
Abstract
Streptococcus pneumoniae is a common cause of pneumonia and infective exacerbations of chronic lung disease, yet there are few data on how adaptive immunity can specifically prevent S. pneumoniae lung infection. We have used a murine model of nasopharyngeal colonization by the serotype 19F S. pneumoniae strain EF3030 followed by lung infection to investigate whether colonization protects against subsequent lung infection and the mechanisms involved. EF3030 colonization induced systemic and local immunoglobulin G against a limited number of S. pneumoniae protein antigens rather than capsular polysaccharide. During lung infection, previously colonized mice had increased early cytokine responses and neutrophil recruitment and reduced bacterial colony-forming units in the lungs and bronchoalveolar lavage fluid compared with control mice. Colonization-induced protection was lost when experiments were repeated in B-cell- or neutrophil-deficient mice. Furthermore, the improved interleukin (IL)-17 response to infection in previously colonized mice was abolished by depletion of CD4+ cells, and prior colonization did not protect against lung infection in mice depleted of CD4+ cells or IL17. Together these data show that naturally acquired protective immunity to S. pneumoniae lung infection requires both humoral and cell-mediated immune responses, providing a template for the design of improved vaccines that can specifically prevent pneumonia or acute bronchitis.
Similar content being viewed by others


INTRODUCTION
Streptococcus pneumoniae is estimated to directly cause over 800,000 deaths annually in children and probably at least a similar number in adults. The largest burden of S. pneumoniae disease morbidity and mortality is caused by pneumonia.1, 2 In addition, S. pneumoniae is a common cause of infective exacerbations of chronic lung disease, thereby indirectly causing extensive morbidity and mortality. For example, S. pneumoniae bronchitis causes up to 25% of infective exacerbations of chronic obstructive pulmonary disease,3 the commonest reason for admission to the hospital in the United Kingdom associated with an in-hospital mortality of 14%.4 Despite this burden of lung disease, vaccination has largely concentrated on prevention of septicemia and meningitis, which although more severe than lung infections are much less common and contribute less to the overall burden of disease.1, 2 The existing polysaccharide capsule-based vaccine used in adults has only weak or no efficacy against S. pneumoniae pneumonia and bronchitis;5 the conjugated polysaccharide capsule antigen vaccine used in children does prevent pneumonia caused by the serotypes included in the vaccine, but it is not clear whether this is due to preventing nasopharyngeal colonization (thought to be a necessary precursor for all disease) or by true improvements in adaptive immune responses within the lung. Overall, the mechanisms of adaptive immunity that specifically protect against S. pneumoniae lung infection are not well understood, and this knowledge gap hinders both identification of subjects at high risk of these infections and the design of novel vaccines that are more efficacious at preventing them.
Although it is an important cause of disease, most S. pneumoniae infections are episodes of asymptomatic colonization of the nasopharynx. Colonization is almost universal in infants up to the age of 2 years and recurs intermittently throughout adult life.6, 7 Human and mouse data demonstrate that colonization episodes are immunizing events that induce cellular, mainly Th17 cell-mediated, and humoral adaptive immune responses to S. pneumoniae.8, 9, 10, 11, 12 In murine models, the protective efficacy of cell-mediated or humoral-mediated immunity varies with the site of subsequent infection. CD4+ cell depletion or neutralization of interleukin (IL)-17 abrogated the effects of experimental colonization on protection against re-colonization of the nasopharynx, indicating adaptive immunity in the nasopharynx is mediated by Th17 CD4-cell-mediated immunity.13 In contrast, protection from septicemia after previous colonization was dependent on antibody alone and was unaffected by CD4 depletion.11 However, the effect of prior colonization on immunity to S. pneumoniae specifically within the lung is poorly understood. Experimental colonization of humans does increase both lung humoral and cellular Th17 immune responses in bronchoalveolar lavage fluid (BALF),8, 9 but cannot assess whether either of these are required for local pulmonary immunity to subsequent infection. Whether or not adaptive immune responses improve lung defences to S. pneumoniae and therefore prevent pneumonia or infective exacerbations of chronic lung disease and the mechanisms involved requires further investigation. Conceptually, both Th17 and antibody could mediate immunity to S. pneumoniae within the lung. Th17 cellular responses have been shown to improve lung immunity against other pathogens by indirectly increasing neutrophil recruitment to the site of infection (so boosting local phagocytic capacity), by increasing production of antimicrobial peptides by epithelial cells, and/or by improving epithelial layer integrity.14, 15, 16 Improved antibody responses in the lung could increase S. pneumoniae opsonization and recognition by resident alveolar macrophages or recruited monocytes or neutrophils. Potentially, improved antibody responses and Th17-mediated increased phagocyte recruitment could be synergistic, combining together to provide more effective immunity than each component separately.
We have used a murine model of nasopharyngeal colonization by the EF3030 capsular serotype 19F strain followed by a lung infection challenge without septicemia to investigate whether colonization boosts naturally acquired adaptive immunity to S. pneumoniae within the lung. We have then used genetic knockout mice and antibody depletion to characterize the mechanisms required for colonization-induced protective immunity to S. pneumoniae lung infection.
RESULTS
EF3030 colonizes the murine nasopharynx and induces a serum antibody response
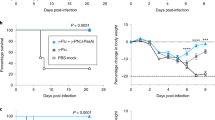
To assess whether EF3030 could colonize the nasopharynx, C57BL/6 mice were experimentally colonized by intranasal inoculation with EF3030 and culled at days 5, 13, and 30. EF3030 was detectable in nasal washes at 5 days and 13 days post colonization but were cleared by 30 days following experimental colonization (Figure 1a), demonstrating that similar to other models of S. pneumoniae murine colonization EF3030 can colonize the nasopharynx of C57BL/6 mice for at least 13 days.11, 17 To assess whether colonization with S. pneumoniae EF3030 was an immunizing event, anti-pneumococcal antibody responses to colonization were measured in day-13 and -30 sera. There was a significant increase in the titer of anti-EF3030 immunoglobulin (Ig)G, but not IgM, in the sera of mice following colonization (Figure 1b,c). No anti-EF3030 IgA response could be detected in sera or BALF from colonized or control mice (data not shown).

