies by antigen staining of affected gut tissue, we identified TG2-specific plasma cells in the lamina propria as well as antibodies in the subepithelial layer, inside the epithelium, and at the brush border. The frequency of TG2-specific plasma cells were found not to correlate with serum antibody titers, suggesting that antibody production at other sites may contribute to serum antibody levels. Upon commencement of a gluten-free diet, the frequency of TG2-specific plasma cells in the lesion dropped dramatically within 6 months, yet some cells remained. The frequency of TG2-specific plasma cells in the celiac lesion is thus dynamically regulated in response to gluten exposure. Laser microdissection of plasma cell patches, followed by antibody gene sequencing, demonstrated that clonal cells were seeded in distinct areas of the mucosa. This was confirmed by immunoglobulin heavy chain repertoire analysis of plasma cells isolated from individual biopsies of two untreated patients, both for TG2-specific and non-TG2-specific cells. Our results shed new light on the processes underlying the B-cell response in celiac disease, and the approach of staining for antigen-specific antibodies should be applicable to other antibody-mediated diseases.
Similar content being viewed by others


INTRODUCTION
Celiac disease (CD) is a multifactorial disorder characterized by an intestinal inflammatory response to ingested cereal gluten proteins.1 The human leukocyte antigen association and the central role of CD4+ T cells in the pathogenesis are thoroughly investigated.2 T cells of the lesion recognize gluten peptides that are deamidated in vivo by the enzyme transglutaminase 2 (TG2).3, 4 Notably, the great majority of CD patients develop an autoantibody response, with TG2 itself being the main autoantigen.5 It is not known whether these antibodies have a role in the pathophysiology of CD, yet anti-TG2 immunoglobulin A (IgA) antibodies are increasingly used as diagnostic tool6 and in the follow-up of the treatment, as upon commencement of a gluten-free diet (GFD) autoantibodies disappear from serum within months.7 Anti-TG2 antibodies are produced by plasma cells (PCs) localized in the lamina propria of the intestinal mucosa,8, 9, 10 but PCs localized elsewhere may also contribute to the antibody production. Korponay-Szabo et al.11 developed a double-color immunofluorescence method that allowed visualizing antibody deposits in correspondence with the subepithelial TG2 layer in the small intestine, as well as in other tissues. This method builds on colocalization between IgA and the TG2 protein, the latter usually identified by means of the CUB7402 murine monoclonal antibody. Importantly, this has been proposed as a tool for early diagnosis12 on affected individuals without signs of villous atrophy13, 14 or with negative/borderline serology.15 Recently, we reported a flow cytometry-based method to describe and enumerate TG2-specific PCs from freshly obtained biopsies of the small intestine of CD patients.10 We found that in untreated CD patients the frequency of these cells was exceptionally high, and that they produced antibodies with limited somatic hypermutation but nonetheless reasonable affinity. In the same work we reported an immunofluorescence-based method to visualize TG2-specific PCs on cryosections. As this material can be stored for prolonged periods of time and requires relatively little amount of specimen, studies on cryosections provide a simple yet very valid alternative for the analysis of intestinal humoral responses. Even though significant discoveries of the anti-TG2 antibody response have been done, its importance in the diagnostic workup of CD and the unknown role in the pathogenesis of the disease suggest that more research on this topic is necessary. In this work, we expand our previous findings on the intestinal anti-TG2 response by investigating and describing the location of TG2-specific PCs and antibodies, the phenotype of these cells as well as the niche supporting them, and the kinetics of their disappearance upon commencement of a GFD, and we discuss a mechanism for a possible pathogenic involvement. Our findings provide a new tool to analyze the antibody response in CD and in general the specificity of antibody responses directly in affected tissues.
RESULTS
TG2-specific plasma cells are abundant and accumulate close to the gut lumen in the celiac lesion
We recently reported that the frequency of TG2-specific cells in the intestinal lesion of untreated CD patients is exceptionally high, averaging 10%. Here, we further investigate this matter by using our novel immunofluorescence technique10 that takes advantage of soluble biotinylated TG2 to visualize intracellular deposits of anti-TG2 antibodies that, in combination with an anti-CD138 antibody, allowed us to directly identify TG2-specific PCs (Figure 1). This method differs from the CUB7402/IgA costaining method11 that reveals the location of the TG2 protein in the tissue, and indirectly the presence of specific antibody deposits because of the colocalization with IgA. At low magnification ( × 10), stained cryosections from untreated CD patients (Figure 1a,b) showed significant plasmacytosis in the lamina propria,16 and the localization of the PCs in the lamina propria was more toward the intestinal lumen than in healthy controls (Figure 1d). Figure 1c is representative of the most common condition observed in GFD-treated patients, with good recovery of the intestinal morphology, markedly decreased plasmacytosis, and the presence of a few residual TG2-specific PCs. In fact, despite the diet, we usually do not observe a complete disappearance of TG2-specific PCs.

Visualization of transglutaminase 2 (TG2)-specific plasma cells (PCs) and antibodies by immunofluorescence on cryosections from celiac disease (CD) patients. (a–d) Low-magnification images of sections from two untreated patients, one gluten-free diet (GFD)-treated patient, and one control individual. TG2-specific plasma cells (identified by costaining for the CD138 plasma cell marker, in red, and TG2-specific antibody deposits as revealed by the biotinylated TG2 antigen, in green) are evident in untreated patients, decreased in the GFD-treated patient, and absent in the control. (e) Higher-magnification images from an untreated patient, further revealing the apical localization of TG2-specific plasma cells (intracellular antibody deposits as revealed by biotinylated TG2, in green, as shown in center and right panels). (f) Colocalization of immunoglobulin A (IgA) and anti-TG2 antibodies (as detected by biotinylated TG2), showing PCs and deposits in the lamina propria, as well as additional deposits in the subepithelial layer and on the brush border. (g) Colocalization of IgA and TG2 (as detected by the anti-TG2 monoclonal antibody (mAb) CUB7402), showing a different pattern in CD patients and controls, with extensive overlap between the two stainings observed in CD patients only.
