
Abstract
Deregulation of host-microbiota interactions in the gut is a pivotal characteristic of Crohn’s disease. It remains unclear, however, whether commensals and/or the dysbiotic microbiota associated with pathology in humans are causally involved in Crohn’s pathogenesis. Here, we show that Crohn’s-like ileitis in TnfΔARE/+ mice is microbiota-dependent. Germ-free TnfΔARE/+ mice are disease-free and the microbiota and its innate recognition through Myd88 are indispensable for tumor necrosis factor (TNF) overexpression and disease initiation in this model. The epithelium of diseased mice shows no major defects in mucus barrier and paracellular permeability. However, TnfΔARE/+ ileitis associates with the reduction of lysozyme-expressing Paneth cells, mediated by adaptive immune effectors. Furthermore, we show that established but not early ileitis in TnfΔARE/+ mice involves defective expression of antimicrobials and dysbiosis, characterized by Firmicutes expansion, including epithelial-attaching segmented filamentous bacteria, and decreased abundance of Bacteroidetes. Microbiota modulation by antibiotic treatment at an early disease stage rescues ileitis. Our results suggest that the indigenous microbiota is sufficient to drive TNF overexpression and Crohn’s ileitis in the genetically susceptible TnfΔARE/+ hosts, whereas dysbiosis in this model results from disease-associated alterations including loss of lysozyme-expressing Paneth cells.
Similar content being viewed by others
INTRODUCTION
A mutually beneficial relationship has been evolutionarily shaped in the mammalian gut between the host and the complex communities of commensal microorganisms, collectively called as the “microbiota”. Misbalanced host-microbiota interactions are considered to underlie the pathogenesis of inflammatory bowel diseases (IBD), chronic inflammatory disorders of the intestine with two major forms, Crohn’s disease and ulcerative colitis.1 Crohn’s disease is manifested primarily in the terminal ileum, a part of the intestinal tract that provides a unique microenvironment for the growth of microbial communities. Contrary to the large bowel, the ileum is characterized by the formation of a single-layered and porous mucus barrier, which is penetrant to bacteria. 2 Antimicrobial peptides secreted by Paneth cells localized at the basal part of the crypts and an extensive network of lymphoid tissues, including Peyer’s patches and isolated lymphoid follicles, provide antimicrobial clearance and immunosurveillance.3, 4 Furthermore, the composition of mucosa-associated microbial populations in the ileum significantly differs from that of the colon both in healthy humans and in IBD patients,5 whereas a series of studies has established that Crohn’s disease is associated with dysbiosis.6 A recent relevant study showed that treatment-naive pediatric Crohn’s disease associates with enrichment of Enterobacteriaceae and depletion of Clades IV and XIVa of Clostridia in the mucosa of the terminal ileum.7 Apart from these microbial alterations associated with disease, genome-wide association studies have indicated several microbial recognition and host defense genes as associated with both Crohn’s disease and ulcerative colitis.8 For example, risk alleles in NOD2 and ATG16L1 genes lead in an additive manner to an abnormal Paneth cell phenotype in Crohn’s disease patients,9 indicative of a possible implication of defective microbial clearance in pathogenesis. Although recent progress in Crohn’s disease genetics and microbiome analysis strengthens the concept that misbalanced host-microbiota interactions underlie pathogenesis and also establish the development of a dysbiotic microbiota as a key feature associated with pathology, the causalities in abnormal host-microbiota interactions with regard to Crohn’s disease pathogenesis remain unknown.
Mouse models of IBD are essential for the understanding of mechanisms implicating host-microbiota interactions in human disease; however, relevant studies have been restricted in colitis models so far.10 TnfΔARE/+ mice chronically overproduce tumor necrosis factor (TNF) and spontaneously develop a fully penetrant Crohn’s-like IBD pathology localized specifically in the terminal ileum and occasionally also in the proximal colon.11 TnfΔARE/+ mice display typical histopathological features of human Crohn’s disease such as massive infiltration of acute and chronic effector cells in the lamina propria (LP), villi blunting, and development of transmural inflammation.11 Local overexpression of TNF in the gut specifically by intestinal epithelial cells (IECs) is sufficient to induce full-blown Crohn’s-like IBD pathology in this model.12 Thus, the TnfΔARE model is an ideal tool for the study of intestinal epithelial responses to luminal bacteria and vice versa, in a disease context sharing many common characteristics with human Crohn’s disease.
Here, we study host-microbiota interactions in murine modeled Crohn’s disease and demonstrate that pathology in TnfΔARE/+ mice depends on the presence of the commensal microbiota that is required for TNF overexpression in the intestine. We also evidence epithelial alterations occurring in established but not in early disease in TnfΔARE/+ mice and show that a dysbiotic microbiota is shaped in diseased mice. Our results suggest that commensals are sufficient to initiate Crohn’s disease pathogenesis in the presence of pro-inflammatory genetic alterations, whereas an overall dysbiotic microbiota with significant alterations at the level of phyla develops in the inflamed mucosa secondarily to disease and is not required for disease initiation.