EF3030 causes colonization of the nasopharynx that induces a systemic antibody response. (a) Colony-forming unit (CFU) in nasal washes of C57BL/6 mice at 5, 13, and 30 days post colonization with 1 × 107 CFU S. pneumoniae EF3030. Each symbol represents data from a single mouse, and horizontal bars represent mean values. (b) Mean (s.e.m.) whole-cell enzyme-linked immunosorbent assay (ELISA) anti-EF3030 immunoglobulin (Ig)G responses in mouse sera 13 and 30 days post colonization with EF3030 compared with uncolonized controls. (c) Mean (s.e.m.) whole-cell ELISA anti-EF3030 IgM responses in mouse sera 30 days post colonization with S. pneumoniae EF3030 compared with uncolonized controls. (d) Mean (s.e.m.) anti-19F polysaccharide IgG in the sera of mice 30 days post colonization with S. pneumoniae EF3030 compared with uncolonized controls. For serology data, n=4 for each group and P-values were obtained using the Student’s unpaired t-test. OD, optical density; PBS, phosphate-buffered saline.
Identification of S. pneumoniae target antigens
Potential targets of the serum IgG response in colonized mice include serotype 19F capsular polysaccharide and/or subcapsular protein antigens. Anti-serotype 19F capsular IgG levels were not significantly raised following colonization above the low levels detected in the sera of uncolonized mice (Figure 1d). Responses to pneumococcal proteins following colonization were assessed by immunoblotting of EF3030 lysates. Sera from sham-colonized mice reacted weakly with a single band at ∼65 kDa, whereas immunoblots using sera from mice colonized with EF3030 reacted resulted in several positive bands (Figure 2a,b). Sera from individual colonized mice reacted to the same sized protein bands, although the strength of the response varied between mice for different antigens. To quantify antibody responses to different proteins, and identify specific protein targets of IgG, responses to colonization were assessed using a Luminex assay of binding to selected recombinant pneumococcal surface proteins. Sham-colonized mice displayed only very low levels of IgG binding to all proteins, whereas sera from EF3030-colonized mice gave significant IgG responses to three proteins, PhtD, PsaA, and PpmA (Figure 2c). These data demonstrate that following colonization with EF3030 mice develop serum IgG responses to a limited number of S. pneumoniae proteins that include the surface proteins PhtD, PsaA, and PpmA rather than to capsular polysaccharide antigen.

S. pneumoniae antigens recognized by serum immunoglobulin (Ig)G responses to EF3030 colonization. (a and b) Immunoblots for IgG binding to whole-cell lysates of S. pneumoniae EF3030 incubated with sera obtained on day 30 post colonization from three individual sham-colonized (a) or EF3030-colonized (b) mice. (c) Luminex assay of IgG binding to recombinant pneumococcal proteins conjugated to fluorescent beads in pooled sera obtained on day 30 post colonization from five sham-colonized (clear columns) or EF3030-colonized (black columns) mice. (d) Luminex assay of IgG binding to recombinant pneumococcal proteins conjugated to fluorescent beads in pooled bronchoalveolar lavage fluid (BALF) obtained on day 30 post colonization from five sham-colonized (clear columns) or EF3030-colonized (black columns) mice. For c and d, MFI represents binding of anti-mouse IgG-phycoerythrin secondary antibody, and error bars represent standard deviations. MFI, mean fluorescent intensity; PBS, phosphate-buffered saline.
Colonization with EF3030 protects against subsequent lung infection
To assess whether colonization protected against subsequent lung infection, colonized and sham-colonized mice were challenged after 30 days using a lung infection model. Mice were inoculated with 50 μl volume inoculum of 2 × 107 EF3030 S. pneumoniae colony-forming units (CFUs) suspended in 50 μl phosphate-buffered saline (PBS) under deep general anesthesia, and bacterial CFU in BALF, lung homogenates, and blood were compared between sham- and EF3030-colonized mice 4, 24, and 72 h post colonization. At 4 h following inoculation, there were no differences in BALF or lung bacterial CFU between colonized or control mice (Figure 3a,b). In contrast, 24 h post inoculation there was a significant reduction in bacterial CFU in both BALF and lungs of colonized mice and 38% of mice had cleared infection compared with 8% of controls (Figure 3c,d). At 72 h post inoculation a significant reduction in bacterial burden remained in the lungs, but not BALF, of colonized mice, and 50% of mice had cleared infection compared with 25% of controls (Figure 3e,f). No CFU were identified from the blood at any time point from either EF3030 or sham-colonized mice, confirming that EF3030 causes local lung infection without development of septicemia (data not shown). These data indicate that prior colonization with EF3030 is protective against subsequent lung infection, significantly reducing bacterial burden in lavage fluid and lungs at later time points only.

EF3030 nasopharyngeal colonization protects against subsequent EF3030 lung infection. (a, c, and e) S. pneumoniae colony-forming unit (CFU) in bronchoalveolar lavage fluid (BALF) or (b, d, and f) lungs of mice previously sham-colonized (phosphate-buffered saline (PBS) colonized, black symbols) or colonized with EF3030 (clear symbols) 4 h (a and b), 24 h (c and d), or 72 h (e and f) following the lung infection challenge on day 30 with 2 × 107 CFU S. pneumoniae EF3030. Each symbol represents data from an individual mouse, and the horizontal bars represent mean values for the group. P-values were obtained using the Student’s unpaired t-test.
BALF from colonized mice have significant anti-S. pneumoniae IgG responses during lung infection
To investigate whether colonization led to a significant pulmonary anti-protein IgG response during subsequent lung infection, EF3030-specific and total IgG were measured in BALF following challenge. When measured using the whole-cell enzyme-linked immunosorbent assay (ELISA) there was a trend toward an increased anti-EF3030 IgG level in BALF from colonized mice compared with controls pre-challenge, but this difference was not statistically significant (Figure 4a). The Luminex assay (Supplementary Table 1 online) demonstrated BALF from EF3030-colonized mice had significant IgG responses to the same three proteins as the serum responses (PhtD, PsaA, and PpmA), with no significant responses in sham-colonized mice (Figure 2d). By 4 h post challenge, both the total IgG and anti-EF3030 IgG levels in lavage fluid were significantly increased compared with non-colonized controls (Figure 4a,b). By 24 h post inoculation, the concentration of EF3030-specific IgG in the lavage fluid were lower, and the differences between EF3030- and sham-colonized mice were lost (Figure 4a,b). Total IgG in BALF was similar at 4 and 24 h, indicating that the reduction in anti-EF3030 IgG at 24 h was not driven by a global reduction in IgG concentration. There was a strong correlation between the concentration of total IgG and albumin in the lavage fluid of colonized mice (Figure 4c), compatible with the hypothesis that IgG accumulates in the BALF when there is increased alveolar permeability during S. pneumoniae lung infection. The identification of increased anti-EF3030 IgG levels in lavage fluid at 4 h suggested a potential role for IgG in mediating the protection from lung infection observed in previously colonized mice.