Interestingly, in patients with active disease we consistently observed that TG2-specific PCs were preferentially found in the lamina propria in proximity of the lumen, as evident from Figure 1a,b and further shown in Figure 1e at higher magnification ( × 40). The single channel images showed that all the TG2+ cells express with the anti-CD138 marker, demonstrating that these are PCs.
Double-color immunofluorescence studies of in situ localization of TG2 and IgA suggested that in CD patients, anti-TG2 antibodies are deposited with TG2 in the subepithelial layer.11 Here by using biotinylated TG2 as bait for specific antibodies, we were able to visualize antibodies not only within PCs (as described above), but also at their extracellular location as deposits, primarily in the subepithelial layer in association with TG2, as expected (Figure 1f). Furthermore, in many cases TG2-specific antibodies seemed to be present in the epithelial cells and at the brush border in CD patients, as shown in Figure 1g. Although it can be argued that visualization of subepithelial deposits by biotin-labeled recombinant TG2 may be sensitive to artifacts because of unspecific binding of the bait to endogenous fibronectin,15 this does not seem to be an issue under the conditions used here. In fact in healthy controls who do not produce anti-TG2 antibodies, but also possess the fibronectin-TG2 subepithelial layer (Figure 1g), only weak binding of the bait to fibronectin was observed (for instance, compare Figure 1d with Figure 1a,b).
TG2-specific plasma cells of the mucosa drop 6 months after commencement of a GFD
The ability to stain antigen-specific PCs in frozen samples by immunofluorescence, as well as the availability of biopsies taken from the same patients at different times and stored in the freezer, allowed us to study the kinetics of the disappearance of TG2-specific PCs at early stages of GFD. We collected frozen cryosections of intestinal specimens from CD patients at diagnosis and after 6, 12, or 24 months of GFD, and enumerated TG2-specific PCs by immunofluorescence. The results are reported in Figure 2a as percentage of TG2-specific cells in the PC compartment. All patients had elevated amounts of these cells at diagnosis (n=15, average 9.6%, range 3.0–18.6%), and this frequency markedly decreased during the period with GFD. After 6 and 12 months of diet, TG2-specific cells dropped to an average of 1.7% (n=6, range 0–4.1%) and 1.4% (n=9, range 0–5.7%) respectively. For 3 patients from whom we were able to obtain an additional specimen after 24 months of GFD, we did not detect any TG2-specific PCs at this time point. However, in these patients TG2-specific PCs were absent or in low amounts already at the first sampling (0 and 0.9% in the 2 patient samples after 6 months, and 0% for 1 patient sampled at 12 months). As in some cases a relatively high frequency of TG2-specific PCs was still detected after 6–12 months of GFD, and as some of the patients included in this study had borderline or positive serology according to endomysial antibodies or IgA anti-TG2 despite GFD (yellow dots in Figure 2a), we asked whether residual presence of TG2-specific PCs correlated with seropositivity. This was not the case at 6 months, as all of the cases at this time point were below cutoff for serum IgA anti-TG2 positivity (according to the Celikey test). Of the 5 patients who were seropositive or borderline seropositive at 12 months, 3 had detectable TG2-specific intestinal plasma cells. These findings raise the question of whether in general the TG2-specific PC frequency in untreated patients correlates with their serum titer. As shown in Figure 2b, we found no correlation between PC frequency and serum titer. This may be because of the presence of patches in the intestinal lesion with different densities of TG2-specific PCs, and therefore one single biopsy specimen may not be representative of the small intestine in its entirety. In an attempt to address this, from one patient we were able to obtain three biopsies sampled from different intestinal locations. Flow cytometry quantification of TG2-specific PCs showed consistent frequency among the three samples (4.5, 4.0, and 4.3%, Figure 2c) and these data, although they need to be substantiated, speak toward a fairly homogenous distribution of autoreactive PCs in biopsies of the upper small bowel of CD patients.

Quantification of TG2-specific plasma cells. (a) Kinetics of the disappearance of transglutaminase 2 (TG2)-specific plasma cells (PCs) at early (6 months) or later (12 and 24 months) stages of gluten-free diet (GFD), as determined using our standard immunofluorescence staining technique showed in Figure 1a–e. Ab, antibody; EMA, endomysial antibody. (b) Lack of correlation between the TG2–immunoglobulin A (IgA) serum titer and the PC count in the biopsy, as assessed at the time of small intestinal biopsy sampling in patients with active disease. (c) Frequency of TG2-specific plasma cells in three different biopsy samples from the same patient, as determined by flow cytometry using multimerized labeled TG2 as bait for antigen-specific cells.
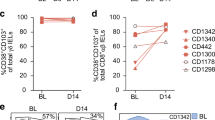
To further understand the factors regulating TG2-specific plasma cell survival, we also investigated the PC expression of TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor), BCMA (B-cell maturation antigen), and BAFF-R (B-cell activating factor receptor) that are receptors for BAFF and APRIL (a proliferation-inducing ligand) involved in B-cell survival. Staining gut tissue sections with TACI- and BAFF-R-specific antibodies showed that TACI was expressed in a subset of PCs, and in some cases its expression was paired to that of BAFF-R (Figure 3a,b). A similar heterogeneity regarding these markers was observed in TG2-specific cells, with no significant difference compared with other PCs (Figure 3c). A significant fraction of PCs appeared to express BCMA (Figure 3d). In order to improve the analysis of BAFF and APRIL receptors, we used a flow cytometry approach that yields a more quantitative and objective set of data. Figure 3e and 3f shows the percentage of IgA PCs expressing BCMA, BAFF-R, or TACI as judged by staining of single-cell suspensions of biopsies of five untreated CD patients. These data confirmed BCMA expression on most plasma cells, whereas BAFF-R and TACI were expressed only by a subgroup of cells and at variable levels. There were no significant differences in the relative expression of these markers between TG2-specific and non-TG2-specific PCs, suggesting that these receptors play a role in the development of the PC compartment in the gut, but it does not appear that the autoimmune B-cell response in CD differs significantly from its physiological counterpart. Similarly, the expression levels of BCMA and BAFF-R were similar in CD patients and healthy controls, whereas TACI appeared to be expressed at slightly higher levels in CD patients, although the difference did not reach statistical significance (P=0.055, Mann–Whitney; Figure 3g).