RESULTS
Crohn’s-like IBD pathology and disease-initiating TNF overexpression in the intestine of TnfΔARE/+ mice depend on the presence of bacteria
To examine the role of the intestinal microbiota in Crohn’s disease pathogenesis, we generated germ-free TnfΔARE/+ mice and compared disease development with TnfΔARE/+ mice reared under specific pathogen-free conditions. Strikingly, germ-free TnfΔARE/+ mice were completely protected from IBD pathology showing no signs of inflammatory infiltration or villi blunting in the terminal ileum as evidenced by histological analysis at the age of 15 weeks, an age when TnfΔARE/+ mice show fully developed IBD pathology.13 In sharp contrast to germ-free TnfΔARE/+ mice, Crohn’s-like pathology was readily evidenced in the ileum of 11-week-old control TnfΔARE/+ mice originating from the same mouse colony and raised under identical but non-sterile, specific pathogen-free housing conditions (Figure 1a,b). These results demonstrate that the development of Crohn’s-like IBD pathology in TnfΔARE/+ mice is microbiota-dependent. The complete microbial dependence of intestinal pathology in TnfΔARE/+ mice suggests a possible implication of the microbiota in the very early events driving pathogenesis in this model, which are related to TNF overproduction. Relevantly, we have previously established that IECs are a major local source of early TNF overexpression in TnfΔARE/+ mice and that mice engineered to overexpress TNF specifically by IECs develop full Crohn’s-like IBD pathogenesis in the ileum.12 We examined the necessity of the intestinal microbiota in ileal TNF overexpression, by performing quantitative PCR analysis in the ileum showing significantly higher TNF expression levels in the ileum of specific pathogen-free compared with germ-free TnfΔARE/+ mice (Figure 1c). These results show that microbial signals are necessary for the induction of aberrant TNF expression in the intestine, a condition sufficient for disease initiation in this model. As innate recognition of the microbiota by several Toll-like receptors initiates signaling cascades through the Myd88 adaptor molecule and activates cytokine production, including TNF,14 we hypothesized that Myd88-mediated innate signaling may have a central role in IBD pathogenesis in the TnfΔARE model. Indeed, TnfΔARE/+Myd88−/− mice examined at the age of 16 weeks, a stage when fully developed disease is observed,13 showed a significantly attenuated IBD pathology compared with TnfΔARE/+ littermate controls, characterized by less inflammatory infiltration in the ileal mucosa and the submucosa and showing no signs of transmural inflammation, as it was the case for TnfΔARE/+ controls (Figure 2a,b). To examine whether Myd88 is required for TNF overexpression in TnfΔARE/+ mice, we isolated peritoneal macrophages from TnfΔARE/+, TnfΔARE/+Myd88−/−, Myd88−/−, and wild-type (WT) control mice and measured TNF secretion in response to lipopolysaccharide stimulation. Macrophages isolated from TnfΔARE/+Myd88−/− mice showed defective lipopolysaccharide-induced TNF secretion compared with TnfΔARE/+ macrophages and reached levels similar to that of WT macrophages (Figure 2c). This suggests that innate response to lipopolysaccharide through Myd88 drives TNF overexpression and subsequently mediates IBD pathogenesis in TnfΔARE/+ mice. These results establish that Toll-like receptor/Myd88-mediated innate recognition is required for TNF-driven IBD pathology development, in contrast to the homeostatic role established for Toll-like receptor/Myd88 signaling in epithelial-injury-induced colitis,15 and also indicate the presence of additional, Myd88-independent, innate pathways mediating TNF-driven murine Crohn’s-like pathogenesis.

Germ-free TnfΔARE/+ mice are completely rescued from TNF overexpression in the intestine and Crohn’s-like IBD pathology. (a) Histological analysis was performed in the terminal ileum of n=6 TnfΔARE/+ mice raised under germ-free conditions at the age of 15 weeks. N=7 TnfΔARE/+ mice raised under SPF conditions (11 weeks) were used as controls. Representative H&E-stained sections are shown. (Scale bars: 100 μm). (b) Histopathological analysis performed in the terminal ileum of germ-free and SPF control TnfΔARE/+ mice examined at the age of 15 and 11 weeks, respectively. Statistical significance was calculated by Kruskal–Wallis one-way analysis of variance. (c) Quantification of TNF gene expression in whole tissue biopsies from the terminal ileum of TnfΔARE/+ mice reared under germ-free and SPF conditions, examined at the age of 15 and 11 weeks, respectively. Statistical significance was calculated with two-tailed Mann–Whitney test. All data shown represent mean±s.e.m. H&E, hematoxylin and eosin; IBD, inflammatory bowel disease; TNF, tumor necrosis factor; SPF, specific pathogen-free.

Myd88 mediates TNF overexpression and IBD pathogenesis in TnfΔARE/+ mice. (a) Crohn’s-like pathology is attenuated in the terminal ileum of 16-week-old TnfΔARE/+Myd88−/− mice as compared with TnfΔARE/+ littermate controls. Representative H&E-stained sections are shown. Scale bars: 50 μm. (b) Histopathological analysis performed in the terminal ileum of TnfΔARE/+Myd88−/− and TnfΔARE/+ control mice at the age of 16 weeks. Statistical significance was calculated with Mann–Whitney test. **P<0.01. (c) Thioglycollate-elicited peritoneal macrophages (see Supplementary Methods online) were isolated from 16-week-old TnfΔARE/+, TnfΔARE/+Myd88−/−, Myd88−/−, and WT control mice (n=3 mice per genotype) and stimulated with LPS for 6 h. TNF secretion was measured by ELISA. Macrophages isolated from TnfΔARE/+Myd88−/− mice show defective LPS-induced TNF secretion. Statistical significance was calculated with unpaired Welch t test. **P<0.01. All data shown represent mean±s.e.m. H&E, hematoxylin and eosin; IBD, inflammatory bowel disease; LPS, lipopolysaccharide; TNF, tumor necrosis factor; WT, wild type.