Bronchoalveolar lavage fluid (BALF) antibody responses in colonized mice after S. pneumoniae lung infection. (a and b) Mean anti-EF3030 immunoglobulin (Ig)G measured by whole-cell enzyme-linked immunosorbent assay (ELISA) (a) or total murine IgG (b) in BALF from sham- (clear columns) or EF3030 (black columns)-colonized mice 0, 4, and 24 h post lung infection challenge on day 30 with 2 × 107 colony-forming unit (CFU) S. pneumoniae EF3030. Results were obtained for five mice per condition and error bars represent s.e.m. P-values were obtained using one-way analyses of variance (ANOVAs) and Tukey’s post-test to compare columns. (c) Linear regression of BALF total murine IgG concentration compared with BALF albumin concentration of EF3030-colonized mice 4 h following EF3030 challenge. OD, optical density; PBS, phosphate-buffered saline.
Colonization causes significant increases in lung cytokine and cellular responses to S. pneumoniae lung infection
To investigate the effects of prior colonization on the inflammatory responses to S. pneumoniae lung infection, lung homogenate concentrations for a range of cytokines known to be relevant in regulation of inflammation were measured 4 and 24 h after infection of EF3030 and sham-colonized mice. By 4 h post challenge, there were significantly higher levels of tumor necrosis factor-α, keratinocyte chemoattractant (KC), IL17, IL23, and IL10 in the lungs of EF3030-colonized mice compared with controls (Figure 5a). At 24 h following challenge, only tumor necrosis factor-α and IL17 showed persistent significant increases in lung homogenate levels from colonized compared with control mice (Figure 5b). The largest differences were seen for IL17, with virtually no IL17 detected in sham-colonized mice at either time point despite significant IL17 levels in colonized mice at either time point. To assess the effects of prior colonization on cellular responses to subsequent challenge, cell numbers in the lavage fluid were quantified following challenge. Both colonized or control mice developed an alveolar neutrophilic inflammatory infiltrate, during EF3030 lung infection (Figure 6a–d). There were no changes in macrophage numbers. Prior colonization had no effect on baseline numbers of neutrophils prior to challenge, but was associated with substantially greater BALF neutrophilia 4 h following challenge compared with sham-colonized mice (Figure 6d). By 24 h post challenge, there were no significant differences in neutrophil numbers between colonized and sham-colonized mice (Figure 6d). These data demonstrate that colonization primed for enhanced pulmonary inflammatory responses and neutrophil recruitment within 4 h of lung infection compared with uncolonized mice.

Lung cytokine responses to lung infection in EF3030-colonized mice. (a and b) Concentrations of tumor necrosis factor (TNF)-α, KC, interleukin (IL)17, IL23, and IL10 measured by enzyme-linked immunosorbent assay (ELISA) in lung homogenates of EF3030-(black columns) or sham-colonized (clear columns) mice 4 h (a) or 24 h (b) following lung infection challenge on day 30 with 2 × 107 colony-forming unit (CFU) S. pneumoniae EF3030. Data are for five individual mice, and presented as mean (s.e.m.). P-values were obtained using the Student’s unpaired t-test. *P<0.05. KC, keratinocyte chemoattractant; NS, not significant; PBS, phosphate-buffered saline.

Cellular immune responses to lung infection challenge after EF3030 colonization. (a and b) Histological lung sections stained with hematoxylin and eosin, from previously uncolonized mice 24 h following intranasal challenge with phosphate-buffered saline (PBS) (a) or S. pneumoniae EF3030 (b), showing a neutrophilic alveolar inflammatory infiltrate (examples identified by black arrows). (c and d) Bronchoalveolar lavage fluid (BALF) macrophage (c) or neutrophil (d) numbers in BALF from sham- (clear columns) or EF303 (black columns)-colonized mice 0, 4, and 24 h following lung infection on day 30 with 2 × 107 colony-forming unit (CFU) S. pneumoniae EF3030. Data were obtained from five mice per group and are presented as means (s.e.m.). P-values were obtained using the Student’s unpaired t-test.
Both antibody and neutrophils are required for improved protection against lung infection after EF3030 colonization
To identify potential distinct effects of humoral and cellular responses on post-colonization protective immunity, infection experiments were repeated in mice after depletion of specific components of the immune response. The importance of serum and BALF IgG anti-S. pneumoniae responses found in colonized mice was investigated by EF3030 colonization followed by a lung infection challenge of B-cell-deficient (μMT) mice. Whole-cell ELISAs confirmed the absence of a specific IgG response to EF3030 in serum obtained from colonized μMT mice (Figure 7a). Twenty-four hours following the lung infection challenge, there were no differences in lung or BALF bacterial CFU in the lavage fluid or lungs between EF3030- or sham-colonized μMT mice (Figure 7b,c), demonstrating an important role for antibody for the improved protection from lung infection following colonization with the S. pneumoniae EF3030 strain. To assess the protective role of the significantly greater neutrophil numbers in BALF 4 h post infection, EF3030- or sham-colonized wild-type mice were treated with anti-Ly6G monoclonal antibody prior to the lung infection challenge, resulting in an eightfold reduction in BALF neutrophils 24 h post infection (Figure 8a). Treatment with anti-Ly6G monoclonal antibody abrogated the protective effect of colonization on lung infection, with no significant difference in lung or BALF CFU between EF3030- and sham-colonized mice (Figure 8b,c). Neutrophil depletion but not antibody deficiency was associated with S. pneumoniae septicemia in a minority of mice (Figures 7d, 8d).

Importance of antibody for protection against lung infection in EF3030-colonized mice. (a) Concentration of anti-EF3030 immunoglobulin (Ig)G measured by whole-cell enzyme-linked immunosorbent assay (ELISA) in the sera of wild-type (WT) or B-cell-deficient (μMT) mice 30 days after colonization with 1 × 107 colony-forming unit (CFU) EF3030. Data are presented as medians with interquartile ranges. (b, c, and d) S. pneumoniae CFU in bronchoalveolar lavage fluid (BALF) (b), lung homogenates (c), and blood (d) of sham- (black symbols) or EF3030-colonized (white symbols) B-cell-deficient μMT mice 24 h following lung infection on day 30 with 2 × 107 CFU EF3030 S. pneumoniae. Each symbol represents data from one mouse, and the horizontal bars mean values for that group. P-values were obtained using the Student’s unpaired t-test. OD, optical density; PBS, phosphate-buffered saline.