Analysis of the expression of BAFF (B-cell activating factor) and APRIL (a proliferation-inducing ligand) receptors on the plasma cell (PC) populations in the lamina propria. (a–f) Visualization of transglutaminase 2 (TG2)-specific PCs by immunofluorescence on gut biopsy cryosections of celiac disease (CD) patients, with characterization of their phenotype and the supporting niche. Images are representative of stainings performed on two or three CD patients and two healthy controls. (e) Quantitative analysis of the expression of BCMA (B-cell maturation antigen), BAFF-R (B-cell activating factor receptor), and TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor) by flow cytometry using monoclonal antibodies and multimerized TG2 antigen. Panels indicate percentage of cells expressing BCMA, BAFF-R, and TACI, shown for TG2-specific and non-TG2-specific IgA PCs. Box plots with minimum and maximum values are shown. The results derive from analysis of five untreated CD patients. (f) Panels showing flow cytometry plots for one of the subjects. Receptor expression by TG2-specific PCs is shown in blue and non-TG2-specific PCs in light red. The gray curves to the left in each flow diagram represent baseline (Fluorescence Minus One (FMO)). (g) Comparison of the expression levels of BCMA, BAFF-R, and TACI by flow cytometry of immunoglobulin A (IgA) PCs from gut biopsies of five untreated CD patients and five healthy controls. Percentage positive cells with box plots giving minimum and maximum values are shown.
Analysis of PCs dissemination in the gut mucosa by laser capture and microdissection
To investigate whether intestinal PC populations expand locally or at a distant site, we performed laser capture and microdissection followed by sequencing of immunoglobulin heavy chain variable (IGHV) genes on patches with such cells from intestinal biopsies of CD patients. To this end, we adapted a published set of primers for a seminested PCR of antibody genes on genomic DNA. Because of the available microscope configuration, the procedure was optimal for dissecting sections stained by immunohistochemistry under transmitted light. Preliminary experiments indicated that staining for the CD138 marker was optimal under these conditions, and we used this marker to stain sections from samples known to have high frequency of TG2-specific PCs (identified using the standard immunofluorescence method shown in Figure 1). We dissected patches comprising 20 to 30 PCs (an example is shown in Figure 4a) from one patient with active CD. In order to increase the variability of the analysis, the same patch was picked from three consecutive slides. We amplified genomic DNA and obtained a PCR product from all of the picks, and built and sequenced libraries. Importantly, several IGHV-5 sequences with few or no mutations were found; IGHV-5 genes had on average 2.2 mutations (n=9) as compared with other genes that averaged 11.4 mutations/sequence (n=33). Although not a formal demonstration, this strongly suggests specificity for TG2 of some of the dissected cells; in fact, as us and others have reported,10, 17 IGHV-5 genes with low degree of somatic hypermutation are otherwise rare among the intestinal PC compartment. Interestingly, in some cases clonal sequences were found that showed shared and unique mutations, thus raising the possibility that such clones mutate and evolve locally. We further extended our analysis and compared sequences found in different patches that were sampled from an additional patient with an active disease. We dissected four PC patches (named A, B, C, and D) that were not adjacent to each other. The same patch was dissected from several consecutive slides. Libraries were obtained from 14 picks, yielding 53 unique sequences representing 26 different clones; 18 were represented by one single sequence and 8 by 2 or more and allowed us to build lineage trees. Table 1 provides a summary of the eight clonal trees that were built with sequences from this patient: whereas a few simple trees could be built from clonal sequences that were found only within one of the patches (example in Figure 4b, with sequences derived from one single patch, clone ID 23 according to Table 1), in many other cases larger trees could be built where several different patches contributed clones (example in Figure 4c, with sequences from three different patches, clone ID 22 according to Table 1).

Laser capture and microdissection (LCM) of plasma cell patches rich in transglutaminase 2 (TG2)-specific ones. Sequences are obtained from PCR performed on genomic DNA from dissected patches in the lamina propria. (a) Example of a pick comprising ∼20 plasma cells. (b, c) Examples of clonal trees built from clonally related sequences derived from the same patch ((b) clone ID 23 from Table 1) or from three different patches ((c) clone ID 22 from Table 1) of the same patient. Data are summarized in Table 1. The letter inside the node indicates the patch from which the sequence comes, whereas different colors indicate sequences derived from different, consecutive cryosections. The number next to the line connecting the inferred sequence (gray node) and actual observed sequences (colored nodes) represents the number of mutations by which the two connected nodes differ.
Analysis of clonal relatedness by high-throughput sequencing of PCs from multiple individual biopsies
This limited sequence analysis suggests that clonally related PCs disseminate to adjacent effector sites in the small intestine following differentiating and maturation in a distal inductive site, as was recently demonstrated in the mouse.18, 19 To further substantiate this notion, we determined the level of clonal sharing between PCs that were isolated from different intestinal biopsies of the same CD patients by high-throughput sequencing analysis of their IGHV genes. Two patients were included and single-cell suspensions from each subject were individually processed from five intestinal biopsies and IgA PCs were sorted into TG2-specific and non-TG2-specific cells as previously described10 using the staining strategy shown in Figure 2c. Next, the IGHV genes were PCR amplified and sequenced. Unique sequences that originated within each individual biopsy were grouped into clones. The number of sorted cells and number of PC clones for each biopsy are given in Supplementary Table S1 online. The degree of clonal relatedness between different biopsies was further determined following clonal clustering between the different biopsies. Among non-TG2-specific PCs, the level of clonal sharing between different biopsies ranged from 10.7 to 22.7% and from 8.2 to 13.8% in patients CD1191 and CD1192, respectively. In comparison, the levels of clonal sharing among TG2-specific PCs were higher and ranged from 17.9 to 73.1% and from 17.8 to 60% in the 2 patients (Figure 5a,b). Genealogy analysis revealed that there existed PCs in all five biopsies that were clonally related and derived from one common ancestor. A representative clonal tree is depicted in Figure 5c.

