
Abstract
A better understanding of the cellular targets of HIV infection in the female genital tract may inform HIV prevention efforts. Proposed correlates of cellular susceptibility include the HIV co-receptor CCR5, peripheral homing integrins, and immune activation. We used a CCR5-tropic pseudovirus to quantify HIV entry into unstimulated endocervical CD4+ T cells collected by cytobrush. Virus entry was threefold higher into cervix-derived CD4+ T cells than blood, but was strongly correlated between these two compartments. Cervix-derived CD4+ T cells expressing CD69, α4β7, or α4β1 were preferential HIV targets; this enhanced susceptibility was strongly correlated with increased CCR5 expression in α4β7+ and CD69+ CD4+ T cells, and to a lesser extent in α4β1+ CD4+ T cells. Direct binding of gp140 to integrins was not observed, integrin inhibitors had no effect on virus entry, and pseudotypes with an env that preferentially binds α4β7 still demonstrated enhanced entry into α4β1+ cells. In summary, a rapid and sensitive HIV entry assay demonstrated enhanced susceptibility of activated endocervical CD4+ T cells, and those expressing α4β7 or α4β1. This may relate to increased CCR5 expression by these cell subsets, but did not appear to be due to direct interaction of α4β7 or α4β1 with HIV envelope.
Similar content being viewed by others


INTRODUCTION
Heterosexual sex is the most common mode of HIV transmission, and women account for almost two thirds of new infections globally.1 Although sexual transmission is inefficient, with a probability of 1/200 to 1/2,000 per episode of penile-vaginal sex,2 the immunopathogenesis of transmission events is incompletely understood. Most sexual exposures do not result in HIV infection, presumably because the virus is repelled by innate mucosal immune defenses. These include an intact genital epithelium, cervical mucus, innate antimicrobial peptides in the genital fluids, and immune cells within the mucosa (neutrophils, αβ and γδ T cells, dendritic cell subsets, and others).3 However, preventing HIV infection is a delicate balance, as HIV preferentially replicates in activated CD4+ T cells4 and inflammation in the female genital tract (FGT) was directly associated with HIV acquisition in young women from South Africa.5 Increased levels of antimicrobial peptides are associated with an improved ability of genital fluids to neutralize HIV in vitro, but in a prospective study, this ability was paradoxically associated with increased HIV acquisition in vivo,6 perhaps because mucosal inflammation is accompanied by increased mucosal HIV target cells7 and reduced epithelial integrity.8, 9
Although mucosal T-cell immune activation enhances HIV susceptibility, there are several other cellular correlates of HIV susceptibility in the FGT. Following gp120 binding to CD4, the HIV co-receptor CCR5 is necessary for viral entry and establishment of infection by sexually transmitted strains of the virus.10 The mucosal integrins α4β7 and/or α4β1 may also have a role in sexual transmission of HIV. α4β7 binds to mucosal vascular addressin cell adhesion molecule-1 (MAdCAM-1) on high endothelial venules and mediates lymphocyte migration to the intestine,11 whereas α4β1 binds VCAM-1 (vascular cell adhesion molecule-1)12 to mediate lymphocyte migration to nonlymphoid tissue and sites of inflammation.13, 14, 15 α4β7 has been shown in some studies,16, 17 although not all,18 to enhance cellular HIV replication in CD4+ T cells derived from peripheral blood mononuclear cells (PBMCs). In cervical explants, addition of α4, β7, and β1 blocking monoclonal antibodies reduced HIV replication of the R5-tropic BaL strain,19 suggesting that α4β7 and/or α4β1 may play a role in the early establishment of HIV infection in the FGT. Collectively, these results suggest that mucosal CD4+ T-cell expression of CCR5, CD69, α4β7, or α4β1 may flag mucosal T-cell subsets with increased susceptibility to HIV.
Measuring inflammatory genital biomarkers or mucosal T-cell expression of putative susceptibility markers provides a surrogate measure of HIV susceptibility. However, as the optimal mucosal surrogate of HIV susceptibility is not known, measuring actual mucosal T-cell HIV infection ex vivo would be the most biologically relevant way to assess HIV susceptibility, and might allow for improved identification and characterization of susceptible cells. Currently, most cervical ex vivo infection assays are performed on biopsies or cervical tissue obtained during hysterectomies.19, 20, 21 However, hysterectomies are rarely performed on young and/or healthy individuals and can only be done once per individual, making them impractical for studying HIV susceptibility in prospective studies. Although sequential ectocervical biopsies or cervical cytobrush specimens can be obtained from healthy individuals, both yield a relatively low number of CD4+ T cells22 and this poses a challenge for measuring HIV infection ex vivo.
Given these concerns, the purpose of this study was to apply a rapid and sensitive assay to quantify HIV entry into freshly isolated and unstimulated cervix-derived CD4+ T cells obtained using cervical cytobrushes, and to identify preferential cellular targets of HIV. Using an R5-tropic β-lactamase (BlaM)-viral protein R (Vpr) HIV pseudovirus, we hypothesized that cervix-derived CD4+ T cells were highly susceptible to HIV entry and that immunologically activated α4β7+ and α4β1+ CD4+ T cells were preferential cellular targets of HIV entry in the FGT.
RESULTS
Study participants
Fifty-four participants were enrolled in the Nairobi-based studies. No participants had genital symptoms or clinical findings (see exclusion criteria in Methods), and asymptomatic genital infections were rare: 7/54 had bacterial vaginosis (BV, Nugent score >6),23 1/54 Neisseria gonorrhoeae, 2/54 Chlamydia trachomatis, 1/54 Trichomonas vaginalis, and 0/54 active syphilis. Of these 54 participants, HIV entry into cervical T-cell targets was assessed in 41 participants (median age, 28 years; range, 19–46 years), and 13 participants were enrolled in studies of mucosal Ki67 expression (30 years; 19–39 years).
Enhanced HIV susceptibility of cervix-derived CD4+ T cells
Cytosolic entry of the BlaM-Vpr HIV pseudovirus was restricted to CD4+ T cells in both blood (Figure 1a,c) and cervix (Figure 1b,d). In blood, median virus entry was 4.5% (interquartile range (IQR) 2.2–6.4%) in CD4+ T cells as compared with 0.22% (IQR 0.0–0.6%) in CD4- T cells (P<0.0001; Figure 1c). Similarly, in cervical CD4+ and CD4- T cells, median HIV entry was 10.4% (IQR 6.0–25.6%) and 0.02% (IQR 0.0–0.1%), respectively (P<0.0001; Figure 1d).