Importance of neutrophils for protection against lung infection in EF3030-colonized mice. (a) Mean (s.e.m.) bronchoalveolar lavage fluid (BALF) neutrophil numbers in EF3030-colonized mice with (1A8, black columns) or without (white columns) pre-treatment with the anti-Ly6G antibody neutrophil-depleting antibody or phosphate-buffered saline (PBS) 24 h after lung infection challenge with 2 × 107 colony-forming unit (CFU) S. pneumoniae EF3030. (b, c, and d) S. pneumoniae CFU in BALF (b), lung homogenates (c), and blood (d) of neutrophil depleted (using anti-Ly66 antibody) either in sham-colonized (black symbols) or EF3030-colonized (white symbols) mice 24 h after lung infection challenge on day 30 with 2 × 107 CFU S. pneumoniae EF3030 in mice. Each symbol represents data from one mouse, and the horizontal bars the mean values for that group. P-values were obtained using the Student’s unpaired t-test.
Depletion of CD4+ cells and IL17 abrogated the protective effects of prior EF3030 colonization against subsequent lung infection
Th17 cells have been implicated in adaptive immunity to colonization by S. pneumoniae,13, 18 and the increase in IL17 and IL23 levels in lung homogenates from EF3030-colonized mice suggested that prior colonization could be boosting Th17 responses during subsequent lung infection. To assess whether CD4+ T cells were important in this model of protection from lung infection, CD4 cells were depleted prior to EF3030 challenge using an anti-CD4 monoclonal antibody. Antibody depletion led to a sixfold reduction in the lung CD4+ cells (Figure 9a,b) and a similar reduction in lung CD3+ CD8− cells (data not shown). CD4+ cell depletion completely abrogated the lung homogenate IL17 response 24 h post challenge for both EF3030- and sham-colonized mice (Figure 9c), indicating that CD4+ T cells rather than γδ cells or other lymphocyte subsets were the likely cellular source of the lung IL17 response seen in EF3030-colonized mice post lung infection. Following CD4+ T-cell depletion, there were no differences between previously colonized and sham-colonized mice in EF3030 BALF and lung CFU 24 h following lung infection (Figure 9d–f). These results suggest that CD4+ cells mediated the protection from lung infection and the increased IL17 response following EF3030 colonization. To confirm that colonization-induced protection is IL17 dependent, EF3030- and sham-colonized mice were depleted of IL17 using a monoclonal antibody prior to EF3030 lung infection. Following IL17 depletion there were no differences in BALF or lung CFU from EF3030- or sham-colonized mice 24 h after challenge (Figure 9g,h). Overall, these data demonstrate a key role for both CD4 cells and IL17, and therefore a Th17 response, in mediating protection against lung infection after S. pneumoniae colonization.

Importance of CD4 cells and interleukin (IL)17 for protection against lung infection in EF3030-colonized mice. (a) Representative flow cytometry dot-plots of splenocytes showing gating on CD3+ CD4+ splenocytes from mice treated with phosphate-buffered saline (PBS) or anti-CD4-depleting antibody (GK1.5) 24 h post infection with S. pneumoniae EF3030. (b) Mean (s.e.m.) total CD4+ cells in the lungs of mice (n=5) treated with GK1.5 (anti-CD4 antibody). (c) Mean (s.e.m.) concentration of IL17 in the lung homogenates of sham-(clear column) or EF3030-colonized (black column) mice 24 h following lung infection challenge on day 30 with 2 × 107 colony-forming unit (CFU) EF3030 S. pneumoniae mice. (d–g) CFU in bronchoalveolar lavage fluid (BALF) (d), lungs (e), and blood (f) at 24 h after intranasal inoculation with 2 × 107 CFU S. pneumoniae EF3030 on day 30 of sham-colonized (black symbols) or EF3030 (clear symbols)-colonized mice pretreated with CD4-depleting antibody (GK1.5, anti-CD4). (g and h) CFU in BALF (g) or lungs (h) at 24 h after intranasal inoculation with 2 × 107 CFU S. pneumoniae EF3030 on day 30 of sham-colonized (black symbols) or EF3030 (clear symbols)-colonized mice pretreated with murine IL17A neutralizing antibody. For d–h, each symbol represents data from an individual mouse and horizontal bars mean values for the group. P-values for b–f were obtained using the Student’s unpaired t-test.
DISCUSSION
Bacterial lung infections, including those caused by S. pneumoniae, are one of commonest cause of global morbidity and mortality.19, 20, 21 However, the existing S. pneumonaie vaccines either do not protect against pneumonia or could rapidly lead to serotype replacement due to the large range of serotypes that cause adult lung disease.5, 22, 23, 24, 25 Identifying adaptive immune mechanisms that can specifically prevent bacterial lung infection is an important global health priority, and this will require an improved understanding of the immune mechanisms involved in protection against S. pneumoniae pneumonia or bronchitis. We have used murine models and a noninvasive 19F strain of S. pneumoniae (EF3030) that causes lung infection without septicemia26 to investigate whether the adaptive immune response to colonization protects against subsequent noninvasive pneumonia. Our data demonstrate that colonization does reduce bacterial CFU during subsequent lung infection challenge, both in lung homogenates and in BALF. Data obtained from control or colonized antibody-deficient mice or mice depleted of neutrophils, CD4 cells, or IL17 showed each of these components was necessary for colonization-induced protection. We chose to compare results between control and colonized mice as the most direct method of assessing whether a specific immune component was required for any protective efficacy of colonization. An alternative approach would be to compare BALF and lung CFU in colonized mice with and without immunomodulation, but this approach has the disadvantages of not showing whether protection has been only partially or completely reduced, and confounding of results for the comparison of μMT vs. C57BL/6 mice by natural antibody. Our results indicate that in this model neither naturally acquired CD4 cell-mediated nor humoral immune responses alone are adequate to provide significant protection against S. pneumoniae lung infection; instead intact CD4-dependent and antibody-dependent immunity were both necessary for improved control of lung infection. These results complement previous data on adaptive immune responses that can protect against S. pneumoniae infection at other anatomical sites. For example, antibody responses to colonization, but not CD4 cells, prevent septicemia associated with pneumonia caused by the invasive S. pneumoniae strain D39.11 In contrast, colonization of the nasopharynx can be prevented by colonization or vaccine-induced CD4 Th17-adaptive immune responses and these are effective independent of antibody-mediated immunity.13, 18
In our model, there was an increased early neutrophil recruitment in previously colonized mice during lung infection. This observation combined with our data showing neutrophils were necessary for the protective effects of colonization suggest a potential model for the synergistic effects of Th17 and antibody: (1) CD4 Th17 responses cause faster recruitment of neutrophils to the lungs during early lung infection; (2) inflammation increases the leak of anti-S. pneumoniae IgG from the sera into the lung, increasing the efficacy of S. pneumoniae opsonization; (3) in combination, the increased recruitment of neutrophils plus improved opsonization by antibody results in a significant synergistic boost to the phagocytic capacity within the lung and leads to better control of bacterial CFU. The early increase in IL17 after challenge in colonized mice could potentially be produced by innate γδ cells rather than CD4 Th17 cells, although if this was the case then perhaps there should have been an IL17 response in uncolonized mice as well. The reduction in the IL17 response at 24 h in colonized mice after CD4 depletion confirms that at least at this time point IL17 was produced by CD4 cells. Comparison of data for uncolonized mice between Figures 3 and 9 suggests CD4 depletion results in an increase in lung and BALF CFU, suggesting a potential effect of CD4 cells on innate immunity that needs further investigation. The relative lack of efficacy of antibody in lung infection compared with septicemia could reflect a lower concentration of antibody in the lung compared with the sera, and/or the possible greater efficiency of the reticuloendothelial system compared with the lung for controlling pathogen numbers. The increase in other pro-inflammatory cytokines during early lung infection in colonized mice could also contribute to improved protection in colonized mice by increasing alveolar leak and therefore IgG recruitment to the site of infection. A stronger tumor necrosis factor-α response might also indicate a role for improved Th1-mediated immunity during lung infection in EF3030-colonized mice, but this has not been investigated in this study. However, Th1-mediated immunity would be predicted to affect macrophage-mediated responses, which predominate at early time points in S. pneumoniae lung infection27 when there were no differences in BALF or lung CFU in our model.