Clonal sharing between plasma cells (PCs) from different duodenal biopsies of patients with active celiac disease. Heat maps denoting the level of clonal relatedness between PCs that were collected from five different small intestinal biopsies from two celiac patients: (a) CD1191 and (b) CD1192. Single-cell suspensions were prepared from small intestinal biopsies, immunoglobulin A (IgA) PCs were further sorted for transglutaminase 2 (TG2)-specific and non-TG2-specific PCs, and their immunoglobulin heavy chain variable (IGHV) genes were sequenced. Clonal relatedness of PCs from distinct biopsies was determined following clonal clustering between the different populations (see Methods for detailed description). The left panels depict results from non-TG2-specific plasma cells, whereas the right panels depict results from TG2-specific plasma cells. The number of PC clones in each biopsy is shown in the diagonal across each diagram. The numbers of clones that are shared between biopsies are displayed and percentages are calculated as a fraction from the total number of clones from the biopsy on the horizontal axis. (c) A representative lineage tree that illustrates clonal sharing between five different biopsies of subject CD1191.
DISCUSSION
In this work we have uncovered several novel aspects of the anti-TG2 response in CD. The emerging picture is that the frequency of TG2-specific PCs cells in the mucosa is dynamically regulated and that the generation of these PCs is taking place outside of the lamina propria with seeding of cells, often clonally related, into distinct sites of the gut mucosa.
Recent data suggest that PCs in the small intestine are longer lived than previously thought. Hapfelmeier et al.20 demonstrated that in mice in a germ-free environment, and thus in the absence of competition, intestinal IgA-secreting cells have the capacity to be long-lived. By colonization with given commensal bacteria, specific PC antibody responses could be raised, but without antigen persistence, the PCs disappeared within a few months, suggesting that intestinal antibody repertoire is characterized by constant attrition and thus represents the dominant antigen currently present in the intestine. Mesin et al.21 demonstrated that PCs from human small intestine biopsies survive and secrete immunoglobulins for many weeks in the biopsy cultures, and interleukin-6 and APRIL were identified to be essential for plasma cell survival and antibody secretion. TG2-specific PCs are abundant in the lesion of patients with active CD,10 and for the maintenance of this high abundance, lengthy survival time appears crucial. We assessed the expression of BAFF and APRIL receptors (BCMA, TACI, and BAFF-R) on PCs. Whereas signaling through TACI induces class switching and antibody production, BAFF-R and BCMA promote B-cell and plasma cell survival, respectively.22, 23, 24, 25, 26, 27 As shown by flow cytometry quantitation, BCMA was expressed on the majority of PCs. In humans, both APRIL and BAFF can bind BCMA, the latter with low avidity. BAFF-R and TACI were expressed by a subset of cells and at variable levels. In general, we did not observe any phenotype suggesting differences between TG2-specific and other PCs. Although it is likely that these receptors play a role in the development of the PC compartment in the gut, it does not appear that the autoimmune B-cell response in CD differs significantly from its physiological counterpart. CD patients and healthy controls appeared to have similar expression levels of BCMA and BAFF-R, whereas we observed a minor (and not statistically significant) increase in the levels of TACI in CD patients.
We found that the number of TG2-specific PCs in the small intestinal mucosa drops dramatically within 6 months when gluten is withdrawn from the diet. IgA and IgM PCs of the gut express cell surface immunoglobulin.21 Thus conceivably, recognition of antigen by surface immunoglobulin could allow PCs to sense antigen and this could explain how PCs in the gut are maintained by antigen persistence and by attrition. Removal of gluten likely does not lead to removal of TG2 antigen available to gut PCs, and hence this is hardly the explanation for the observed drop of TG2-specific PCs. Possibly, however, it could relate to cessation of presentation of antigen by PCs to T cells that would affect the survival niche for the PCs. Is has been suggested that in CD, gluten-specific T cells may provide help to TG2-specific B cells via hapten-carrier-like complexes of gluten and TG2.28, 29 If such gluten–TG2 complexes can be taken up via surface immunoglobulin of TG2-specific PCs and the PCs present gluten antigen via scarcely expressed human leukocyte antigen-DQ molecules, removal of gluten from the diet would affect T-cell activation and thereby survival of TG2-specific PCs.
Importantly, we observed that some TG2-specific PCs were detectable at low frequency even at late time points. One subject still had 4% of the PCs being TG2 specific after 6 months of GFD. This subject was the one with the highest frequency of TG2-specific PCs at diagnosis (18%). It would be interesting to analyze earlier and/or closer time points and to compare them with serum antibody titers.7 However, because of the current guidelines for the CD diagnostic workup, such samples are difficult to obtain. Notwithstanding, it appears clear that many TG2-specific PCs disappear rapidly after removal of the diet and few remain in the mucosa. In future studies it will be interesting to analyze in depth differences in phenotype and transcription factors between PCs that disappear quickly and PCs that remain when CD patients start the GFD.
It is well known that TG2-specific antibodies disappear from the circulation within months after the commencement of the GFD.7, 30 It is thus surprising that we did not observe a clear correlation between reduction of the serum titer and disappearance of the PCs at the intestinal level in the GFD-treated patients. TG2-specific PCs were present in 5 out of 6 samples collected after 6 months of GFD, despite seronegativity. Several factors are relevant to this observation. First, in situ detection of anti-TG2 antibodies can precede appearance in serum and hence the two are not necessarily correlated. Moreover, compliance to a strict GFD, or even inadvertent gluten intake (gluten being increasingly present in processed foods), may suffice to trigger local, limited secretion that does not match serology results. Furthermore, we did not observe any correlation between the antibody serum titer and the frequency of TG2-specific PCs in the lesion. It is likely that the absolute amount, rather than the frequency, of TG2-specific PCs could correlate with serum titer. The absolute amounts of PCs will depend on the degree of plasmacytosis, the size of the affected small intestine, the volume of circulating blood, and the degree of spillover from the intestinal compartment. We did not analyze for these factors, although preliminary data from one patient showed comparable amounts of TG2-specific PCs in individual biopsies sampled at the same time. Finally, it cannot be excluded that at least some of the TG2 antibodies in the blood are produced at sites outside of the gut, like lymph nodes, spleen, and bone marrow, or from circulating plasmablasts, and although this topic is arduous to address because of the challenges in collecting proper samples, it represents a scenario that has implications that possibly go beyond CD.