Enhanced HIV susceptibility of cervix-derived CD4+ T cells compared with blood. HIV entry into blood- and cervix-derived CD4+ and CD4- T cells infected ex vivo with a CCR5-tropic BlaM-Vpr pseudovirus. Gating on from left to right, singlets, live cells, lymphocytes, and CD4+ and CD4- T cells in top rows in (a) blood and (b) cervix. Bottom rows in a and b show representative cytosolic HIV entry or blue vs. green signal in CD4+ and CD4- T cells in mock- or virus-treated samples. Overall % HIV entry into CD4+ and CD4- T cells in (c) 24 blood and 20 (d) cervical samples. (e) Comparison and (f) correlation of HIV entry into CD4+ T cells derived from 18 matched blood and cervical samples. Graph shows median values, and statistical comparisons are done by Wilcoxon rank-pairs test. Correlation was performed by Spearman’s correlation. *P=0.001 and **P=0.0003. BlaM, β-lactamase; Vpr, viral protein R.
We hypothesized that cervical CD4+ T cells would demonstrate enhanced HIV susceptibility compared with blood, as these cells were more activated and express higher levels of CCR5 (Supplementary Figure S1 online).24 In keeping with this, HIV entry was 2.8-fold higher into CD4+ T cells derived from the cervix (median 11.8%, IQR 6.1–26.6%) than from the blood (median 4.2%, IQR 2.0–6.4%, P=0.0003; Figure 1e). Asymptomatic sexually transmitted diseases or asymptomatic BV were diagnosed in four participants, but enhanced virus entry into cervix-derived CD4+ T cells compared with blood was seen regardless of genital coinfection or BV status (P=1.0). As CD4+ T cells were responsible for a smaller proportion of flow cytometry events in cervical samples (mean, 1.7 × 103 CD4+ T cells per participant) than blood (mean, 1.7 × 105 CD4+ T cells per participant), in order to ensure that lower viral entry in blood CD4+ T cells did not simply reflect a lower stoichiometric ratio of input BlaM-Vpr virus per CD4+ T cell, HIV entry was then assessed in six samples matched for actual CD4+ T-cell input number from each compartment (Supplementary Figure S2). Again, HIV entry into cervix-derived CD4+ T cells (median 5.5%, IQR 1.0–11.8%) was 7.7-fold higher than CD4+ T cells from blood (median 0.9%, IQR 0.2–1.2% P=0.03). Overall, there was a consistent increase in HIV entry into cervix-derived CD4+ T cells. However, there was a strong positive correlation between HIV entry into blood- and cervix-derived CD4+ T cells (r=0.748, P=0.0002; Figure 1f) within a given individual, suggesting shared determinants of susceptibility between these two compartments.
Next we compared expression of Ki67, a marker of metabolic activity and cell proliferation, between blood and cervical compartments, as metabolically active cells are more likely to sustain productive HIV infection. Ki67 expression was 1.9-fold higher in CD4+ T cells derived from cervix (median 4.6%, IQR 2.7–5.2%) compared with blood (median 2.4%, IQR 1.6–2.8%; P=0.005; Supplementary Figure S3). These results demonstrate that cervix-derived CD4+ T cells are highly susceptible to HIV, as they not only demonstrate preferential viral entry but also have a higher proportion of actively cycling cells.
BlaM-Vpr HIV entry is dependent on CCR5 and virus/cell fusion but not CXCR4
The gp160 env in our construct was described as R5-tropic,25 and hence to test whether the increased blue/green signal observed in virus-treated samples was a consequence of CCR5-dependent viral fusion with CD4+ T cells, the CCR5 inhibitor maraviroc was titrated on PBMCs (Figure 2a,b). A dose-dependent inhibition of HIV entry was observed, with 34% inhibition (IQR 23–59%) at 10 nM maraviroc, 89% inhibition (IQR 78–96%) at 100 nM maraviroc, and 94% inhibition (IQR 90–98%) at 1 μM maraviroc (Figure 2a). Furthermore, median HIV entry dropped from 11.7% (IQR 4.4–15.0%) to 0.0% (IQR 0–1.5%) in cervical CD4+ T cells treated with 1 μM maraviroc (P=0.03; Figure 2c). As expected, treatment with the CXCR4 inhibitor AMD3100 had no effect on HIV entry into CD4+ T cells, either in the cervix (median 8.9%, IQR 1.1–13.8% vs. 8.6%, IQR 2.0–17.5% in untreated; P=0.69; Figure 2d) or blood (median 2.9%; IQR 1.9–4.8% vs. 2.2%; IQR 1.6–4.5% in untreated; P=0.07; Figure 2b).

Effect of HIV-entry inhibitors on cytosolic entry of a CCR5-tropic BlaM-Vpr HIV pseudovirus into CD4+ T cells in blood and the cervix. Peripheral blood mononuclear cells (PBMCs) were titrated with (a) 0–1,000 nM maraviroc and treated with (b) 1 μM maraviroc, 1 μM Fuzeon (T20), or 250 nM AMD3100 before addition of BlaM-Vpr pseudovirus. Cervical cells were treated with (c) mock, virus, or virus plus 1 μM maraviroc or (d) 250 nM AMD3100. All inhibitors were incubated with cells for 30 min at room temperature before addition of virus. Graphs show median values, and statistical comparisons are done by Wilcoxon rank-pairs test. N=6. *P=0.03 and **P=0.003. BlaM, β-lactamase; NI, no inhibitor; NS, not significant; Vpr, viral protein R.
Next, we tested whether the increased blue/green fluorescence in virus-treated samples was a consequence of fusion-dependent cytosolic entry of HIV rather than de novo expression of BlaM-Vpr. Treatment with T20 (Fuzeon), which prevents fusion of viral and host membranes, resulted in near-complete ablation of viral entry in PBMCs (median 2.2%, IQR 1.6–4.5% vs. 0.1%, IQR 0.0–0.2%, P=0.03; Figure 2b). However, the reverse transcriptase inhibitor lamivudine, which blocks the postviral entry step of reverse transcription of viral RNA into DNA, had no effect on HIV entry (data not shown). Collectively, these results demonstrate that the increased blue/green signal in blood- and cervix-derived T cells treated with the R5-tropic BlaM-Vpr pseudovirus was dependent on CD4, CCR5, and fusion of viral and host membranes, and not on de novo expression of BlaM, suggesting that this assay can measure physiologically relevant HIV entry.