Perhaps surprisingly, our data demonstrate that protein rather than capsular polysaccharide antigens are the targets for the humoral-mediated immunity in the EF3030 model. Using a Luminex bead assay28, 29 we have identified three of the limited number of proteins recognized by sera from colonized mice as PpmA, PhtD, and PsaA. Western blotting indicates there are IgG responses following EF3030 colonization to several additional proteins, but these may not be included on the Luminex assay or have significant allelic variation between EF3030 and the strains, which supplied the Luminex proteins (e.g., the choline-binding proteins).30, 31 All three identified proteins are surface expressed and cause antibody responses in other murine models of colonization or whole-cell vaccination as well as in a human model of colonization.8, 12, 32, 33, 34 These proteins are also highly conserved and so might induce protection against heterologous strains, but are subcapsular that maybe one reason why these antigens alone induce too weak an adaptive response to protect against lung infection. Potentially strong anti-capsular antibody responses maybe more effective for mucosal immunity, as suggested by data showing the conjugated, but not the unconjugated, vaccine can prevent colonization and pneumonia with vaccine serotypes.5, 35, 36 One curious result for which we have no explanation was the fall in total whole-cell IgG levels between days +13 and +30 post colonization. Recently S. pneumoniae proteins that stimulate a Th17-mediated response to a whole-cell vaccine (including the lipoproteins Sp0148 and Sp2108) have been identified.18, 32 However, identifying antigen targets of cell-mediated immune responses is technically challenging, and which antigens stimulated the cell-mediated response to EF3030 colonization will need further investigation.
The incidence of S. pneumoniae lung infections is closely associated with age, increasing almost exponentially after 65 years.37 The reasons for this marked increase are probably multifactorial but could include immune senescence.38 Both cellular and humoral naturally acquired adaptive immune responses develop during childhood in response to S. pneumoniae nasopaharyngeal colonization,28, 39, 40, 41 and epidemiological data suggest these immune responses protect against invasive infections.42 Immunosenescence could cause these colonization-induced adaptive immune responses against S. pneumoniae to weaken with increasing age, resulting in an increased incidence of S. pneumoniae lung infection. Lung-adaptive immune responses to S. pneumoniae may also be affected by chronic lung disease such as chronic obstructive pulmonary disease, asthma, and bronchiectiasis, where immune dysregulation within the lung is likely to be considerable and could contribute to the increased risk of pneumonia and infective exacerbations associated with these diseases.3, 43 By identifying the mechanisms of immunity to S. pneumoniae lung infection in a murine model, our data indicate what aspects of adaptive immunity need to be assessed to characterize whether immune defects could account for the increased incidence of S. pneumoniae infections in the elderly or in patients with chronic lung disease. Furthermore, our data indicate that novel vaccines that specifically target prevention of lung infection rather than septicemia or meningitis should combine both a cellular Th17 CD4 and a humoral response. Importantly, the three protein antigens we have identified as targets for the humoral response after colonization have individually been shown to be effective vaccine candidates.44, 45, 46 These antigens plus approaches designed to stimulate a Th17 response to additional protein antigens47 could provide a novel vaccine strategy to prevent the substantial morbidity and mortality attributable to S. pneumoniae lung infections.
In summary, we have demonstrated that in our murine model both humoral- and cell-mediated immunity combined are required for naturally acquired protective immunity to S. pneumoniae lung infection. We believe these data provide a template for identifying patients at risk of S. pneumoniae lung infection, and for the design of improved vaccines that can specifically prevent pneumonia and bronchitis. Similarly, combined cell-mediated and antibody-mediated immunity could also be important for protection against a range of other extracellular pathogens that cause infections at mucosal sites.
METHODS
Bacterial strains and methods. The S. pneumoniae strain EF3030 was a kind gift from David Briles (University of Birmingham, Alabama). S. pneumoniae was cultured overnight at 37 °C in 5% CO2 on Columbia agar (Oxoid, Thermo Scientific, Basingstoke, UK) supplemented with 5% horse blood (TCS Biosciences, Buckingham, UK). Working stocks were made by transferring one colony of S. pneumoniae to Todd–Hewitt broth supplemented with 0.5% yeast extract Todd–Hewitt yeast, grown to an optical density (OD) of 0.4 (∼108 CFU ml−1) and stored at −80 °C in 10% glycerol as single-use aliquots. CFUs were confirmed by colony counting of log10 serial dilutions of bacteria cultured overnight on 5% Columbia blood agar.