Previously, we reported that TG2-specific PCs did not express the proliferation marker Ki-67, suggesting that their abundance is not a direct consequence of sustained local expansion.10 However, the Ki-67 marker accumulates upon sustained cell division, and it may not be expressed if cells undergo cell proliferation at a slow, homeostatic rate. It should also be noted that immunofluorescence may not be sensitive enough for this type of analysis, and that flow cytometry approaches could provide better readouts. Cell division is also necessary for accumulation of mutations in antibody genes. Although not commonly acknowledged, there is very clear evidence for mutations occurring outside of germinal centers, at least in some contexts in mice.31 In addition, it has been suggested that precursors of intestinal antibody-forming cells may undergo somatic hypermutation and/or local expansion directly in the lamina propria,32 although evidence remains to be substantiated. To determine whether intestinal PCs proliferate locally or at a distant site we performed laser capture and microdissection of TG2-specific plasma cell patches from two patients followed by antibody gene sequencing. We obtained and then compared sequences obtained from several different patches; moreover, the same patch was dissected, when possible, from consecutive slides, thus creating a tridimensional grid of the patch and increasing the variability of the analysis. The rationale for the analysis of this experiment is that finding clonal sequences that differ by one or few mutations within the same patch, and not in patches from distant areas, would be a good indicator that the mutations occurred locally; if this were not the case, clonal sequences would not be confined exclusively to local areas. Some of the clonal trees were generated from sequences obtained exclusively from one patch, thus possibly suggesting some degree of local proliferation and mutation. However, there were larger trees with sequences from several different patches, suggesting that these clones acquired mutations and likely expanded at a different site before dissemination in the mucosa. To further substantiate this notion, we undertook analysis of the antibody repertoire among TG2-specific and non-TG2-specific PCs from five individual biopsies of two patients with untreated CD. We indeed observed clonal sharing among cells of the different biopsies, particularly among the TG2-specific PCs where the degree of sharing ranged from 20% to 60–70%. These findings suggest that the PCs of the duodenum clonally expand before seeding into the mucosa, although the fact that small trees could be built exclusively from sequences found in one patch indicates that the possibility of local expansion and mutation, at least to some extent, cannot be completely ruled out. In general, our findings are consistent with recent observations in mouse models.18, 19 We interpret the extensive degree of clonal sharing between TG2-specific PCs of individual biopsies likely to reflect an expansion of dominant clones at inductive sites that is followed by seeding of PCs into large areas of the small intestine.
Although anti-TG2 antibody deposits are usually visualized by the classical CUB7402/IgA staining,11 we found that deposits can be observed using the antigen as bait. By staining with biotin-TG2 in combination with an anti-IgA (Figure 1f), we confirmed the presence of subepithelial deposits, but also detected TG2-specific antibodies inside epithelial cells and on the brush border. In fact, most IgA produced by intestinal PCs are dimeric, with two molecules bound together by the J-chain polypeptide. The J-chain interacts with the polymeric Ig receptor at the basal side of epithelial cells, resulting in transport across the epithelium and into the lumen. It is conceivable that anti-TG2 antibodies bind the antigen in the subepithelial layer and are co-transported together with it across the epithelium; this is further suggested by the presence of TG2 itself (colocalizing with IgA) in the epithelium and on the brush border, as shown in Figure 1g and as suggested by figures from a number of other publications.12, 33, 34, 35 This allows us to speculate on a model that builds upon previous observations from Matysiak-Budnik et al.,36 who suggested that anti-gliadin IgA antibodies are exported in the gut lumen, where they bind gluten and transport it across the epithelium back to the lamina propria, via a retrotranscytosis mechanism mediated by the transferrin receptor (CD71). In addition to anti-gliadin IgA, it may also be that TG2 at the brush border can sample gluten peptides by enzymatically acting on them, and these are retrotranscytosed with anti-TG2 IgA (or IgM37). TG2 retains its enzymatic activity when bound to antibodies.10 In addition, it has been shown that the majority of peptides representing preferred substrate for TG2 harbor known gluten T-cell epitopes,38 and that epitopes frequently recognized by patient T cells showed a significant higher level of deamidation compared with those less frequently recognized.39 Other groups have proposed mechanisms that could mediate this or similar transport systems; Rauhavirta et al.40 also implicated a role for celiac IgA that necessitates TG2 enzymatic activity, and Lebreton et al.41 substantiated some of these finding, further reintroducing CD71 in the game of play and also suggesting a role for the enzymatic activity of TG2. These are attractive models that require further study, including the development of an objective quantitative measurement system for antibody deposits in relation to its significance in CD.
In summary, the findings described here shed new light on the characteristics of the anti-TG2 B-cell response in the intestinal mucosa of patients affected by celiac disease. Importantly, the general approach can be applied to similar conditions where the role and especially the origin of (auto)antibodies is unclear. For instance, dermatitis herpetiformis and gluten ataxia (the skin and central nervous system manifestations of gluten sensitivity, respectively, occurring in up to 10% of CD patients) are characterized by the local presence of TG3-IgA and TG6-IgA deposits.42, 43 Relevant diseases where local presence of autoantibodies is thought to be important but is scarcely understood include multiple sclerosis (nervous system), rheumatoid arthritis (joints), and others. The approaches described here may help to further our knowledge of these daunting disorders.