The inflamed ileum of TnfΔARE/+ mice is characterized by epithelial alterations involving loss of lysozyme-expressing Paneth cells and defective antimicrobial responses
High-resolution endoscopy performed in the terminal ileum of diseased TnfΔARE/+ mice showed intestinal villi with increased diameter and an irregular shape, an observation consistent with the increased inflammatory infiltration in the LP in this model (Figure 3a). The intestinal epithelium of diseased TnfΔARE/+ mice is also characterized by the accumulation of αE+CD8αβ+ effector intraepithelial T-lymphocytes (Figure 3b). We have previously shown that these cells home to the ileum in a αEβ7 integrin-dependent manner where they predominate over the resident non-conventional TCRγδ and TCRαβCD8αα T-lymphocytes and express interferon (IFN)-γ and TNF.16 To examine the possible effects of cytokine-expressing immune effectors on intestinal epithelial functions, we characterized barrier properties, epithelial cell type populations, and gene expression in the intestinal epithelium of diseased TnfΔARE/+ mice. TNF and IFN-γ have been suggested to promote a leaky paracellular transport of solutes across the intestinal epithelium.17 We examined the possible presence of defects in paracellular epithelial barrier permeability in the terminal ileum of diseased TnfΔARE/+ mice by measuring blood to lumen 51Cr–EDTA clearance. No difference was found between TnfΔARE/+ mice and WT littermates in the paracellular permeability for this small molecule (364 Da) in the ileum (Figure 3c). In addition, no significant difference was found in the lumen to blood permeability of orally administered 20 kDa fluorescein isothiocyanate-dextran (Figure 3d). Although malfunctions in the paracellular epithelial barrier in TnfΔARE/+ mice cannot be generally excluded based on these experiments, these argue against the presence of major defects permitting the passage of luminal molecules. At the level of transcellular ion transport and ion channel function, we have previously reported defects in bicarbonate secretion and fluid absorption in the ileum of TnfΔARE/+ mice, which may affect luminal pH and gut microbiota composition in these mice.18 The reduced fluid absorption in the ileum is compensated in the colon of TnfΔARE/+ mice,18 thus no diarrhea is observed (Figure 3e). Quantification of Goblet cells per villus by Periodic acid-Schiff staining showed no difference between diseased TnfΔARE/+ and WT control mice (Figure 3f). Furthermore, mucin 2 secretion by Goblet cells was also found similar between TnfΔARE/+ mice and WT controls (Figure 3g). These results suggest that no major defects occur in the formation of the mucus barrier in the inflamed ileum of TnfΔARE/+ mice. In contrast, Paneth cell quantification by immunostaining using anti-lysozyme antibodies revealed significantly reduced lysozyme-expressing Paneth cell numbers per crypt in the ileum of TnfΔARE/+ mice at the age of 8 and 16 weeks, an intermediate and a full-blown disease stage, respectively (Figure 4a). Sixteen-week-old TnfΔARE/+ mice show significantly reduced Paneth cells compared with 8-week-old TnfΔARE/+ mice, indicative of an association between pathology stage and Paneth cell numbers. In addition, 8-week-old TnfΔARE/+ mice showed significantly reduced Paneth cell numbers in crypts surrounded by extensive submucosal inflammation as compared with crypts located in areas where inflammation is less extensive or restricted in the mucosa (Figure 4b). These results show that disease progression in TnfΔARE/+ mice is associated with Paneth cell loss as assessed by immunostaining against lysozyme. To examine whether adaptive immune system effectors are required for this effect, we examined Paneth cell numbers in alymphoid TnfΔARE/+Rag1−/− mice. TnfΔARE/+Rag1−/− mice show strongly attenuated pathology characterized by persistent infiltration of neutrophils in the LP and villi blunting.13 Quantification of Paneth cells by immunostaining against lysozyme in 16-week-old TnfΔARE/+Rag1−/− mice showed significantly attenuated Paneth cell loss in their ileal crypts when compared with TnfΔARE/+ mice, to values approximating those of WT animals (Supplementary Figure S1a, c). In agreement with the previously described infiltration of neutrophils in the latter,13 lysozyme-producing inflammatory cells were observed in the LP of both TnfΔARE/+ and TnfΔARE/+Rag1−/− mice (Supplementary Figure S1a). Thus, adaptive immune system effectors are essential for Paneth cell loss in TnfΔARE/+ mice, whereas innate inflammatory infiltrates appear not to be sufficient to mediate Paneth cell loss. Furthermore, lysozyme immunostainings in 16-week-old TnfΔARE/+Ifng−/− mice, which also display an attenuated pathology,13 showed a significantly attenuated Paneth cell loss compared with TnfΔARE/+ littermate controls (Supplementary Figure S1b, c). These results indicate a role of IFN-γ-secreting adaptive immune effector cells in Paneth cell reduction in TnfΔARE/+ mice. Whether this is a direct effect of IFN-γ via its receptor on Paneth cells needs further investigation. Paneth cell-specific targeting of IFN-γ receptor may clarify this issue.

Epithelial barrier function in the ileum of diseased TnfΔARE/+ mice. (a) Endoscopic pictures from the terminal ileum of 12-week-old TnfΔARE/+ mice and WT controls. Representative of n=2 independent experiments. (b) Quantification of IELs isolated from the ileum of 12-week-old TnfΔARE/+ mice and WT controls (n=5 each). Unpaired two-tailed Student t-test (*** P<0.001). (c) Epithelial paracellular permeability, as measured by blood to lumen 51Cr–EDTA clearance in the terminal ileum of 12-week-old TnfΔARE/+ mice (n=4) and WT controls (n=4). No significant difference (ns) was found between the two groups by two-way ANOVA. (d) Epithelial barrier permeability of 12-week-old TnfΔARE/+ mice (n=7) and WT controls (n=6), as measured by lumen to blood passage of orally delivered 20kDa FITC-dextran. (e) Stool water content of TnfΔARE/+ mice (n=24) and WT littermates (n=18) at the age of 16 weeks. No significant (ns) differences were found by unpaired two-tailed Student t-test. (f, g) Goblet cell numbers and mucus secretion show no major defects in the inflamed ileum of 12-week-old TnfΔARE/+ mice compared with littermate WT controls (n=3 per group) as shown by PAS staining (e) and mucin 2 immunostaining (f). Unpaired two-tailed Student t-test. Scale bars: 50 μm. ANOVA, analysis of variance; FITC, fluorescein isothiocyanate; IEL, intraepithelial lymphocyte; PAS, Periodic acid-Schiff; R.E., relative expression; TNF, tumor necrosis factor; WT, wild type.