Murine models of colonization and lung infection. C57BL/6 or CD1 mice were anesthetized with aerosolized isoflurane (4%) (MiniRad, Bio-Rad Laboratories, Hercules, CA) and colonized with 1 × 107 CFU S. pneumoniae or PBS as control12 via intranasal instillation of 10 μl S. pneumoniae suspended in PBS. At designated time points post infection the nares were washed with 400 μl PBS.12 S. pneumoniae CFU in nasal washings were enumerated by plating onto blood agar, and was discriminated from other bacteria by alpha-hemolysis and optochin sensitivity (100 μg ml−1, Sigma-Aldrich, St Louis, MO). For challenge experiments, 30 days following colonization mice were inoculated by intranasal installation of 2 × 107 EF3030 S. pneumoniae CFU suspended in 50 μl PBS under aerosolized halothane (4%) general anesthesia.48 Mice were culled at designated time points post infection, and blood, BALF, and lung samples collected as previously described.33, 49 Lungs were homogenized through a 40-μm filter in 3 ml PBS. CFU in BALF, lung homogenates, and blood were counted by serial dilution in PBS and plating on blood agar plates. Total cell numbers in lavage fluid were enumerated with a hemocytometer (Cronus Technologies, Houston, TX) after trypan blue staining (Sigma-Aldrich). Lavage fluid (100 μl) was spun on to polysine microscope slides (Shandon Cytopsin, Thermo Scientific) and stained by rapid Romanowsky staining (Diff-quick, Thermo Scientific) for differential cell counts under a light microscope (Zeiss, Oberkochen, Germany). Supernatants of lung homogenates were stored at −80 °C for cytokine measurement. Cellular or cytokine depletions were performed as follows: (a) neutrophil depletion: 600 μg anti-ly6G mAb (1A8, BioXcell, Kuala Lumpur, Malaysia) was administered by intraperitoneal injection 24 h prior to challenge in a volume of 200 μl, and neutrophil depletion was confirmed by counting cells in the lavage fluid following challenge: (b) CD4+ T-cell depletion: 50 μg anti-CD4 mAb (GK1.5, BioXcell) was administered by intraperitoneal injection 48 and 24 h prior to challenge, and CD4+ T-cell depletion confirmed by flow cytometry (see below): (c) IL17 depletion IL17 was neutralized by intraperitoneal administration of 100 μg of anti-mouse IL-17A (BioXcell) 24 h and immediately prior to challenge. μMT mice were a kind gift from Claudia Mauri (University College London).
In vitro assessment of antibody responses. Immunoblots of 10 μl of whole-cell EF3030 lysates using mouse sera (1 in 100) with anti-mouse IgG–horseradish peroxidase conjugate (1:5,000) were performed as previously described,11, 33 and developed with horseradish peroxidase substrate (GE Healthcare, Buckinghamshire, UK) and imaged in the dark using hyperfilm (GE Healthcare). Multiplex (Luminex, Austin, TX) bead assay of serological responses to 18 recombinant pneumococcal proteins (described in Supplementary Table 1) were performed as described previously.12, 33 In brief, 50 μl diluted mouse sera (1%) and BALF (50%) were added to 50 μl bead mix, incubated at room temperature for 35 min, after which beads were washed twice in PBS+1% bovine serum albumin before resuspension in 50 μl buffer and addition of 50 μl secondary antibody (anti-mouse IgG conjugated to phycoerythrin, 1:50). After incubation for 35 min at room temperature and 800 r.p.m. the beads were washed once (PBS+1% bovine serum albumin) and the fluorescence of each antigen-coupled bead measured using a Bio-Plex machine (Bio-Rad Laboratories). Bacterial whole-cell ELISAs were performed using the EF3030 strain as previously described.11 Briefly, each well of a 96-well plate (Maxisorb, Nunc, Thermo Scientific) was coated overnight with 50 μl of bacterial suspension (OD580 1.0), washed four times (200 μl PBS+0.05% tween) before addition of 100 μl blocking buffer (PBS+0.05% tween+1% bovine serum albumin) and incubation for 1 h at 37 °C. Serially diluted sera, nasal washes, or BALF were added to wells in duplicates (50 μl per dilution), incubated for 2 h at room temperature before addition of the secondary antibody diluted (1:10,000), followed by incubation for 2 h at room temperature, washing and addition of 100 μl of para-nitrophenylphosphate (Sigma-Aldrich) substrate (1 mg ml−1) to each well. The plates were then incubated in the dark for 20 min, after which 100 μl 3 N sodium hydroxide (NaOH) was added to each well to terminate the reaction, and absorbance read at 405 nm, subtracting readings at 630 nm (Versamax, Molecular Devices, Austin, TX).
Cytokine ELISAs. The concentrations of different cytokines in lung homogenates were measured using the following kits according to the manufacturer’s instructions: IL17, Quantikine ELISA kit (R&D systems, Oxford, UK); tumor necrosis factor-α and IL10, Duo Set ELISA (R&D systems); IL23, CytoSet ELISA (Invitrogen, Grand Island, NY); KC, KC ELISA set (Insight Biotechnology, Wembley, UK).
Flow cytometry. Flow cytometric assessment of cell populations of murine lungs were performed as previously described.11 Briefly, lungs were collected into ice cold PBS and single-cell preparations made by homogenization through a 40-μm filter followed by lysis of red blood cells by resuspension in 5 ml red blood-cell lysis buffer for 5 min (Santa-Cruz Biotechnology, Santa Cruz, CA). Washed cells (1 × 106) were added to each well of a 96-well plate, washed, and resuspended in 50 μl of specific antibodies (CD4, CD8, CD3) diluted 1:50 in PBS+1% bovine serum albumin For multiple antibody staining, single-stained controls and “fluorescent minus one” controls were included. Plates were incubated for 20 min at 4 °C, and then washed twice in 200 μl PBS, resuspended in 100 μl of 4% paraformaldehyde, and kept in the dark at 4 °C before analysis using a FACS Calibur flow cytometer (BD Bioscience, San Jose, CA) and FlowJo software (Miltenyi Biotec, San Diego, CA). At least 10,000 lymphocytes were acquired per sample on the basic of size (forward scatter) and granularity (side scatter).
Histology. Lungs from mice were collected into 4% paraformaldehyde, in PBS and left to fix for 4 h. Lungs were then incubated overnight in 15% sucrose then transferred to 70% ethanol for storage at 4 °C until processing. Lungs were processed in paraffin wax overnight using an automated tissue-processor (Leica Microsystems, Milton Keynes, UK). Samples were then embedded into paraffin blocks. Three-μm lung sections were prepared on a rotary microtome (Shandon), and mounted onto glass slides. Slides were stained with hematoxylin and eosin using an Tissue-Tek automated stainer (Sakura, Tokyo, Japan), then overlaid with a glass coverslip. Slides were imaged using a NanoZoomer digital pathology system (Hammatsu, Welwyn, UK).