METHODS
Patient material. Adult CD patients were diagnosed according to standard criteria, including human leukocyte antigen genotyping, anti-TG2 serum titer, and histological analysis of small intestinal biopsy.44 The same criteria were used to exclude CD diagnosis in controls. Ethical approval for study of Norwegian subjects was obtained from the Regional Ethics Committee in South-Eastern Norway (project S-97201). Each study subject gave written informed consent. Duodenal biopsy specimens were obtained by gastroduodenoscopy. Tissue sections of biopsies of seven CD patients with active disease, from three CD patients treated with a GFD and two healthy control subjects were studied. In addition, biopsies from five untreated CD patients and five healthy controls were processed and used for flow cytometry analysis and PCs of biopsies from two untreated CD patients were processed for high-throughput sequencing. Ethical approval for study of the Finnish subjects was obtained from the Ethics Committee of Tampere University Hospital, and each subject provided written informed consent. Samples from 15 CD patients and 4 nonceliac controls suffering from dyspepsia were studied. From these subjects small-bowel mucosal biopsies were taken either with an adult-size Watson capsule from the proximal jejunum or upon endoscopy with forceps from the distal duodenum. For each subject, part of the samples were snap-frozen and embedded in optimal cutting temperature compound (OCT, Tissue-Tec, Miles, Elkhart, IN) for storing at −70 °C, whereas the rest of the biopsy specimens were paraffin embedded. Serum IgA class antibodies against TG2 were detected by enzyme-linked immunosorbent assay using human recombinant TG2 as antigen, with a cutoff line of 5.0 U ml−1 (Celikey, Phadia, Freiburg, Germany).
Immunofluorescence on intestinal biopsy cryosections. Immunofluorescence experiments were done as reported before.10 The technique is based on the ability of biotinylated TG2, followed by labeled streptavidin, to selectively bind TG2-specific antibody deposits inside plasma cells. Briefly, biopsy specimens stored at −70 °C were thawed and rinsed with phosphate-buffered saline (PBS). Primary reagents were incubated in 1.2% bovine serum albumin in PBS for 45 min at room temperature (RT). Sections were extensively rinsed with PBS, and secondary reagents were incubated in 12% bovine serum albumin in PBS for 30 min at RT. Sections were rinsed with PBS and mounted. The following antibodies and reagents were used: mouse anti-CD138 (1:25, AbD Serotec, Kidlington, UK), recombinant human TG2 produced in Escherichia coli and biotinylated as reported before10 (5 μg ml−1), mouse anti-TG2 monoclonal antibody CUB7402 (1:1,000, Neomarkers, Fremont, CA), anti-TACI (1:200, sc-7332 N-19), anti-BCMA (1:200, sc-11743N-16), and anti-BAFF-R (1:150, eBioscience 8A7, San Diego, CA) primary antibodies. Secondary antibodies were used as follows: Alexa Fluor 546/488/649-conjugated anti-goat, anti-mouse, and anti-rabbit polyclonal antibodies (1:1,000, Life Technologies, Carlsbad, CA), streptavidin-cy2 (1:1,000, GE Healthcare Biosciences, Pittsburgh, PA), streptavidin-cy3 (1:2,000, GE Healthcare Biosciences), anti-mouse-cy3 (1:1,500, Southern Biotech, Birmingham, AL), and anti-IgA fluorescein isothiocyanate (1:40, Dako). DAPI (4',6-diamidino-2-phenylindole) was used to counterstain nuclei.
Flow cytometry. The frequency of TG2-specific PCs in small intestinal biopsies was determined using flow cytometry as previously reported.10 The expression levels of BCMA, BAFF-R, and TACI on intestinal PCs that either stained positive or negative for TG2 were also examined using flow cytometry of single-cell suspensions of biopsies with Bir-A biotinylated TG2 multimerized on fluorescently labeled streptamers. In short, recombinant human TG2 was expressed in Sf9 insect cells by baculovirus with a Bir-A recognition site at its N-terminus. TG2 was biotinylated with Bir-A enzyme (Avidity, Aurora, CO), and incubated with allophycocyanin-labeled streptamers (IBA, Gottingen, Germany) in a 4:1 molar ratio for 1 h to generate TG2-multimers. Cryopreserved single-cell suspensions that were prepared from gut biopsies were thawed and stained with allophycocyanin-labeled TG2-multimer for 45 min on ice. IgA fluorescein isothiocyanate (Southern Biotech), Brilliant Violet 570 CD3, CD14, and CD38 PerCp-Cy5.5 (BioLegend, San Diego, CA), CD19 Pacific Blue (BD Biosciences), CD27 PE-Cy7 (eBioscience), and either BCMA, BAFF-R, or TACI (all phycoerythrin (PE) conjugated, BioLegend) were added for the last 20 min for staining of specific cellular lineage markers. FMO control (i.e., Fluorescence Minus One) was used to properly interpret the expression of BCMA, BAFF-R, and TACI by staining single-cell suspensions of each subject with a combination of all antibodies and TG2-multimers except the PE-conjugated antigens. Samples were acquired on LSRFortessa (BD Biosciences, San Jose, CA) and the data were analyzed by FlowJo, version 10.0.7 (Ashland, OR).
Immunohistochemistry and laser capture and microdissection. For laser capture and microdissection, 7 μm thick cryosections from specimens of patients were cut on 2 μm PEN-membrane slides (Leica, Buffalo Grove, IL). Sections were fixed in acetone for 10 min and stored at −80 °C. For laser capture and microdissection, a Leica LMD6500 instrument equipped with an optical microscope was used, and sections were therefore stained by immunohistochemistry. Briefly, slides were thawed and rinsed in PBS. Primary reagents (mouse anti-CD138 or recombinant human biotinylated TG2 produced in E. coli, as reported above) were diluted in 1% bovine serum albumin in PBS and added to the slide for 45 min at RT in a moist chamber. Slides were extensively rinsed and secondary reagents (goat anti-mouse IgG-HRP, 1:500, Santa Cruz (Dallas, TX), or SA-HRP, 1:500, Southern Biotech) in 1% bovine serum albumin in PBS were added for 30 min at RT. After extensive washing with PBS, development was performed by adding 3-amino-9-ethylcarbazole substrate (Sigma, St Louis, MO) and incubating for 2 to 20 min until proper development occurred. Slides were carefully washed with water to remove all salts, dried at RT, and then stored at 4 °C until necessary. Dissections were performed in the caps of 0.5 ml tubes, to which 10 μl of digestion buffer (50 mM Tris-HCl, 50 mM KCl, 0.63 mM EDTA, 0.22% Igepal, 0.22% Tween-20, and 0.8 mg ml−1 proteinase K) were added. After dissection of plasma cell patches in the caps, these were closed and the tubes spinned. The samples were added with a drop of mineral oil, digested at 55 °C for 2 h, followed by inactivation of proteinase K at 90 °C for 5 min, and used for PCR amplification of antibody genes directly from genomic DNA.