Loss of Paneth cells and altered expression of antimicrobials in the ileum of diseased TnfΔARE/+ mice. (a) Immunohistochemistry for lysozyme for the quantification of Paneth cells per crypt in the terminal ileum of TnfΔARE/+ mice at the age of 8 and 16 weeks and littermate WT controls (n=3 per group). Two-tailed Mann–Whitney test (***P<0.001). (b) Paneth cell numbers are significantly reduced as the local IBD histopathological score increases in the crypts of 8-week-old TnfΔARE/+ mice (n=3). A total of n=108 well-oriented crypts were examined. Indicative lysozyme immunostaining pictures are shown for each disease stage. (Scale bars: 33 μm). Statistical significance was calculated by two-tailed Mann–Whitney test (**P<0.01). (c) Relative mRNA expression of Defa21 in the last 2 cm of the terminal ileum of WT and TnfΔARE/+ mice at different ages (n=4–8 per group). Two-tailed Student t-test. (d) Relative expression of antimicrobials Cr2, Defa29, and Lyzc1 in whole tissue extracts from the last 2 cm of the ileum of WT and TnfΔARE/+ mice at 18 weeks (n=6 per group). Two-tailed Student’s t-test. (e) Relative expression of Tnf in whole tissue extracts from the last 2 cm of the ileum of WT and TnfΔARE/+ mice at different ages (n=4–8 per group). Two-tailed Student t-test. Each dot represents an animal. All data shown represent mean±s.e.m. IBD, inflammatory bowel disease; R.E., relative expression; TNF, tumor necrosis factor; WT, wild type.
To examine the functional consequences of Paneth cell loss in the epithelium of TnfΔARE/+ mice, we tested antimicrobial responses because these cells are a major source of antimicrobial peptides in the small intestine. quantitative PCR analysis performed in the ileum of 8-week-old TnfΔARE/+ mice and littermate WT controls for a panel of antimicrobial genes showed significantly decreased expression levels for the alpha defensin genes (Defa) Defa21 and Defa29, the complement receptor type 2 (CR2) and the lysozyme-encoding gene Lyzc1 in TnfΔARE/+ mice (Figure 4c,d). These results suggest that IBD pathology in TnfΔARE/+ mice is associated with diminished antimicrobial gene expression, in agreement with the loss of Paneth cells observed in these mice. To compare the above findings with inflammation levels, we measured gene expression levels of Tnf, the major driver of inflammation and pathogenesis in TnfΔARE/+ mice. In agreement with our previous results on an early local Tnf overexpression before disease onset in this model,12 TnfΔARE/+ mice showed increased Tnf expression in the ileum compared with their WT littermates at 4, 8, and 18 weeks of age (Figure 4e).
Dysbiosis, increased abundance of segmented filamentous bacteria, and increased epithelial attachment of bacteria in the terminal ileum of diseased TnfΔARE/+ mice
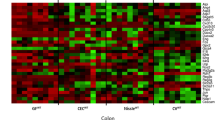
Antimicrobial peptides can affect the composition of the intestinal microbiota.19 To determine whether alterations in the expression of antimicrobial peptides and/or bicarbonate secretion defects observed in TnfΔARE/+ mice altered the composition of the microbiome, we analyzed the mucosa-associated microbiome in the distal ileum of TnfΔARE/+ and co-housed WT control littermates by 16S rDNA sequencing.20 Before disease onset, at 4 weeks of age, no significant difference in alpha diversity, beta diversity, or the relative abundance of the different phyla was found between WT and TnfΔARE/+ mice (Figure 5). After 8 weeks of age, a progressive increase in the relative abundance of Firmicutes was observed with a concomitant significant decrease in Bacteroidetes at 18 weeks of age. This progression was accompanied by a significant decrease in alpha diversity (species richness) and significant separation of WT mice from TnfΔARE/+ mice by weighted UniFrac analysis (Figure 5). These results indicate that the composition of the microbiome in the ileum of diseased TnfΔARE/+ mice and co-housed WT control littermates is markedly different. Detailed analysis of the 16S rDNA sequencing data revealed the presence of 139 operational taxonomic units (OTUs) present across the different samples after filtering (average read fraction 92±7%). At 4 and 8 weeks of age, none of these OTUs was significantly different between co-housed TnfΔARE/+ mice and WT littermate controls (Figure 6, Supplementary Figure S2). At 18 weeks of age, a significant dysbiosis was observed between co-housed TnfΔARE/+ mice and WT littermate controls. Out of the 139 OTUs detected, 23 were significantly different between WT and TnfΔARE/+ mice. In TnfΔARE/+ mice, 20 out of 21 OTUs that were decreased in abundance were of the phyla Bacteroidetes. Two OTUs (greengenes id: 376862, 425767) were significantly increased in TnfΔARE/+ mice compared with co-housed WT control littermates (Figure 6, Supplementary Figure S2). These two OTUs both belonged to the phylum Firmicutes and the genus Candidatus Arthromitus or Candidatus Savagella,21 informally known as segmented filamentous bacteria (SFB). These results show that at 18 weeks of age, compared with co-housed WT littermate controls, TnfΔARE/+ mice have an increased relative abundance of SFB in the terminal ileum.

Biome analysis of TnfΔARE/+ mice. (a) Relative abundance of phyla present in the terminal ileum of WT and TnfΔARE/+ mice at 4 (WT, n=6; TnfΔARE/+, n=9), 8 (WT, n=9; TnfΔARE/+, n=7), and 18 (WT, n=19; TnfΔARE/+, n=20) weeks of age. Data shown represent the most abundant phyla, while low abundant and unclassified OTUs were grouped in “Other”, * represents a significant (q<0.05) change in abundance between age-matched WT and TnfΔARE/+ mice. (b) Alpha diversity in TnfΔARE/+ mice at 4, 8, and 18 weeks of age. Statistical significance was calculated with Mann–Whitney test. (c) Weighted Unifrac distances between TnfΔARE/+ and co-housed WT littermate controls at 4, 8, and 18 weeks of age. Statistical significance was calculated with Adonis test. OTU, operational taxonomic unit; TNF, tumor necrosis factor; WT, wild type.

Increased abundance of Candidatus Arthromitus in TnfΔARE/+ mice. (a) In total, 139 OTUs were detected across all samples. Pearson hierarchical clustering of the abundance profiles of the 23 OTUs that were significantly altered in the terminal ileum of TnfΔARE/+ mice compared with co-housed WT littermate controls at 4, 8, and 18 weeks; 4 weeks: WT (n=6), TnfΔARE/+ (n=9); 8 weeks: WT (n=9), TnfΔARE/+ (n=7); 18 weeks: WT (n=19), TnfΔARE/+ (n=20). Two OTUs, 376862 and 425767, were significantly enriched at 18 weeks; both represent the genus Candidatus Arthromitus. Each column represents a different mouse. Left colored boxes indicate the family/genus taxonomy classification (T). Right colored boxes indicate if the comparison (P) between WT and TnfΔARE/+ shows significant difference (blue, P<0.05). (b) LOESS regression of the median read counts of the S24-7/- and Clostridiaceae/Candidatus Arthromitus OTUs shown in a. Shaded region represents standard error. (c) Boxplots showing the relative abundance of the two OTUs (376862, 425767) enriched in TnfΔARE/+mice at 18 weeks of age. OTU, operational taxonomic unit; TNF, tumor necrosis factor; WT, wild type.