As our BlaM-Vpr HIV included an HIV envelope that was previously described as R5-tropic, we assessed whether CCR5+ CD4+ T cells were preferential targets of HIV entry (Figure 3a). HIV entry into CCR5+ CD4+ T cells was substantially enhanced compared with CCR5- CD4+ T cells in both blood (P<0.0001; Figure 3b and Table 1) and the cervix (P<0.0001; Figure 3c and Table 1). Within an individual, however, HIV entry into bulk cervix-derived CD4+ T cells correlated with the frequency of cervix-derived CCR5+ CD4+ T cells (r=0.705, P=0.0005; Figure 3d) and the mean fluorescent intensity of CCR5 (r=0.465, P=0.04; Supplementary Figure S4A), but not with CD4 mean fluorescent intensity (r=−0.069, P=0.77; data not shown). This suggests that although CD4 and CCR5 are required for cellular viral entry (Figures 1d and 2c), the expression level of CCR5 is a critical determinant of HIV entry into cervix-derived CD4+ T cells. We also observed a strong correlation between HIV entry into bulk cervix-derived CD4+ T cells and the frequency of CCR5+ CD4+ T cells in blood (r=0.637, P=0.007; Figure 3e), probably because of the strong correlation of CCR5 expression in CD4+ T cells between the two compartments (r=0.650, P=0.006; Supplementary Figure S4B). These data further support the notion that CCR5 is a strong correlate of susceptibility to HIV, and suggest that expression level of CCR5 in blood may be a reasonable surrogate of the susceptibility of cervical CD4+ T cells to HIV.

HIV entry into cervix-derived CD4+ T cells correlates with CCR5 expression. (a) Gating strategy for CCR5+ and CCR5- CD4+ T cells in blood and cervix. Comparison of HIV entry into CCR5- vs. CCR5+ CD4+ T cells in (b) blood and (c) cervix. Correlation between HIV entry into cervix-derived CD4+ T cells and % CCR5+ CD4+ T cells in (d) cervix and (e) blood. All correlations were performed by Spearman’s correlation. In d and e, N=19 and 17, respectively. *P<0.0001.
Preferential HIV entry into activated cervical CD4+ T cells
We next assessed HIV entry into CD4+ T cells that expressed several putative correlates of HIV susceptibility, specifically the activation marker CD69, and the integrins α4β7 and α4β1. HIV entry into blood-derived CD69+ CD4+ T cells was 1.6-fold greater than CD69- CD4+ T cells (P<0.0001) (Figure 4a,b, Table 1), and CCR5 expression on these CD69+ CD4+ T cells was increased 2.4-fold (P=0.0002; Table 2). In the cervix, HIV entry into CD69+ CD4+ T cells (Figure 4a,c) was increased 1.9-fold (P=0.001; Figure 4c and Table 1), and CCR5 expression was 3.6-fold higher (P<0.0001; Table 2). These data demonstrate that activated CD4+ T cells are preferential targets of HIV entry in both blood and cervix, potentially because of increased CCR5 expression.

CD69 and CCR5 expression identifies CD4+ T cells susceptible to HIV. (a) Gating strategy for CD69+ and CD69- CD4+ T cells in blood and cervical compartments. Comparison of HIV entry between CD69+ and CD69- CD4+ T cells in (b) 24 blood and (c) 20 cervical samples. Comparison of HIV entry between CCR5- CD69-, CCR5- CD69+, CCR5+ CD69-, and CCR5+ CD69+ CD4+ T cells in (d) 24 blood and (e) 19 cervical samples. Graphs show median values, and statistical comparisons are done by Wilcoxon rank-pairs test. *P<0.05, **P<0.01, and ***P<0.0005.
Next, we assessed the impact of coexpression of CCR5 and CD69 on HIV entry to determine the stronger correlate of susceptibility. In blood, in comparison with CCR5- CD69- CD4+ T cells (median 4.1%, IQR 2.1–5.2%, P<0.0001), CCR5+ CD69+ CD4+ T cells were most susceptible to HIV entry (median 12.9%, IQR 8.3–22.4%) followed by CCR5+ CD69- (median 8.1%, IQR 6.3–15.7%, P=0.0003), and CCR5- CD69+ (median 6.9%, IQR 3.3–8.9%, P=0.0002) CD4+ T cells, respectively (Figure 4d). The same associations were seen in the cervix: CCR5+ CD69+ CD4+ T cells were most susceptible (median 22.8%, IQR 14.0–28.4%) followed by CCR5+ CD69- (median 16.7%, IQR 7.6–29.2%, P=0.24), CCR5- CD69+ (median 10.4%, IQR 4.4–14.4%, P=0.002), and CCR5- CD69- (median 7.0%, IQR 3.4–13.5%, P=0.0003) CD4+ T cells (Figure 4e). HIV entry into CCR5+ CD69- CD4+ T cells was 1.2- and 1.8-fold higher compared with CCR5- CD69+ CD4+ T cells in blood (P=0.001) and the cervix (P=0.04), respectively; this suggests that CCR5 is a stronger correlate of HIV entry and that the enhanced HIV entry into activated CD4+ T cells may be largely because of the higher expression of CCR5.
Integrins and CD4+ T-cell susceptibility
Next, we assessed the association of integrin expression with cellular HIV entry. First, we demonstrated that nearly all of blood- and cervix-derived α4+β7- CD4+ T cells expressed the integrin β1 (median 99.8% IQR 96.9–100.0% and median 94.7% IQR 90.6–100.0% respectively; Figure 5a,b); therefore, in subsequent Nairobi-based work, the α4+β7- CD4+ T-cell subset was defined as being α4β1+.

α4β7 and α4β1 expression identifies CD4+ T cells susceptible to HIV. Gating strategy for integrin β1 expression in α4+β7- CD4+ T cells from (a) blood and (b) cervical samples obtained from 10 HIV-negative participants in Toronto, Canada. Gating strategy is shown for α4-β7-, α4+β7+ (α4β7+), and α4+β7- (α4β1+) CD4+ T cells derived from (c) blood and (d) cervix. Percent expression of α4β1 and α4β7 in (e) blood and cervix-derived CD4+ T cells and percent HIV entry into α4-β7-, α4β7+, and α4β1+ CD4+ T cells in (f) blood and (g) cervix. In c–g, N=24 for blood and 20 for cervical samples obtained from HIV-negative participants from Nairobi, Kenya. Graphs show median values and statistical comparisons are done by Wilcoxon rank-pairs test. *P<0.05, **P<0.005, and ***P<0.0001.