Statistics. Data are presented as group means±s.e.m. Student’s unpaired T-test was used to compare the mean of two groups or analysis of variance for comparisons between multiple groups, using the Tukey’s post-test to compare selected groups.
Ethics statement. All animal experiments were approved by the UCL Biological Services Ethical Committee and the UK Home Office (Project License PPL70/6510). Experiments were performed according to UK national guidelines for animal use and care, under UK Home Office license.
References
O'Brien, K.L., Wolfson, L.J., Watt, J.P., Henkle, E., Deloria-Knoll, M., McCall, N. et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374, 893–902 (2009).
Fitzwater, S.P., Chandran, A., Santosham, M. & Johnson, H.L. The worldwide impact of the seven-valent pneumococcal conjugate vaccine. Paediatr. Infect. Dis. J. 31, 501–508 (2012).
Garcha, D.S., Thurston, S.J., Patel, A.R., Mackay, A.J., Goldring, J.J., Donaldson, G.C. et al. Changes in prevalence and load of airway bacteria using quantitative PCR in stable and exacerbated COPD. Thorax 67, 1075–1080 (2012).
Roberts, C.M., Lopez-Campos, J.L., Pozo-Rodriguez, F. & Hartl, S. European hospital adherence to GOLD recommendations for chronic obstructive pulmonary disease (COPD) exacerbation admissions. Thorax 68, 1169–1171 (2013).
Moberley, S., Holden, J., Tatham, D.P. & Andrews, R.M. Vaccines for preventing pneumococcal infection in adults. Cochrane Database Syst. Rev. 1, CD000422 (2013).
Turner, P., Turner, C., Jankhot, A., Helen, N., Lee, S.J., Day, N.P. et al. A longitudinal study of Streptococcus pneumoniae carriage in a cohort of infants and their mothers on the Thailand-Myanmar border. PLoS One 7, e38271 (2012).
Hussain, M., Melegaro, A., Pebody, R.G., George, R., Edmunds, W.J., Talukdar, R. et al. A longitudinal household study of Streptococcus pneumoniae nasopharyngeal carriage in a UK setting. Epidemiol. Infect. 133, 891–898 (2005).
Ferreira, D.M., Neill, D.R., Bangert, M., Gritzfeld, J.F., Green, N., Pennington, S.H. et al. Controlled human infection and re-challenge with Streptococcus pneumoniae reveals the protective efficacy of carriage in healthy adults. Am. J. Respir. Crit. Care Med. 187, 855–864 (2013).
Wright, A.K., Bangert, M., Gritzfeld, J.F., Ferreira, D.M., Jambo, K.C., Wright, A.D. et al. Experimental human pneumococcal carriage augments IL-17A-dependent T-cell defence of the lung. PLoS Pathog. 9, e1003274 (2013).
McCool, T.L., Cate, T.R., Moy, G. & Weiser, J.N. The immune response to pneumococcal proteins during experimental human carriage. J. Exp. Med. 195, 359–365 (2002).
Cohen, J.M., Khandavilli, S., Camberlein, E., Hyams, C., Baxendale, H.E. & Brown, J.S. Protective contributions against invasive Streptococcus pneumoniae pneumonia of antibody and Th17-cell responses to nasopharyngeal colonisation. PloS One 6, e25558 (2011).
Cohen, J.M., Chimalapati, S., de Vogel, C., van Belkum, A., Baxendale, H.E. & Brown, J.S. Contributions of capsule, lipoproteins and duration of colonisation towards the protective immunity of prior Streptococcus pneumoniae nasopharyngeal colonisation. Vaccine 30, 4453–4459 (2012).
Zhang, Z., Clarke, T.B. & Weiser, J.N. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Invest. 119, 1899–1909 (2009).
Kao, C.Y., Chen, Y., Thai, P., Wachi, S., Huang, F., Kim, C. et al. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J. Immunol. 173, 3482–3491 (2004).
Chen, K., McAleer, J.P., Lin, Y., Paterson, D.L., Zheng, M., Alcorn, J.F. et al. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35, 997–1009 (2011).
Aujla, S.J., Chan, Y.R., Zheng, M., Fei, M., Askew, D.J., Pociask, D.A. et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14, 275–281 (2008).
Richards, L., Ferreira, D.M., Miyaji, E.N., Andrew, P.W. & Kadioglu, A. The immunising effect of pneumococcal nasopharyngeal colonisation; protection against future colonisation and fatal invasive disease. Immunobiology 215, 251–263 (2010).
Moffitt, K.L., Gierahn, T.M., Lu, Y.J., Gouveia, P., Alderson, M., Flechtner, J.B. et al. T(H)17-based vaccine design for prevention of Streptococcus pneumoniae colonization. Cell Host Microbe 9, 158–165 (2011).
WHO Pneumonia Fact Sheet (2013).
Patel, I.S., Seemungal, T.A., Wilks, M., Lloyd-Owen, S.J., Donaldson, G.C. & Wedzicha, J.A. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 57, 759–764 (2002).
Levine, O.S., O'Brien, K.L., Knoll, M., Adegbola, R.A., Black, S., Cherian, T. et al. Pneumococcal vaccination in developing countries. Lancet 367, 1880–1882 (2006).
Huss, A., Scott, P., Stuck, A.E., Trotter, C. & Egger, M. Efficacy of pneumococcal vaccination in adults: a meta-analysis. CMAJ 180, 48–58 (2009).
Hansen, J., Black, S., Shinefield, H., Cherian, T., Benson, J., Fireman, B. et al. Effectiveness of heptavalent pneumococcal conjugate vaccine in children younger than 5 years of age for prevention of pneumonia: updated analysis using World Health Organization standardized interpretation of chest radiographs. Paediatr. Infect. Dis. J. 25, 779–781 (2006).
Bewick, T., Sheppard, C., Greenwood, S., Slack, M., Trotter, C., George, R. et al. Serotype prevalence in adults hospitalised with pneumococcal non-invasive community-acquired pneumonia. Thorax 67, 540–545 (2012).
Griffin, M.R. & Grijalva, C.G. Distribution of pneumococcal serotypes in adult pneumococcal pneumonia cases: filling the evidence gap to inform vaccination policies. J. Infect. Dis. 208, 1734–1736 (2013).
Briles, D.E., Hollingshead, S.K., Paton, J.C., Ades, E.W., Novak, L., van Ginkel, F.W. et al. Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J. Infect. Dis. 188, 339–348 (2003).
Sun, K. & Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 14, 558–564 (2008).