The dysbiosis associated with the loss of Paneth cells and defects in antimicrobial genes expression observed in diseased TnfΔARE/+ mice may affect not only the composition but also the number of mucosa-associated bacteria. We performed fluorescence in situ hybridization analysis using a eubacterial probe on terminal ileal sections obtained from TnfΔARE/+ and WT littermate controls. We observed an increased number of bacteria in direct contact with IECs in TnfΔARE/+ mice compared with WT controls (Figure 7a,b). To confirm these results, we quantified the number of bacteria by pan-bacterial 16S rDNA quantitative PCR analysis. While the levels of bacterial 16S rDNA in the stool were not significantly different between WT and TnfΔARE/+ mice (Figure 7c), an increased number of mucosa-associated bacteria was detected in TnfΔARE/+ compared with WT animals (Figure 7d). Similar results were obtained using SFB-specific primers,22 i.e., TnfΔARE/+ mice showed an increase in the levels of mucosa-associated SFB (Figure 7e). These results suggest that in diseased TnfΔARE/+ mice, an increased number of bacteria, likely SFB, are in close contact with IECs.

Increased number of mucosa-associated bacteria in the terminal ileum of TnfΔARE/+ mice. (a, b) Visualization of bacterial localization in the distal ileum of WT and TnfΔARE/+ mice at 18 weeks. Transversal sections of the distal ileum were hybridized with a eubacterial 16S probe (red) and counterstained with Pan-keratin antibody to visualize the intestinal epithelial cells (green) and DAPI for nuclear staining (blue). Notice the increased attachment of bacteria to the epithelium of TnfΔARE/+ mice when compared with WT animals. Shown are representative images (a) and quantification of five mice per group (b). (c, d) Bacterial content in the stool (c) and in the distal ileum mucosa (d) of WT (n=4) and TnfΔARE/+ mice (n=5) at 18 weeks of age determined by pan-bacterial 16S qPCR amplification relative to host ubiquitin. (e) Relative expression of mucosa-associated SFB in the terminal ileum of WT and TnfΔARE/+ mice at 18 weeks of age. Each dot presents an animal. Data are expressed as mean±s.e.m. Statistical significance was calculated with two-tailed Student t-test. Scale bars=50 μm. DAPI, 4',6-diamidino-2-phenylindole; qPCR, quantitative PCR; R.E., relative expression. SFB, segmented filamentous bacteria; TNF, tumor necrosis factor; WT, wild type.
Modulation of the microbiota by antibiotic treatment reduces ileitis in TnfΔARE/+ mice
To determine whether we can affect Crohn’s-like disease progression by modulating the microbiota at an early disease stage, we treated TnfΔARE/+ mice with water or with 0.5 g l−. vancomycin in the drinking water. Antibiotic treatment was initiated at 6 weeks of age, an early disease stage when the first signs of pathology are observed, and maintained until analysis at 20 weeks of age, when full-blown disease is expected (Figure 8a). Histopathological analysis showed severe Crohn’s-like disease in TnfΔARE/+ mice treated with water (n=5). In contrast, none of the TnfΔARE/+ mice treated with antibiotics (n=5) showed signs of a Crohn’s-like disease (Figure 8b and d). These results establish the critical role of the microbiota in the progression of Crohn’s-like IBD and also highlight the therapeutic potential of microbiota modulation by antibiotics when administered at the early stages of chronic intestinal inflammation.

Antibiotics treatment rescues ileitis in TnfΔARE/+ mice. (a) Diagram of antibiotic treatment. TnfΔARE/+ mice at 6 weeks of age were treated with water (n=5) or vancomycin (n=5). Animals were killed at 20 weeks of age. Inflammatory infiltrates in the ileum of TnfΔARE/+ mice were completely abrogated in animals treated with vancomycin (c) when compared with water-treated controls (b). All mice (n=5 per group) were analyzed histologically and representative H&E-stained sections are shown. (d, e) Immunostaining of CD45 and Pan-keratin in the terminal ileum of TnfΔARE/+ mice treated with water (n=5) (d) or vancomycin (n=5) (e), killed at 20 weeks of age. (f) Ileitis incidence in 20-week-old TnfΔARE/+ mice treated with water (n=5) or vancomycin (n=5). H&E, hematoxylin and eosin; TNF, tumor necrosis factor.
DISCUSSION
The results presented in this study collectively provide a mechanistic interpretation of the complex host-microbiota dialogue in Crohn’s disease pathogenesis modeled in TnfΔARE/+ mice. First, we show that Crohn’s-like IBD pathology in the TnfΔARE model is microbiota-dependent, originating from TNF overexpression in the intestine, which is driven by a normal, indigenous microbiota. Second, we show that Crohn’s-like IBD pathology in TnfΔARE/+ mice is associated with dampened antimicrobial responses and reduction of Paneth cells, mediated by adaptive immune system effectors. We further establish that secondarily to disease, these conditions shape a dysbiotic gut microbiota in TnfΔARE/+ mice which is not sufficient to transfer disease to co-housed WT mice but also involves increased abundance of epithelial-adhering SFB, which may further promote disease.