PCR and analysis of IGHV genes from plasma cell picks. A seminested PCR was performed in 50 μl using the high-fidelity PfuTurbo DNA polymerase enzyme (Agilent, Santa Clara, CA) as per the manufacturer’s instructions. Primers used in the primary and secondary PCRs were as described by Kuppers.45 The VH1, VH3, and VH5 primers were used as a mix in the primary PCR, and individually in the secondary PCR. Antisense primers in the J regions were used as originally described in the protocol.45 PCR products were purified and cloned in the Zero Blunt TOPO PCR cloning kit for sequencing (Life Technologies) as per the manufacturer’s instructions, and individual colonies were sequenced at the Keck DNA sequencing facility at Yale. Analysis of the sequences was performed as follows: preprocessing was carried out using pRESTO (http://clip.med.yale.edu/pRESTO) as previously described.46, 47 V(D)J germline segments were determined using IMGT/HighV-QUEST48 and divided into clonally related groups based on common V gene, J gene, and junction region length. Lineage trees were constructed for each clonal group with the dnapars application of PHYLIP.46
High-throughput sequencing of IGHV genes of isolated plasma cells. Single-cell suspensions were prepared from individual biopsies taken from two CD patients with active disease and cryopreserved until used. TG2 multimerized on PE-labeled streptavidin was used for identifying TG2-specific or non-TG2-specific PCs from single-cell suspensions as previously described,10 and cells were sorted directly into 25 μl catch buffer (10 mM dithiothreitol and 2 U μl−1 RNAsin (Promega, Madison, WI) in PBS). For complementary DNA (cDNA) synthesis, 1 μg of random hexamers (Roche, Basel, Switzerland), 0.45% v/v NP-40, 1 μl RNAsin 40 U μl−1 (Promega), and diethylpyrocarbonate-treated water (to a final volume of 50 μl) were added; the mix was incubated at 65 °C for 5 min and put on ice. Next, 20 μl 5 × RT buffer, 7 μl dithiothreitol 100 mM, 9 μl of 10 mM dNTP-Mix (Promega), 1.5 μl RNAsin 40 U μl−1 (Promega), 1.8 μl superscript III (Life Technologies), and 14 μl diethylpyrocarbonate-treated water were added. The cDNA was synthesized at 42 °C for 10 min, 25 °C for 10 min, 50 °C for 60 min, and 94 °C for 5 min. The Ig heavy chain VH region was amplified between framework (FR) 2 and J from cDNA using two consecutive PCRs. For the first PCR, six forward primers, i.e., FR2 VH1–6, and a single IGHJ reverse primer were used.49, 50 Both forward and reverse primers included six nucleotides that were used as indexes for the different subsets and that were further followed by six random nucleotides and partial Illumina adapters (San Diego, CA). The first PCR was performed using AmpliTaq Gold polymerase (Life Technologies) as follows: 95 °C for 7 min, 25 PCR cycles (95 °C for 30 s, 60 °C for 45 s, 72 °C for 90 s), and 72 °C for 10 min. A second PCR was designed so that it included Illumina adapter sequences using Qiagen Multiplex PCR as follows: 95 °C for 15 min, 10 PCR cycles (95 °C for 30 s, 60 °C for 45 s, 72 °C for 90 s), and 72 °C for 10 min. Final amplicons libraries were extracted from agarose gel (Qiagen, Venlo, The Netherlands) and paired-end sequencing of 250 base pairs was performed using Illumina MiSeq. Primers sequences are given in Supplementary Table S2. Raw sequencing data were processed by pRESTO, and unique sequences that originated within each individual biopsy were grouped into clones as previously described.51
References
Sollid, L.M. Molecular basis of celiac disease. Annu. Rev. Immunol. 18, 53–81 (2000).
Jabri, B. & Sollid, L.M. Mechanisms of disease: immunopathogenesis of celiac disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 516–525 (2006).
Molberg, O. et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 4, 713–717 (1998).
van de Wal, Y. et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J. Immunol. 161, 1585–1588 (1998).
Dieterich, W. et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 3, 797–801 (1997).
Rostom, A. et al. The diagnostic accuracy of serologic tests for celiac disease: a systematic review. Gastroenterology 128 (4 Suppl 1), S38–S46 (2005).
Sugai, E. et al. Dynamics of celiac disease-specific serology after initiation of a gluten-free diet and use in the assessment of compliance with treatment. Dig. Liver Dis. 42, 352–358 (2010).
Marzari, R. et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J. Immunol. 166, 4170–4176 (2001).
Picarelli, A., Maiuri, L., Frate, A., Greco, M., Auricchio, S. & Londei, M. Production of antiendomysial antibodies after in-vitro gliadin challenge of small intestine biopsy samples from patients with coeliac disease. Lancet 348, 1065–1067 (1996).
Di Niro, R. et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat. Med. 18, 441–445 (2012).
Korponay-Szabo, I.R. et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut 53, 641–648 (2004).
Koskinen, O. et al. Gluten-dependent small bowel mucosal transglutaminase 2-specific IgA deposits in overt and mild enteropathy coeliac disease. J. Pediatr. Gastroenterol. Nutr. 47, 436–442 (2008).
Kaukinen, K. et al. Small-bowel mucosal transglutaminase 2-specific IgA deposits in coeliac disease without villous atrophy: a prospective and randomized clinical study. Scand. J. Gastroenterol. 40, 564–572 (2005).
Tosco, A. et al. Immunoglobulin A anti-tissue transglutaminase antibody deposits in the small intestinal mucosa of children with no villous atrophy. J. Pediatr. Gastroenterol. Nutr. 47, 293–298 (2008).
Salmi, T.T. et al. Endomysial antibody-negative coeliac disease: clinical characteristics and intestinal autoantibody deposits. Gut 55, 1746–1753 (2006).
Green, P.H. & Cellier, C. Celiac disease. N. Engl. J. Med. 357, 1731–1743 (2007).
Benckert, J. et al. The majority of intestinal IgA+ and IgG+ plasmablasts in the human gut are antigen-specific. J. Clin. Invest. 121, 1946–1955 (2011).