Although expression of the integrins α4β7 and α4β1 was higher in CD4+ T cells derived from blood than the cervix (median 12.4% vs. 5.2%, P=0.0008; and 25.0% vs. 18.8%, P=0.02, respectively; Figures 5c–e), the expression of either integrin was associated with enhanced HIV susceptibility in both compartments. In blood, HIV entry was 3.8-fold higher into α4β7+ (P<0.0001) and 7.3-fold higher into α4β1+ (P<0.0001) CD4+ T cells compared with α4-β7- CD4+ T cells (Figure 5f and Table 1), whereas α4β7+ and α4β1+ CD4+ T cells in the cervix demonstrated 1.7-fold (P=0.003) and 1.8-fold (P=0.0002) higher HIV entry, respectively (Figure 5g and Table 1). Expression of CCR5 was higher in both α4β7+ and α4β1+ CD4+ T-cell subsets compared with α4-β7- CD4+ T cells in blood and the cervix (all P≤0.002; Table 2), but differences in CCR5 expression did not completely explain enhanced HIV susceptibility. In blood, although α4β1+ CD4+ T cells had similar CCR5 expression to α4β7+ CD4+ T cells, HIV entry was nearly twofold higher in the former subset (Tables 1 and 2 and Figure 5f). In the cervix, CCR5 expression was ∼1.4-fold lower in α4β1+ CD4+ T cells compared with α4β7+ CD4+ T cells (P=0.0006), but HIV entry levels were similar (P=0.54, Tables 1 and 2 and Figure 5g). The cause of the enhanced HIV entry into α4β1+ CD4+ T cells is not known, but these data suggest that although CCR5 is a critical determinant of HIV entry, CCR5-independent mechanisms may also alter HIV entry into α4β1+ and/or α4β7+ CD4+ T cells. Importantly, although both α4β7+ and α4β1+ CD4+ T cells were preferential cellular targets of HIV in the cervix, α4β1+ CD4+ T cells were 3.6-fold more abundant (Figure 5e), suggesting that α4β1+ CD4+ T cells may be important early cellular target of HIV in the FGT.
Enhanced HIV susceptibility of CD4+ T cells expressing α4β7 and α4β1 integrins is not mediated by direct envelope–integrin interaction
To further elucidate the mechanism of enhanced HIV entry into α4β7+ and α4β1+ CD4+ T cells, we tested whether recombinant Q259d2.17 gp140 env could bind to integrins on T cells in a CD4-independent manner. First we confirmed proper folding of Q259d2.17 gp140 by immunoprecipitation of gp140 with soluble CD4 dimer and various antibodies that target different regions of envelope including the V1/V2 glycan peptide, V3 loop, and the CD4-binding site (data not shown). To assess CD4-independent binding of α4β7 and α4β1 to gp140, CD4+ T cells were removed from PBMCs by positive selection and the remaining cells were stimulated with all-trans retinoic acid (ATRA) to upregulate expression of β7 (as described elsewhere26). We first demonstrated that mucosal vascular addressin cell adhesion molecule-1 bound to ATRA-stimulated CD8+ CD4- T cells (Figure 6a) expressing high levels of α4, β7, and β1 (Figure 6b), confirming that our assay conditions were conducive for ligand binding to integrins. In contrast, Q259d2.17 gp140 did not bind to ATRA-stimulated CD8+ CD4- T cells despite high levels of expression of α4, β7, and β1 (Figure 6a,b). However, gp140 bound to the small proportion (<2%) of CD4+ CD8- T cells that remained after CD4 depletion (Figure 6a), further confirming that the gp140 envelope was properly folded and capable of binding to CD4. Similar results were observed in ATRA-unstimulated blood-derived T cells (data not shown) and in cervical cytobrush-derived cells, where HIV gp140 bound to CD4+ CD8- T cells but not to CD8+ CD4- T cells (Supplementary Figure S5). These data suggest that enhanced HIV susceptibility of CD4+ T cells expressing α4β7 or α4β1 was not mediated by direct binding between the HIV envelope and α4β7 or α4β1.

Enhanced HIV susceptibility of CD4+ T cells expressing α4β7 and α4β1 integrins is not mediated by direct envelope–integrin interaction. (a) CD4-depleted peripheral blood mononuclear cells (PBMCs, left panel) cultured for 6 days in all-trans retinoic acid (ATRA)-conditioned media were incubated with phosphate-buffered saline (PBS; Mock), Q259d2.17 gp140-FLAG, or MAdCAM-1-Fc and stained with FITC-anti-FLAG or FITC-anti-human IgG Fc as appropriate (right panel). (b) ATRA (RA)-stimulated and unstimulated cells stained with antibodies against α4, β7, and β1. Representative figure from three independent experiments is shown in a and b. Blood-derived CD4+ T cells from 9 participants were preincubated with integrin blockers (c) 10 μg HP2/1 or (d) 33 nM Act-1 or isotype control and then infected with Q259d2.17 pseudotypes. (e) Fold difference in virus entry into α4β1+ vs. α4β7+ blood-derived CD4+ T cells from 10 participants was compared using BlaM-Vpr pseudotypes that expressed Q259d2.17 or 92Ug037 envelopes. BlaM, β-lactamase; FMO, fluorescence minus one; NS, not significant; Vpr, viral protein R.
We further tested whether α4β7 and/or α4β1 directly promote HIV infection by preincubation of blood-derived CD4+ T cells with the α4β7 inhibitor Act-1 or the α4 inhibitor HP2/1. We targeted the common α−subunit of α4β7 and α4β1 using HP2/1 because antagonistic antibodies against the α4β1 heterodimer are not available. Neither inhibitor had any impact on virus entry: median virus entry after Act-1 incubation was 3.1% (IQR=1.9–4.9% vs. 3.8%, IQR=2.9–4.8% for isotype control; Figure 6c, P=0.64), and entry after HP2/1 incubation was 3.7% (IQR=2.4–4.7% vs. 3.8%, IQR=1.8–5.1% for isotype control; (Figure 6d, P=0.9). We confirmed that Act-1 bound to α4β7 on T cells as preincubation with unlabeled Act-1 reduced staining with Act-1-PE (data not shown). These results suggest that although expression of α4β7 and α4β1 correlates with HIV susceptibility, these integrins may not play a direct role in HIV entry into CD4+ T cells.
Thus far, the data presented were obtained using BlaM-Vpr pseudotypes containing the Q259d2.17 env, where integrin binding had not been previously assessed. Therefore, we sought to confirm our results using pseudotypes expressing the R5-tropic env 92Ug037, previously shown to have threefold higher binding to α4β7 compared with α4β1.16 However, despite the use of pseudotypes expressing the 92Ug037 env that preferentially binds α4β7, we still observed approximately twofold greater virus entry into α4β1+ CD4+ T cells compared with α4β7+ CD4+ T cells (Figure 6e), whereas CCR5+ and CD69+ CD4+ T cells remained preferential targets of HIV entry (Supplementary Figure S6). Collectively, these data demonstrate that the integrin binding properties of HIV envs do not influence virus entry into CD4+ T cells, at least in our system.
DISCUSSION
HIV infection after sexual exposure is thought to be caused by the infection of a small number of highly susceptible mucosal target cells,27 with subsequent slow local expansion leading to eventual systemic dissemination.28, 29 However, the low risk of HIV acquisition after sexual exposure has hampered the study of mucosal HIV transmission/susceptibility in human cohorts, and the identification of optimal HIV targets in the FGT. Therefore, we applied the BlaM-Vpr HIV entry assay as a means to rapidly quantify viral entry into the cytosol of freshly isolated and unstimulated endocervical CD4+ T cells, and identified several preferential cellular targets of HIV. Cervix-derived CD4+ T cells were more susceptible to HIV entry than those from blood, although within an individual, HIV entry was strongly correlated between the two compartments. Activated (CD69+) CD4+ T cells and those expressing the integrins α4β7 and α4β1 were preferential targets for HIV entry in both the cervix and blood. However, rather than increased susceptibility of the latter subsets being due to any direct interaction of these integrins with the HIV envelope, our results suggest that increased virus entry was likely because of increased CCR5 expression in these subsets and potentially other as yet undefined factors.