Concerning our first conclusion that Crohn’s-like IBD pathogenesis in TnfΔARE/+ mice is microbiota-dependent, studies performed in other animal models of IBD, mainly in colitis models, have been controversial so far. Thus, an essential role in colitis pathogenesis has been shown for the microbiota in IL-10−/− mice and in HLA-B27 transgenic rats, which were completely rescued from colitis under germ-free conditions.23, 24 In contrast, germ-free conditions attenuated but did not prevent spontaneous pathology in the IL-2−/− mouse model of colitis.25 The role of the microbiota in murine ileitis was examined so far only in the SAMP1/YitFc model in which germ-free conditions led to an attenuated phenotype but did not prevent pathogenesis.26 Consistently with the fact that adaptive immune effectors are a primary cause of disease in this model bearing transferable pathogenic activity,27 the persistence of the SAMP1/YitFc ileal pathology under germ-free conditions has been attributed to defects in immunoregulation.26 These studies indicate the presence of microbiota-independent mechanisms sufficient for IBD pathogenesis, particularly when adaptive immune responses are deregulated. In contrast, the microbiota is indispensable for the development of ileitis in the TnfΔARE model where innate and adaptive immune responses form a continuum. Although aberrant adaptive immune responses are an important component of the fully developed IBD pathology in TnfΔARE/+ mice,16 ileal pathology is observed in TnfΔARE/+Rag1−/− mice, involving infiltration of neutrophils and villi blunting.11, 13 Thus, germ-free studies in mouse models of IBD collectively suggest that the microbiota is indispensable for pathogenesis when deregulated inflammatory mechanisms predominate. By contrast, when aberrant adaptive immune responses are the major driver of disease, the microbiota appears to be dispensable for pathology initiation.
Our second conclusion is that Crohn’s-like pathogenesis in TnfΔARE/+ mice relies on the commensal microbiota, whereas global changes in microbial communities, referred to as dysbiosis, are secondary to disease. This conclusion stems from our findings that significant alterations in gut microbiota composition at the level of phyla and at the level of the individual OTUs detected are observed only in late disease in TnfΔARE/+ mice and thus appear not to be essential for the initiation of IBD pathogenesis. By contrast, the commensal microbiota is highly potent in inducing local TNF expression in the intestine of WT mice and TNF overexpression in the intestine of TnfΔARE/+ mice at an early time-point, before disease onset. An important question raised by these findings is whether TNF transcriptional activation in the gut depends on specific bacterial taxa within the indigenous microbiota. Bacteria within commensals which are highly potent in inducing TNF transcriptional activation would function as pathobionts in the context of TNF-dependent ileitis in TnfΔARE/+ mice,28 in a similar manner to the role played by Prevotellaceae in colitis susceptibility in Nlrp6−/− mice for example.29 The identification of such pathobionts could provide alternatives for localized targeting of TNF secretion in the gut. Thus, commensals or specific pathobionts within commensals, and not necessarily an overall dysbiotic microbiota, may be highly relevant to the early events initiating Crohn’s disease, particularly in the presence of genetic variants, which, either directly or indirectly, promote pro-inflammatory activation and local TNF expression in the gut.
Our present results, however, do not exclude a possible disease-exacerbating function of the dysbiotic microbiota developed in diseased TnfΔARE/+ mice. In the T-bet−/−RAG2−/− mouse model of colitis, the dysbiotic microbiota has been shown to be colitogenic and, interestingly, this activity is transferable even to WT mice.30 Although the dysbiotic microbiota of TnfΔARE/+ mice is not sufficient to transfer pathology in co-housed WT mice, the findings in the T-bet−/−RAG2−/− model indicate that the inflamed intestine may privilege the development of particularly harmful bacterial strains that may also bear full pathogenic potential. The increased abundance of SFB and their epithelial attachment in the inflamed ileum of TnfΔARE/+ mice may contribute to disease perpetuation in this model. SFB have been shown to both induce Th17 cells in the LP and tightly adhere to the surface of epithelial cells in the terminal ileum of mice bearing Th17 cells.31 We have previously shown that TnfΔARE/+ mice display increased Th17 responses in the ileal LP compared with WT controls.16 It is likely that the increased SFB abundance and their epithelial attachment in the ileum, reported here, underlie the increased Th17 responses in TnfΔARE/+ mice.
The dysbiosis and the increased bacterial attachment on the epithelium observed in TnfΔARE/+ mice secondarily to pathogenesis can be largely explained by the loss of Paneth cells and the associated disturbances in the expression of antimicrobial peptide genes. Paneth cells have a key role in intestinal antimicrobial surveillance, host defense, and homeostasis and the involvement of Paneth cell dysfunction in IBD pathophysiology is under intense investigation, in light of genome-wide association studies results indicating abnormalities in Paneth cell function as risk factors for disease.3 Paneth cells in Crohn’s disease patients have been shown to strongly overexpress TNF, a fact indicating that these cells have the potential to introduce inflammatory signals in the intestinal mucosa under some conditions.32 Reduced Paneth cell alpha defensins are observed in the ileum of Crohn’s disease patients, though not in association with inflammation grade.33 In addition, in agreement with early reports,34 patients with active Crohn’s disease have recently been shown to display reduced numbers of Paneth cells in their crypts, a defect suggested to result from increased necroptotic Paneth cell death.35 Whether the loss of lysozyme+ cells in TnfΔARE/+ mice reflects Paneth cell death or a dysfunction of live Paneth cells needs to be addressed in future studies. It is likely that the loss of lysozyme+ Paneth cells in TnfΔARE/+ mice is caused by IFN-γ and TNF, both overproduced in their inflamed ileum. TNF has been shown to trigger Paneth cell necroptosis in mice bearing epithelial-specific caspase-8 ablation,35 whereas acute exogenous TNF administration in normal mice leads to Paneth cell dysfunction and loss of cellular integrity.36 In addition, IFN-γ produced during mucosal responses against the protozoan parasite Toxoplasma gondii leads to the reduction of Paneth cells.37 IFN-γ is also a trigger for Paneth cell degranulation, epithelial extrusion, and death, in organoids and in vivo upon anti-CD3 antibody injection.38 However, the physiological context of murine ileitis is markedly different from the above models; thus, the requirement of TNF and IFN-γ for Paneth cell loss should be directly examined by Paneth cell-specific ablation of the respective receptors in TnfΔARE/+ mice.