Bergqvist, P. et al. Re-utilization of germinal centers in multiple Peyer's patches results in highly synchronized, oligoclonal, and affinity-matured gut IgA responses. Mucosal Immunol. 6, 122–135 (2013).
Lindner, C. et al. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J. Exp. Med. 209, 365–377 (2012).
Hapfelmeier, S. et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328, 1705–1709 (2010).
Mesin, L., Di Niro, R., Thompson, K.M., Lundin, K.E. & Sollid, L.M. Long-lived plasma cells from human small intestine biopsies secrete immunoglobulins for many weeks in vitro. J. Immunol. 187, 2867–2874 (2011).
Avery, D.T. et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Invest. 112, 286–297 (2003).
He, B. et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat. Immunol. 11, 836–845 (2010).
Litinskiy, M.B. et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 3, 822–829 (2002).
Magri, G. et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat. Immunol. 15, 354–364 (2014).
Moore, P.A. et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science 285, 260–263 (1999).
O'Connor, B.P. et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 199, 91–98 (2004).
Sollid, L.M., Molberg, O., McAdam, S. & Lundin, K.E. Autoantibodies in coeliac disease: tissue transglutaminase—guilt by association? Gut 41, 851–852 (1997).
Maki, M. Autoantibodies as markers of autoimmunity in coeliac disease pathogenesis. Proceedings of the Sixth International Symposium on Coeliac Disease Held at Trinity College, Dublin, in July 1992, (Feighery C., O'Farelly C., eds) 246–252 Oak Tree Press: Dublin, Ireland, (1994).
Sulkanen, S. et al. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterology 115, 1322–1328 (1998).
William, J., Euler, C., Christensen, S. & Shlomchik, M.J. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 297, 2066–2070 (2002).
Yuvaraj, S. et al. Evidence for local expansion of IgA plasma cell precursors in human ileum. J. Immunol. 183, 4871–4878 (2009).
Esposito, C. et al. Expression and enzymatic activity of small intestinal tissue transglutaminase in celiac disease. Am. J. Gastroenterol. 98, 1813–1820 (2003).
Koskinen, O., Villanen, M., Korponay-Szabo, I., Lindfors, K., Maki, M. & Kaukinen, K. Oats do not induce systemic or mucosal autoantibody response in children with coeliac disease. J. Pediatr. Gastroenterol. Nutr. 48, 559–565 (2009).
Stenman, S.M. et al. Secretion of celiac disease autoantibodies after in vitro gliadin challenge is dependent on small-bowel mucosal transglutaminase 2-specific IgA deposits. BMC Immunol. 9, 6 (2008).
Matysiak-Budnik, T. et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J. Exp. Med. 205, 143–154 (2008).
Borrelli, M. et al. High density of intraepithelial gammadelta lymphocytes and deposits of immunoglobulin (Ig)M anti-tissue transglutaminase antibodies in the jejunum of coeliac patients with IgA deficiency. Clin. Exp. Immunol. 160, 199–206 (2010).
Dorum, S. et al. The preferred substrates for transglutaminase 2 in a complex wheat gluten digest are Peptide fragments harboring celiac disease T-cell epitopes. PLoS One 5, e14056 (2010).
Dorum, S., Qiao, S.W., Sollid, L.M. & Fleckenstein, B. A quantitative analysis of transglutaminase 2-mediated deamidation of gluten peptides: implications for the T-cell response in celiac disease. J. Proteome Res. 8, 1748–1755 (2009).
Rauhavirta, T. et al. Epithelial transport and deamidation of gliadin peptides: a role for coeliac disease patient immunoglobulin A. Clin. Exp. Immunol. 164, 127–136 (2011).
Lebreton, C. et al. Interactions among secretory immunoglobulin A, CD71, and transglutaminase-2 affect permeability of intestinal epithelial cells to gliadin peptides. Gastroenterology 143, 698–707 e1-4 (2012).
Sardy, M., Karpati, S., Merkl, B., Paulsson, M. & Smyth, N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J. Exp. Med. 195, 747–757 (2002).
Hadjivassiliou, M., Aeschlimann, P., Strigun, A., Sanders, D.S., Woodroofe, N. & Aeschlimann, D. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann. Neurol. 64, 332–343 (2008).
AGA Institute. AGA Institute Medical Position Statement on the Diagnosis and Management of Celiac Disease. Gastroenterology 131, 1977–1980 (2006).
Kuppers, R. Molecular single-cell PCR analysis of rearranged immunoglobulin genes as a tool to determine the clonal composition of normal and malignant human B cells. Methods Mol. Biol. 271, 225–238 (2004).
Stern, J.N. et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 6, 248ra107 (2014).
Vander Heiden, J.A. et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932 (2014).
Alamyar, E., Duroux, P., Lefranc, M.P. & Giudicelli, V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol. 882, 569–604 (2012).
Boyd, S.D. et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci. Transl. Med. 1, 12ra23 (2009).
van Dongen, J.J. et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 17, 2257–2317 (2003).
Yaari, G. et al. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front. Immunol. 4, 358 (2013).
Acknowledgements
We thank Moriah Cohen for the help generating clonal trees. This work received support by the European Commission (grant ERC-2010-Ad-268541 to L.M.S.), the South-Eastern Norway Regional Health Authority (to L.M.S.), the Research Council of Norway (partially through its Centres of Excellence funding scheme, grant 179573/V40, to L.M.S.), the Academy of Finland (to K.K.), the Competitive State Research Financing of the Expert Area of Tampere University Hospital (grant 9R018 to M.M. and K.K., and grant 9R034 to M.M.), the Sigrid Juselius Foundation (to K.K.), as well as by grants from the NIH (grant R01 AI-43603 to M.J.S., grant R01 AI-104739 to S.H.K., and grant P01 AI-061093 to A.C.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Di Niro, R., Snir, O., Kaukinen, K. et al. Responsive population dynamics and wide seeding into the duodenal lamina propria of transglutaminase-2-specific plasma cells in celiac disease. Mucosal Immunol 9, 254–264 (2016). https://doi.org/10.1038/mi.2015.57
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.57