In keeping with prior studies,4, 30 cervix-derived CD4+ T cells demonstrated enhanced immune activation and increased CCR5 expression compared with blood. Our data support the hypothesis that this enhances HIV susceptibility at the mucosa, as cervical CD4+ T cells were highly susceptible to cellular HIV entry. In addition, the increased proportion of metabolically active (Ki67+) CD4+ T cells present in the cervix suggests that cervical CD4+ T cells may be more likely to sustain productive HIV infection after virus entry, as ex vivo infected Ki67+ CD4+ T cells in the cervix demonstrated increased de novo simian immunodeficiency virus (SIV) production compared with Ki67- CD4+ T cells.28, 31 In summary, the enhanced HIV susceptibility of cervix-derived CD4+ T cells compared with blood is consistent with differences in expression of several correlates of HIV susceptibility between the two compartments such as CCR5, CD69, and Ki67; this suggests that HIV entry detected using the BlaM-Vpr assay is a good proxy of physiologically relevant alterations in mucosal HIV susceptibility. Furthermore, the strong positive correlation that was observed between HIV entry into cervix-derived CD4+ T cells and CCR5 expression by and/or HIV entry into blood-derived CD4+ T cells suggests that at least in some contexts, differences in CCR5 expression by and/or HIV entry into blood-derived CD4+ T cells may be a useful end point to assess cervical HIV susceptibility.
Increased HIV susceptibility at the mucosa may relate to changes in the phenotype of putative HIV target cells and/or be due to an increase in the number of mucosal target cells.4 For instance, HSV-2 infection increases the risk of HIV acquisition by approximately threefold,32 and is associated with an increase in the overall number and expression of CCR5 and CD69 by CD4+ T cells in the cervix.33 Furthermore, specific genital CD4+ T-cell subsets may be preferential targets of productive HIV,28, 34 including activated CD4+ T cells34 and T helper type 17 cells.35 The frequency of T helper type 17 cells is increased in the mucosa, and these cells not only express higher levels of α4β7 and CCR5,24 but are depleted from the cervix very early in HIV infection.24 Therefore, in this study, the preferential entry of a sexually transmitted R5-tropic BlaM-Vpr HIV pseudovirus into CD69+, CCR5+, and α4β7+ CD4+ T cells demonstrates that enhanced cellular viral entry may be an important mechanism for enhanced HIV susceptibility in activated cervical CD4+ T cells and T helper type 17 cells. This is likely because of increased expression of CCR5 in α4β7+ CD4 T+ cells, and potentially to other unknown factors, rather than a direct effect of α4β7 on HIV entry. HIV susceptibility at the genital mucosa may also be associated with the number of target cells (CD4+ T cells), and the protection of macaques from SIV infection after intravenous administration of Act-1 may be in part due to the substantial reduction in cervical CD4+ T cell numbers that were observed early after Act-1 administration.36 Finally, our data suggest that α4β1+ CD4+ T cells may be preferential cellular targets for HIV in the FGT. The α4β1+ CD4+ T cells are common in the endocervix, and in the murine model, this integrin is important in genital CD4+ T-cell homing and pathogen clearance during C. trachomatis infection.13 Consistent with prior studies,24, 37 we reported higher expression of α4β7 in blood than cervix, possibly because of downregulation of the integrin after mucosal homing. Further work will be needed to define the mechanism(s) underlying increased HIV susceptibility in α4β7+ and α4β1+ CD4+ T cells and should also investigate the role of these integrins in the homing of CD4+ T cells to the genital tract in the context of sexually transmitted diseases and alterations in the genital microbiota.
Previously, it has been difficult to define cervix-derived CD4+ T-cell subsets that are preferentially infected by HIV ex vivo because of the low number of immune cells obtained by minimally- invasive sampling methods such as endocervical cytobrush sampling or ectocervical biopsies.22 Assessment of cellular viral entry using the reporter enzyme BlaM fused to Vpr addresses the critical issue of sensitivity because BlaM causes multiple rounds of cleavage of CCF2-AM, thereby amplifying the blue/green signal from ex vivo infected cells. However, our assessment of ex vivo HIV entry does have limitations. We observed some entry of an R5-tropic HIV pseudovirus into apparently CCR5- CD4+ T cells. This occurred despite the absolute requirement of CCR5 for virus entry into blood and cervix-derived CD4+ T cells as demonstrated by the ablation of virus entry by CCR5 (but not CXCR4) blockade. In addition, the complete inhibition of HIV entry by the fusion inhibitor T20 (Fuzeon) demonstrates that HIV entry was not because of nonspecific effects. The low levels of virus entry into CCR5- CD4+ T cells is in keeping with data from the macaque SIV model, where memory CD4+ T cells that appeared to be CCR5- by flow cytometry harbored similar SIV levels to CCR5+ cells, and were subsequently shown to express 20-fold higher levels of CCR5 mRNA than naive CD4+ T cells.38, 39 The most likely explanation for maraviroc-inhibited virus entry into CD4+ T cells that appear to be CCR5- by flow is that these cells are in fact expressing low levels of CCR5, below the threshold of detection. This may also account for the observation that percent HIV entry into CD4+ T cells sometimes exceeds the percent CCR5 expression, although other factors on the cell surface, such as G protein-coupled receptors,40 can also modulate HIV entry. Nevertheless, the detection of cellular HIV targets that appear to be CCR5- by flow cytometry may in fact constitute an advantage of the BlaM-Vpr assay, as it can identify cells that might otherwise not be considered as HIV targets.
In order to assess ex vivo HIV infection, we used pseudotypes produced in 293T cells that display different envelope glycosylation patterns to primary CD4+ T cells. Infection of a small number of unstimulated, cytobrush-derived CD4+ T cells by a T cell-derived virus, although more physiological, could not be detected in our hands by p24 enzyme-linked immunosorbent assay or intracellular staining (data not shown); the latter would require expansion and activation of cytobrush-derived cells that would affect our ability to assess the impact of in vivo parameters (immune activation, genital infections, and so on) on HIV susceptibility. Nonetheless, Cavrois et al.41 previously used a replication-competent BlaM-Vpr HIV provirus to demonstrate a direct correlation between HIV entry and de novo viral replication. Furthermore, we found that HIV entry was much higher into cervical than blood-derived CD4+ T cells, directly correlating with cellular immune activation and CCR5 expression, strongly suggesting that the BlaM-Vpr HIV entry assay measures physiologically relevant HIV entry and is an appropriate tool to assess HIV susceptibility of cervical cells obtained by cytobrush.