Lebon, A., Verkaik, N.J., Labout, J.A., de Vogel, C.P., Hooijkaas, H., Verbrugh, H.A. et al. Natural antibodies against several pneumococcal virulence proteins in children in the pre-pneumococcal vaccine-era: the Generation R Study. Infect. Immun. 79, 1680–1687 (2011).
de Voer, R.M., van der Klis, F.R., Engels, C.W., Rijkers, G.T., Sanders, E.A. & Berbers, G.A. Development of a fluorescent-bead-based multiplex immunoassay to determine immunoglobulin G subclass responses to Neisseria meningitidis serogroup A and C polysaccharides. Clin. Vaccine Immunol. 15, 1188–1193 (2008).
Iannelli, F., Oggioni, M.R. & Pozzi, G. Allelic variation in the highly polymorphic locus pspC of Streptococcus pneumoniae. Gene 284, 63–71 (2002).
Hollingshead, S.K., Becker, R. & Briles, D.E. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 68, 5889–5900 (2000).
Roche, A.M. & Weiser, J.N. Identification of the targets of cross-reactive antibodies induced by Streptococcus pneumoniae colonization. Infect. Immun. 78, 2231–2239 (2010).
Chimalapati, S., Cohen, J., Camberlein, E., Durmort, C., Baxendale, H., de Vogel, C. et al. Infection with conditionally virulent Streptococcus pneumoniae Deltapab strains induces antibody to conserved protein antigens but does not protect against systemic infection with heterologous strains. Infect. Immun. 79, 4965–4976 (2011).
Overweg, K., Kerr, A., Sluijter, M., Jackson, M.H., Mitchell, T.J., de Jong, A.P. et al. The putative proteinase maturation protein A of Streptococcus pneumoniae is a conserved surface protein with potential to elicit protective immune responses. Infect. Immun. 68, 4180–4188 (2000).
Paradiso, P.R. Pneumococcal conjugate vaccine for adults: a new paradigm. Clin. Infect. Dis. 55, 259–264 (2012).
Guevara, M., Barricarte, A., Gil-Setas, A., Garcia-Irure, J.J., Beristain, X., Torroba, L. et al. Changing epidemiology of invasive pneumococcal disease following increased coverage with the heptavalent conjugate vaccine in Navarre, Spain. Clin. Microbiol. Infect. 15, 1013–1019 (2009).
van der Poll, T. & Opal, S.M. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556 (2009).
Baxendale, H.E. & Brown, J.S. Mechanisms of immune protection to pneumococcal infection in the young and the elderly In: Immunosenescence (Thiel, A., ed) 85–126 Springer Basel AG: Basel, Switzerland, (2012).
Goldblatt, D., Hussain, M., Andrews, N., Ashton, L., Virta, C., Melegaro, A. et al. Antibody responses to nasopharyngeal carriage of Streptococcus pneumoniae in adults: a longitudinal household study. J. Infect. Dis. 192, 387–393 (2005).
Zhang, Q., Leong, S.C., McNamara, P.S., Mubarak, A., Malley, R. & Finn, A. Characterisation of regulatory T cells in nasal associated lymphoid tissue in children: relationships with pneumococcal colonization. PLoS Pathog. 7, e1002175 (2011).
Glennie, S.J., Banda, D., Mulwafu, W., Nkhata, R., Williams, N.A. & Heyderman, R.S. Regulation of naturally acquired mucosal immunity to Streptococcus pneumoniae in healthy malawian adults and children. PLoS One 7, e51425 (2012).
Lipsitch, M., Whitney, C.G., Zell, E., Kaijalainen, T., Dagan, R. & Malley, R. Are anticapsular antibodies the primary mechanism of protection against invasive pneumococcal disease? PLoS Med. 2, e15 (2005).
Hurst, J.R., Vestbo, J., Anzueto, A., Locantore, N., Mullerova, H., Tal-Singer, R. et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N. Engl. J. Med. 363, 1128–1138 (2010).
Wang, S., Li, Y., Shi, H., Scarpellini, G., Torres-Escobar, A., Roland, K.L. et al. Immune responses to recombinant pneumococcal PsaA antigen delivered by a live attenuated Salmonella vaccine. Infect. Immun. 78, 3258–3271 (2010).
Briles, D.E., Ades, E., Paton, J.C., Sampson, J.S., Carlone, G.M., Huebner, R.C. et al. Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect. Immun. 68, 796–800 (2000).
Khan, M.N. & Pichichero, M.E. Vaccine candidates PhtD and PhtE of Streptococcus pneumoniae are adhesins that elicit functional antibodies in humans. Vaccine 30, 2900–2907 (2012).
Moffitt, K.L., Malley, R. & Lu, Y.J. Identification of protective pneumococcal T(H)17 antigens from the soluble fraction of a killed whole cell vaccine. PLoS One 7, e43445 (2012).
Brown, J.S., Gilliland, S.M. & Holden, D.W. A Streptococcus pneumoniae pathogenicity island encoding an ABC transporter involved in iron uptake and virulence. Mol. Microbiol. 40, 572–585 (2001).
Yuste, J., Khandavilli, S., Ansari, N., Muttardi, K., Ismail, L., Hyams, C. et al. The effects of PspC on complement-mediated immunity to Streptococcus pneumoniae vary with strain background and capsular serotype. Infect. Immun. 78, 283–292 (2010).
Acknowledgements
This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centre’s funding scheme. R.W. was supported by a UCL Impact PhD Fellowship. J.M.C. was supported by a NIHR Academic Clinical Lectureship and received funding from the Academy of Medical Sciences. R.J.J. was supported by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Wilson, R., Cohen, J., Jose, R. et al. Protection against Streptococcus pneumoniae lung infection after nasopharyngeal colonization requires both humoral and cellular immune responses. Mucosal Immunol 8, 627–639 (2015). https://doi.org/10.1038/mi.2014.95
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.95
This article is cited by
-
Suppressive effect of therapeutic antibiotic regimen on antipneumococcal Th1/Th17 responses in neonatal mice
Pediatric Research (2023)
-
A novel Streptococcus pneumoniae human challenge model demonstrates Treg lymphocyte recruitment to the infection site
Scientific Reports (2022)
-
Toll-like receptor 10 has a role in human macrophage response against Streptococcus pneumoniae
Cell and Tissue Research (2022)
-
Novel LysM motifs for antigen display on lactobacilli for mucosal immunization
Scientific Reports (2021)
-
Clarithromycin impairs tissue-resident memory and Th17 responses to macrolide-resistant Streptococcus pneumoniae infections
Journal of Molecular Medicine (2021)