A study published while our paper was under peer-review39 showed that TnfΔARE ileitis is microbiota-dependent and associated with dysbiosis; results that are in agreement with our results. Schaubeck et al. also concluded that TnfΔARE ileitis is transmissible by disease-associated but not healthy microbiota. This conclusion was based on the fact that germ-free TnfΔARE/+ mice developed ileitis upon caecal content transfer from diseased but not from non-responder (healthy) TnfΔARE/+ mice present in their colony; transfer from WT mice was not tested. The authors did not explain, however, why diseased TnfΔARE/+ mice did not transmit ileitis to the co-housed non-responders of the same litter, a fact that argues against a transmissible phenotype. Therefore, the evidence presented in the work by Schaubeck et al. was not firmly conclusive as to whether ileitis is transmissible in the TnfΔARE model by disease-associated but not healthy microbiota and that intestinal pathology is secondary to dysbiosis.
Our study establishes a mouse model of Crohn’s disease in which the indigenous intestinal microbiota is causatively involved in pathogenesis under genetic conditions favoring TNF overexpression. In this model, dysbiosis is a result and not a cause of intestinal pathology. Future studies may clarify whether in humans, the indigenous microbiota has the potential to have a similar pathogenic role in the presence of genetic conditions that favor inflammatory activation in the gut or affect the antimicrobial defenses that normally maintain commensals at check.
METHODS
Mice, design of in vivo experiments, and study approval
Mice were bred and maintained on a C57BL6/J genetic background in the animal facilities of BSRC “Alexander Fleming” (for Figures 2, 3, 4a–b) and the Icahn School of Medicine at Mount Sinai (for Figure 4c–e and Figures 5, 6, 7, 8) under specific pathogen-free conditions. As an exception, TnfΔARE/+Ifng−/− mice were maintained on a mixed C57BL/6J × 129Sv genetic background in the animal facilities of BSRC “Alexander Fleming”. TnfΔARE/+ mice,11 Myd88−/− mice,40 Rag1−/− mice,41 and Ifng−/− mice42 have been previously described as indicated. For all in vivo experiments, littermate and sex-matched mice were used. Mice were randomly assigned to experimental groups on a first-come basis. No mice were excluded from analyses performed. TnfΔARE/+ mice were analyzed before disease onset (4 weeks) and/or at an intermediate (8 weeks), established (12 weeks), or full-blown (≥16 weeks) disease stage, based on the previously described disease course for this model in the animal facilities of BSRC “Alexander Fleming”.13 A similar TnfΔARE/+ disease course was observed in the animal facilities of Icahn School of Medicine at Mount Sinai as shown in Supplementary Figure S3.
All mice were used in accordance with the guidance of the Institutional Animal Care and Use Committee of BSRC “Alexander Fleming” and the guidelines of the Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai.
Generation of germ-free TnfΔARE/+ mice
Germ-free mice were generated and raised at the Instituto Gulbenkian de Ciência, Oeiras, Portugal. C57BL6J TnfΔARE/+ and WT mice were transferred to germ-free conditions upon rederivation by caesarean section, as described in the relevant EMMA protocol (https://doi.org/strains.emmanet.org/protocols/GermFree_0902.pdf). Briefly, 20 days post-coitum donor females were killed and their uteri transferred to a sterile chamber through a reservoir filled with 1% VirkonS (Sigma-Aldrich, Steinheim, Germany). Pups were extracted and adopted by C3H/HeN Germ-Free foster mothers. The microbiological status of the isolator and the animals were monitored 3 weeks after transfer and every 3 weeks onward. Germ-free and specific pathogen-free control TnfΔARE/+ mice were raised and maintained under strictly identical conditions, except the microbiological status. These include food pellets, drinking water, humidity, and temperature.
Histopathological analysis of TnfΔARE/+ mice
Histological analysis of TnfΔARE/+ mice crossed with Myd88−/−, Rag1−/−, and Ifng−/− mice and of germ-free TnfΔARE/+ mice was performed in the terminal ileum as described previously.12 The proximal and distal colon was also examined. Briefly, the tissue was embedded in paraffin and stained with hematoxylin and eosin. Semi-quantitative assessment of intestinal pathology was performed in a blinded manner. The following scoring system was used: 0=normal, 1=villi blunting and mucosal inflammation, 2=villi blunting, extensive mucosal inflammation, and submucosal inflammation, 3=extensive submucosal inflammation, 4=transmural inflammation.
Pan-bacterial PCR amplification
Total genomic DNA was isolated from tissue using the DNeasy Blood & Tissue Kit (QIAGEN, Valencia, CA). Quantitative PCR analysis and SFB primer sequences are described elsewhere.43 Relative quantity was calculated by the ΔCt method and normalized by the presence of mouse ubiquitin.
Biome analysis
DNA was obtained from distal ileum (2 cm) using the DNeasy Blood and Tissue Kit (QIAGEN). Bacterial 16S rRNA genes were amplified using the primers as described.44 Sequencing was performed with an Illumina (San Diego, CA) MiSeq sequencer for sequencing as previously described.44 Detailed methods for DNA Extraction, 16S rDNA Amplification, Multiplex sequencing, and Analysis of 16S rDNA Sequences are provided in the Supplementary Methods.
Antibiotic treatment
Mice were treated for the indicated time with 0.5 g l−1 vancomycin (Western Medical Supply, Arcadia, CA) ad libitum in the drinking water for the indicated time.
Statistical analyses
For graphs and statistical analyses indicated, we used GraphPad Prism, SigmaPlot, or R statistical environment. Sampling distributions were tested for normality with a threshold of alpha=0.05 (GraphPad Prism). For P<0.05 in the normality test, differences in means were tested for statistical significance with two-tailed Mann–Whitney U test (Wilcoxon rank-sum test). For P>0.05 in the normality test, F test or Bartlett's test were used for homoscedasticity. For similar variances (F test, P>0.05), unpaired two-tailed Student’s t-test was applied; for unequal variances (F test, P<0.05), unpaired two-tailed Welch’s t-test was applied. P-values <0.05 were considered as statistically significant.
References
Xavier, R.J. & Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007).
Johansson, M.E., Larsson, J.M. & Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl Acad. Sci. U S A 108 (Suppl 1), 4659–4665 (2011).