In conclusion, a rapid and sensitive assay to quantify HIV entry into unstimulated endocervical CD4+ T cells demonstrated enhanced susceptibility of cervix-derived CD4+ T cells, with preferential HIV entry into activated endocervical CD4+ T cells and those expressing the integrins α4β7 or α4β1. Enhanced susceptibility of the latter subsets did not appear to be mediated by direct integrin–envelope interaction, and was related in part to increased CCR5 expression. Future studies using the BlaM-Vpr assay will be useful to identify novel cellular targets of HIV and/or assess the impact of clinical interventions such as treatment of sexually transmitted diseases or BV on genital HIV susceptibility.
METHODS
Ethics statement. Informed written consent was obtained from all participants before enrolment, and the study was approved by Institutional Review Boards at both Kenyatta National Hospital (Nairobi, Kenya) and St Michael’s Hospital (Toronto, ON, Canada), as well as the Universities of Manitoba and Toronto (Canada). The described studies were conducted according to the principles expressed in the Declaration of Helsinki.
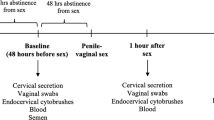
Study participants. Female participants were recruited from two outpatient clinics in Nairobi, Kenya. Participants had no vaginal discharge, no clinical signs/symptoms of genital inflammation, were pre-menopausal, and were not actively menstruating. HIV rapid testing was performed according to Kenyan national guidelines (using Determine, Inverness Medical, Waltham, MA), and only HIV-uninfected participants were enrolled. BV was diagnosed by Gram’s stain based on Nugent criteria,23 C. trachomatis and N. gonorrhoeae were screened by PCR (Roche, Pleasonton, CA), syphilis by serology (Diagnostics Worldwide, Brooklyn, NY), and T. vaginalis by In-Pouch culture (Biomed Diagnostics, White City, OR) A semistructured questionnaire capturing a range of behavioral and reproductive health data was administered to all participants.
For analysis of surface expression of integrins α4, β7, and β1 on CD4+ T cells, female participants were recruited from a Colposcopy Clinic at St Michael’s Hospital, Women’s Health Care Centre in Toronto, Canada. Recruited participants self-reported to be HIV negative, were not actively menstruating at the time of sample collection, and did not present with clinical signs and symptoms of genital inflammation. As our goal in this study was to develop the BlaM-Vpr assay for use in cytobrush-derived endocervical CD4+ T cells, formal HIV or sexually transmitted disease testing was not performed and samples were not collected from any specific stage of the menstrual cycle.
Sample collection and processing. Heparinized blood (10 ml) was collected by venipuncture, and two cervical cytobrushes by insertion of the cytobrush into the endocervical os and rotation through 360°. Cytobrushes were then transferred to a conical vial containing complete media (RPMI-1640, fetal bovine serum, antibiotic/antimycotic cocktail containing clindamycin, streptomycin, polymyxin B, amphotericin B, and gentamycin) and transported on ice to the laboratory for processing. PBMCs were isolated by Ficoll density separation and reconstituted at 107 cells per ml in complete media. Cervical immune cells were removed from the cytobrushes by agitation, washed in complete media, filtered through a 100-μm cell strainer, and reconstituted in 200 μl complete media for use in the HIV entry assay.
BlaM-Vpr HIV entry assay. The BlaM-Vpr HIV entry assay has been described in detail elsewhere.41, 42 Briefly pseudovirions incorporating BlaM fused to the Vpr are used to infect blood and cervical cells by spinoculation. Samples are then loaded with a cell-permeant dye, CCF2-AM, that is susceptible to cleavage by BlaM. CCF2 is a fluorescence resonance energy transfer substrate that emits green light at 520 nm when excited by a 405-nm laser. Upon cleavage by BlaM, blue light is emitted at 447 nm. The change in blue/green emission can be monitored by flow cytometric analysis and flags cells with cytosolic entry of virus.
As our primary goal was to develop an infection assay relevant for sexual transmission of HIV, the envelope used for pseudotyping the BlaM-Vpr virions was early transmitted, CCR5-tropic and Clade A, the most common clade in Kenya,43 where we performed our mucosal studies. As described elsewhere, the Q259d2.17 envelope had been cloned from an HIV isolate that was obtained from a Kenyan woman, 1 week after HIV seroconversion. BlaM-Vpr pseudovirions were produced by transfection of 293T cells with 20 μg HIV backbone lacking envelope Q23 Δ env gfp nef,44 10 μg early-transmitted R5-tropic Clade A envelope Q259d2.17env,25 10 μg pCMV-BlaM-Vpr (Addgene, Cambridge, MA), 5 μg of pAdvantage (Promega, Madison, WI), and 135 μl of the transfection reagent Polyjet (FroggaBio, Toronto, ON, Canada). At 48 h after transfection, supernatant was collected, filtered through a 0.45 μm filter, and incubated for 15 h at 4 °C with PEG-it to allow formation of virus-PEG complexes as per the manufacturer’s instructions (SystemBio, Mountain View, CA). Virus-PEG-it complexes were precipitated at 1,500 g for 30 min at 4 °C and concentrated 100-fold in phosphate-buffered saline and stored at −80 °C. Viral stocks were titrated using the BlaM-Vpr HIV entry assay on reference PBMCs obtained from an HIV-negative donor and by p24 enzyme-linked immunosorbent assay (NCI, Frederick, MD). Input BlaM-Vpr pseudovirus used in experiments below was equivalent to ∼60% of maximum viral entry in the reference PBMCs or 37 ng by p24 enzyme-linked immunosorbent assay. Where specified, pseudotypes expressing Clade A, CCR5-tropic env 92Ug037 were produced using the same protocol.
For ex vivo infections, 106 PBMCs were incubated with BlaM-Vpr pseudovirus or phosphate-buffered saline (negative control; mock infection). Processed cervical cell suspension from each participant was divided in two and similarly incubated with either virus or mock. For testing of HIV entry inhibitors, cervical cells were split into three equal aliquots and preincubated with the appropriate inhibitor or vehicle for 30 min at room temperature followed by addition of virus/phosphate-buffered saline. For testing of various integrin-blocking antibodies, 10 μg HP2/1 (Beckman Coulter, Brea, CA), 33 nM Act-1 (NIH AIDS Reagent Program),16, 18 or isotype control were incubated with PBMCs for 30 min at room temperature, and then infected with BlaM-Vpr pseudovirions. Samples were spinoculated at 1,200 g for 2 h at 17 °C in 48-well flat-bottom plates to allow viral interaction with cells followed by 2 h of incubation at 37 °C, 5% CO2 to allow cell entry.41 Samples were washed twice in CO2-independent media (Invitrogen, Carlsbad, CA) and loaded with 1 μM CCF2-AM (Invitrogen) for 1.5 h. Samples were washed once in CO2-independent media and incubated for 12 h in CO2-independent media supplemented with 10% fetal bovine serum, antibiotic cocktail, and solution D (Invitrogen), an anion transport inhibitor to prevent leakage of CCF2-AM dye. Samples were then stained with fluorescent-labeled antibodies (as described below) and analyzed using a BD LSR-II Flow Cytometer (Becton Dickinson Bioscience, Franklin Lakes, NJ).
For analysis of HIV entry within each individual, the same flow cytometry gating strategy used for blood was applied to cervix, with the exception of the gates denoting HIV entry, as these were not consistent between the two compartments. Hence, an uninfected mock/phosphate-buffered saline control was included for both blood and cervical samples and used to gate on change in blue/green fluorescence (viral entry) between virus- and mock-treated samples.
Flow cytometry. Surface staining of cells with labeled monoclonal antibodies was performed for 30 min at 4 °C, and then acquired and analyzed using a BD LSR-II Flow Cytometer. Antibodies used for staining were (α4) CD49d-PE, integrin β7-PE-Cy5, CD3-A700, CCR5-APC-Cy7 (Becton Dickinson Bioscience), LIVE DEAD Far Red (Invitrogen), CD4-ECD (Beckman Coulter), and CD29 (integrin β1) eFluor 710 and CD69-PE-Cy7 (EBioscience, San Diego, CA).
Intracellular staining for Ki67 could not be performed on the same cervical samples used for the HIV entry assay because cell permeabilization, which is required for detection of Ki67, caused leakage of CCF2-AM. Hence, Ki67-FITC (Becton Dickinson Bioscience) staining was performed on cervical samples from separate participants at 4 °C for 30 min after staining for aforementioned cell surface markers including LIVE DEAD Far Red stain. All cellular events were recorded during flow cytometric analysis of cytobrush samples.
Gp140 production and purification. Mammalian codon-optimized Q259d2.17 gp140 env with a FLAG-tag was cloned into a mammalian expression vector pKF-Foldon. HEK293T cells were stably transfected with the vector using Fugene 6 according to the manufacturer’s recommendation (Promega) and selected for puromycin resistance. Supernatant was collected from stably transfected cells and purified using anti-FLAG-agarose beads as per the manufacturer’s instructions (Sigma, Oakville, ON, Canada). Purified protein was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proper folding of the purified protein was assessed by immunoprecipitation for 1.5 h at room temperature with several antibodies including PG9, 2G12, F105, 447-52D, b12 against various regions of HIV env, and with soluble CD4 dimer (CD4-IgG2). Antibodies used for immunoprecipitation were obtained from the NIH AIDS Reagent Program. This was followed by incubation with Protein G Dynabeads as per the manufacturer’s instructions (Life Technologies, Burlington, ON, Canada) and western blot for anti-FLAG M2 antibody (Sigma).
Integrin-binding assay. PBMCs of HIV-negative donors were obtained as previously described. To assess CD4-independent binding of HIV env to surface integrins, CD4+ T cells were depleted by positive selection (Stemcell, Vancouver, BC, Canada). The remaining cells were cultured for 6 days in RPMI, 10% fetal bovine serum supplemented with 10 nM ATRA (Sigma), 1 μg ml−1 OKT3 anti-human CD3 monoclonal antibody (Biolegend, San Diego, CA), and interleukin-2 (10 U ml−1; NIH AIDS Reagent Program). Cells were tested for expression of α4, β7, and β1 before and after ATRA stimulation using aforementioned antibodies to ensure upregulation of β7 expression before performing the integrin-binding assay. As described elsewhere, CD4-depleted PBMCs that were either ATRA stimulated or unstimulated were incubated with 10 μg FLAG-gp140 or 10 μg MAdCAM-1-Fc in buffer containing 10 mM HEPES, 150 mM NaCl, 100 μM CaCl2 (Sigma), 1 mM MnCl2 (Sigma), 0.5% bovine serum albumin (Sigma), and 0.09% sodium azide (Sigma).45 Cells were stained with FITC-anti-FLAG (Sigma) or FITC-anti-human IgG Fc (Biolegend) as required and then analyzed by flow cytometry.
Statistical analysis. Intraindividual differences in percent HIV entry and CCR5 and Ki-67 expression on CD4+ T-cell subsets were assessed using the Wilcoxon matched pairs signed-rank test. Correlations were determined using Spearman’s rank correlation. All statistical tests were run on Prism 6 (La Jolla, CA) on Mac OS. Flow cytometry data were analyzed in FlowJo v. 8.6.6. (Ashland, OR).
References
UNAIDS. Global report: UNAIDS report on the global AIDS epidemic 1–110 (2012).
Haase, A.T. Perils at mucosal front lines for HIV and SIV and their hosts. Nat. Rev. Immunol. 5, 783–792 (2005).
Hladik, F. & McElrath, M.J. Setting the stage: host invasion by HIV. Nat. Rev. Immunol. 8, 447–457 (2008).
McKinnon, L.R. & Kaul, R. Quality and quantity: mucosal CD4+ T cells and HIV susceptibility. Curr. Opin. HIV AIDS 7, 195–202 (2012).
Passmore, J.S. Genital tract inflammation and susceptibility to HIV infection in women from the CAPRISA 004 microbicide trial of tenofovir gel (Webcast at: http://aidsvac.capitalreach.com/console/player/19096?mediaType=audio In: AIDS Vaccine 2012. Boston, MA: 2012.
Levinson, P. et al. Levels of innate immune factors in genital fluids: association of alpha defensins and LL-37 with genital infections and increased HIV acquisition. AIDS 23, 309–317 (2009).
Kaul, R. et al. Genital levels of soluble immune factors with anti-HIV activity may correlate with increased HIV susceptibility. AIDS 22, 2049–2051 (2008).
Nazli, A. et al. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog. 6, e1000852 (2010).
Valere, K. Alpha-defensins increase HIV transcytosis: role in STI-mediated enhanced HIV transmission (Abstract #211) In: Conference on Retroviruses and Opportunistic Infections (CROI). Seattle, WA: 2015.
Margolis, L. & Shattock, R. Selective transmission of CCR5-utilizing HIV-1: the ‘gatekeeper’ problem resolved? Nat. Rev. Microbiol. 4, 312–317 (2006).
Gorfu, G., Rivera-Nieves, J. & Ley, K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr. Mol. Med. 9, 836–850 (2009).
Elices, M.J. et al. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell 60, 577–584 (1990).
Davila, S.J., Olive, A.J. & Starnbach, M.N. Integrin α4β1 is necessary for CD4+ T cell-mediated protection against genital Chlamydia trachomatis infection. J. Immunol. 192, 4284–4293 (2014).
Mazo, I.B. et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 22, 259–270 (2005).