Bevins, C.L. & Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9, 356–368 (2011).
Pearson, C., Uhlig, H.H. & Powrie, F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol 33, 289–296 (2012).
Frank, D.N. St, Amand, A.L., Feldman, R.A., Boedeker, E.C., Harpaz, N. & Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. U S A 104, 13780–13785 (2007).
Kostic, A.D., Xavier, R.J. & Gevers, D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–1499 (2014).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe 15, 382–392 (2014).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Vandussen, K.L. et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn's disease. Gastroenterology 146, 200–209 (2014).
Chassaing, B. & Darfeuille-Michaud, A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140, 1720–1728 (2011).
Kontoyiannis, D., Pasparakis, M., Pizarro, T.T., Cominelli, F. & Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 (1999).
Roulis, M., Armaka, M., Manoloukos, M., Apostolaki, M. & Kollias, G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc. Natl Acad. Sci. USA 108, 5396–5401 (2011).
Kontoyiannis, D. et al. Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn's-like inflammatory bowel disease. J. Exp. Med. 196, 1563–1574 (2002).
Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 10, 131–144 (2010).
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Apostolaki, M. et al. Role of beta7 integrin and the chemokine/chemokine receptor pair CCL25/CCR9 in modeled TNF-dependent Crohn's disease. Gastroenterology 134, 2025–2035 (2008).
Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 (2009).
Xiao, F. et al. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3(-) secretion in murine ileocolonic inflammation. Inflamm. Bowel Dis 18, 101–111 (2012).
Salzman, N.H. et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–83 (2010).
Caporaso, J.G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336 (2010).
Thompson, C.L., Vier, R., Mikaelyan, A., Wienemann, T. & Brune, A. 'Candidatus Arthromitus' revised: segmented filamentous bacteria in arthropod guts are members of Lachnospiraceae. Environ. Microbiol. 14, 1454–1465 (2012).
Barman, M. et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect. Immun. 76, 907–915 (2008).
Sellon, R.K. et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224–5231 (1998).
Taurog, J.D. et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J. Exp. Med. 180, 2359–2364 (1994).
Schultz, M. et al. IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am. J. Physiol 276 (6 Pt 1), G1461–G1472 (1999).
Bamias, G. et al. Commensal bacteria exacerbate intestinal inflammation but are not essential for the development of murine ileitis. J. Immunol 178, 1809–1818 (2007).
Kosiewicz, M.M. et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn's disease. J. Clin. Invest 107, 695–702 (2001).
Chow, J., Tang, H. & Mazmanian, S.K. Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr. Opin Immunol 23, 473–480 (2011).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Garrett, W.S. et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45 (2007).
Ivanov, I.I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Lala, S. et al. Crohn's disease and the NOD2 gene: a role for paneth cells. Gastroenterology 125, 47–57 (2003).
Wehkamp, J. et al. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc. Natl Acad. Sci. USA 102, 18129–18134 (2005).
Lewin, K. The Paneth cell in disease. Gut 10, 804–811 (1969).
Gunther, C. et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011).
Van Hauwermeiren, F. et al. TNFR1-induced lethal inflammation is mediated by goblet and Paneth cell dysfunction. Mucosal Immunol 8, 828–840 (2015).
Raetz, M. et al. Parasite-induced TH1 cells and intestinal dysbiosis cooperate in IFN-gamma-dependent elimination of Paneth cells. Nat. Immunol. 14, 136–142 (2013).
Farin, H.F. et al. Paneth cell extrusion and release of antimicrobial products is directly controlled by immune cell-derived IFN-gamma. J. Exp. Med. 211, 1393–1405 (2014).
Schaubeck, M. et al. Dysbiotic gut microbiota causes transmissible Crohn's disease-like ileitis independent of failure in antimicrobial defence. Gut [e-pub ahead of print 17 April 2015].
Adachi, O. et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9, 143–150 (1998).
Mombaerts, P., Iacomini, J., Johnson, R.S., Herrup, K., Tonegawa, S. & Papaioannou, V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992).
Dalton, D.K., Pitts-Meek, S., Keshav, S., Figari, I.S., Bradley, A. & Stewart, T.A. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 259, 1739–1742 (1993).
Hartman, A.L. et al. Human gut microbiome adopts an alternative state following small bowel transplantation. Proc. Natl Acad. Sci. USA 106, 17187–17192 (2009).
Caporaso, J.G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Acknowledgements
We thank S. Lalos and A. Katevaini for technical assistance in histology and J. Bom for animal care and rederivation of mice to germ-free conditions. We would also like to acknowledge the contribution of our colleague Maria Apostolaki (deceased 10 December 2010) in the design of this study and the original Myd88 knock-out results. This work was funded by ERC project MCs-inTEST (340217), Innovative Medicines Initiative (IMI)-funded project BeTheCure (115142-2) to G.K. and the NIH grant P01 DK072201 and CCFA research grant (#330239) to S.A.L. Mice axenization was supported by the EC FP7 funded project INFRAFRONTIER-I3 (312325) at the Gulbenkian Institute, EMMA Portugal. Endoscopy experiments have been supported by the Deutsche Forschungsgemeinschaft (SPP1656, SFB 796, KFO 257 CEDER) and the IMI, acronym BeTheCure (115142) to C.B. Measurements of paracellular permeability at Hannover Medical School were financed by the Sonderforschungsbereich SFB 621/C9 and DFG grant SE460/13-4 to U.S. We also thank the InfrafrontierGR Infrastructure, co-financed by the ERDF and NSRF 2007–2013, for animal housing, histopathology, and imaging services.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Roulis, M., Bongers, G., Armaka, M. et al. Host and microbiota interactions are critical for development of murine Crohn’s-like ileitis. Mucosal Immunol 9, 787–797 (2016). https://doi.org/10.1038/mi.2015.102
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.102
This article is cited by
-
Harnessing murine models of Crohn's disease ileitis to advance concepts of pathophysiology and treatment
Mucosal Immunology (2022)
-
Experimental Intestinal Stenosis Alters Crohn’s Disease-Like Intestinal Inflammation in Ileitis-Prone Mice
Digestive Diseases and Sciences (2022)