Bauer, M. et al. Beta1 integrins differentially control extravasation of inflammatory cell subsets into the CNS during autoimmunity. Proc. Natl. Acad. Sci. USA 106, 1920–1925 (2009).
Arthos, J. et al. HIV-1 envelope protein binds to and signals through integrin α4β7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 9, 301–309 (2008).
Nawaz, F. et al. The genotype of early-transmitting HIV gp120s promotes α (4) β(7)-reactivity, revealing α (4) β(7) +/CD4+ T cells as key targets in mucosal transmission. PLoS Pathog. 7, e1001301 (2011).
Parrish, N.F. et al. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin α4β7. PLoS Pathog. 8, e1002686 (2012).
Tjomsland, V. et al. Blocking of integrins inhibits HIV-1 infection of human cervical mucosa immune cells with free and complement-opsonized virions. Eur. J. Immunol. 43, 2361–2372 (2013).
Hladik, F. et al. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 26, 257–270 (2007).
Sperling, R. et al. Differential profiles of immune mediators and in vitro HIV infectivity between endocervical and vaginal secretions from women with Chlamydia trachomatis infection: a pilot study. J. Reprod. Immunol. 99, 80–87 (2013).
McKinnon, L.R. et al. Optimizing viable leukocyte sampling from the female genital tract for clinical trials: an international multi-site study. PLoS One 9, e85675 (2014).
Nugent, R.P., Krohn, M.A. & Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 29, 297–301 (1991).
McKinnon, L.R. et al. Characterization of a human cervical CD4+ T cell subset coexpressing multiple markers of HIV susceptibility. J. Immunol. 187, 6032–6042 (2011).
Long, E.M., Rainwater, S.M.J., Lavreys, L., Mandaliya, K. & Overbaugh, J. HIV type 1 variants transmitted to women in Kenya require the CCR5 coreceptor for entry, regardless of the genetic complexity of the infecting virus. AIDS Res. Hum. Retroviruses 18, 567–576 (2002).
Cicala, C., Arthos, J. & Fauci, A.S. HIV-1 envelope, integrins and co-receptor use in mucosal transmission of HIV. J. Transl. Med. 9, S2 (2011).
Talbert-Slagle, K. et al. Cellular superspreaders: an epidemiological perspective on HIV infection inside the body. PLoS Pathog. 10, e1004092 (2014).
Zhang, Z. Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286, 1353–1357 (1999).
Haase, A.T. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu. Rev. Med. 62, 127–139 (2011).
Meditz, A.L. et al. CCR5 expression is elevated on endocervical CD4+ T cells in healthy postmenopausal women. J. Acquir. Immune Defic. Syndr. 59, 221–228 (2012).
Zhang, Z.-Q. Roles of substrate availability and infection of resting and activated CD4 T cells in transmission and acute simian immunodeficiency virus infection. Proc. Natl. Acad. Sci. USA 101, 1–6 (2004).
Freeman, E.E. et al. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20, 73–83 (2006).
Shannon, B. et al. Impact of asymptomatic herpes simplex virus type 2 infection on mucosal homing and immune cell subsets in the blood and female genital tract. J. Immunol. 192, 5074–5082 (2014).
Saba, E. et al. HIV-1 sexual transmission: early events of HIV-1 infection of human cervico-vaginal tissue in an optimized ex vivo model. Mucosal Immunol. 3, 280–290 (2010).
Rodriguez-Garcia, M., Barr, F.D., Crist, S.G., Fahey, J.V. & Wira, C.R. Phenotype and susceptibility to HIV infection of CD4. Mucosal Immunol. 7, 1375–1385 (2014).
Byrareddy, S.N. et al. Targeting α4β7 integrin reduces mucosal transmission of simian immunodeficiency virus and protects gut-associated lymphoid tissue from infection. Nat. Med. 20, 1397–1400 (2014).
Goode, D. et al. HSV-2-driven increase in the expression of α4β7 correlates with increased susceptibility to vaginal SHIVSF162P3 infection. PLoS Pathog. 10, e1004567 (2014).
Kader, M. et al. Alpha4(+)beta7(hi)CD4(+) memory T cells harbor most Th-17 cells and are preferentially infected during acute SIV infection. Mucosal Immunol. 2, 439–449 (2009).
Mattapallil, J.J. et al. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434, 1093–1097 (2005).
Shimizu, N. et al. Broad usage spectrum of G protein-coupled receptors as coreceptors by primary isolates of HIV. AIDS 27, 761–769 (2009).
Cavrois, M., de Noronha, C. & Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotech. 20, 1151–1154 (2002).
Cavrois, M., Neidleman, J., Bigos, M. & Greene, W.C. Fluorescence resonance energy transfer-based HIV-1 virion fusion assay. Methods Mol. Biol. 263, 333–344 (2004).
Buonaguro, L., Tornesello, M.L. & Buonaguro, F.M. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J. Virol. 81, 10209–10219 (2007).
Humes, D. & Overbaugh, J. Adaptation of subtype a human immunodeficiency virus type 1 envelope to pig-tailed macaque cells. J. Virol. 85, 4409–4420 (2011).
Cicala, C. & Arthos, J. in Methods in Molecular Biology, Humana Press 1087, 3–12 (2013).
Acknowledgements
We acknowledge the gracious support of the study participants in Nairobi and the Kenya AIDS Control Project (KACP) staff who contributed to patient care, data management, administration, and laboratory diagnostics. We also thank Sergey Yegorov, Kamnoosh Shahabi, and Connie Kim for helpful discussions and proofreading of this manuscript. We also appreciate the support of Julie Overbaugh and Stephanie Rainwater from the University of Washington for kindly providing HIV plasmid constructs. This study was supported by the Canadian Institutes of Health Research (grant OCH-131579 to R.K.; THA-11900 to M.A.O.; and studentship to V.R.J.) and by the Ontario HIV Treatment Network (salary award to R.K.).
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Joag, V., McKinnon, L., Liu, J. et al. Identification of preferential CD4+ T-cell targets for HIV infection in the cervix. Mucosal Immunol 9, 1–12 (2016). https://doi.org/10.1038/mi.2015.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.28
This article is cited by
-
CD4+ T cell memory
Nature Immunology (2023)
-
Th22 cells are efficiently recruited in the gut by CCL28 as an alternative to CCL20 but do not compensate for the loss of Th17 cells in treated HIV-1-infected individuals
Mucosal Immunology (2021)
-
The contraceptive medroxyprogesterone acetate, unlike norethisterone, directly increases R5 HIV-1 infection in human cervical explant tissue at physiologically relevant concentrations
Scientific Reports (2019)
-
Resident memory T cells are a cellular reservoir for HIV in the cervical mucosa
Nature Communications (2019)
-
Hormonal Contraceptives Differentially Suppress TFV and TAF Inhibition of HIV Infection and TFV-DP in Blood and Genital Tract CD4+ T cells
Scientific Reports (2017)
