
Abstract
TRPM8 is the molecular sensor for cold; however, the physiological role of TRPM8+ neurons at mucosal surfaces is unclear. Here we evaluated the distribution and peptidergic properties of TRPM8+ fibers in naive and inflamed colons, as well as their role in mucosal inflammation. We found that Trpm8−/− mice were hypersusceptible to dextran sodium sulfate (DSS)-induced colitis, and that Trpm8−/− CD11c+ DCs (dendritic cells) showed hyperinflammatory responses to toll-like receptor (TLR) stimulation. This was phenocopied in calcitonin gene–related peptide (CGRP) receptor-deficient mice, but not in substance P receptor-deficient mice, suggesting a functional link between TRPM8 and CGRP. The DSS phenotype of CGRP receptor-deficient mice could be adoptively transferred to wild-type (WT) mice, suggesting that CGRP suppresses the colitogenic activity of bone marrow–derived cells. TRPM8+ mucosal fibers expressed CGRP in human and mouse colon. Furthermore, neuronal CGRP contents were increased in colons from naive and DSS-treated Trpm8−/− mice, suggesting deficient CGRP release in the absence of TRPM8 triggering. Finally, treatment of Trpm8−/− mice with CGRP reversed their hyperinflammatory phenotype. These results suggest that TRPM8 signaling in mucosal sensory neurons is indispensable for the regulation of innate inflammatory responses via the neuropeptide CGRP.
Similar content being viewed by others

Introduction
The mammalian transient receptor potential (TRP) ion channel family consists of at least 28 members that are grouped into six subfamilies based on amino-acid sequence homology: TRPC1–7 (canonical), TRPV1–6 (vanilloid), TRPM1–8 (melastatin), TRPP1–3 (polycystic), TRPML1–3 (mucolipin), and TRPA1 (ankyrin). Individual TRP channel subunits consist of six putative transmembrane spanning segments, with the pore-forming loop between the fifth and sixth domains. Both the N- and C termini are located intracellularly and are involved in modulation of channel activity and the formation of multimers. The TRP channels can form homo- or heterotetramers and display a great diversity in activation mode, ion selectivity, and physiological function. Almost all TRP channels are permeable to calcium, albeit some with low specificity with the permeability ratio of Ca2+ relative to Na+ (PCa/PNa) typically ranging from 0.3 to 100.1 In the gastrointestinal tract, TRP channels are involved in various sensory functions including chemo-, thermo-, and mechanosensation.2 TRP cation channel, subfamily M, member 8 (TRPM8) is a member within the subset of temperature-sensitive TRP channels and is the principal receptor involved in cold sensation. TRPM8-deficient mice fail to respond to noxious cold stimuli and do not benefit from cold analgesia.3 In addition to thermal stimulation, cooling agents such as menthol or icilin can activate TPRM8.4, 5 Primary afferent neurons that express TRPM8 innervate the surfaces of the mammalian body, including the skin, oral cavity,6 bladder,7 and lungs.8 TRPM8+ afferents in the skin are involved in the regulation of central physiological functions, including body temperature regulation and cold behavior.9 Vagal afferents from the proximal gastrointestinal tract were also shown to express TRPM8 and to respond to cold and icilin.10 Importantly, we recently identified TRPM8+ spinal (that is, extrinsic) afferents in multiple layers of the colon.11 In this context, TRPM8 signaling is likely to contribute to visceral perception but may also have potential effects in the regulation of inflammatory responses.
The concept of neurogenic inflammation with regard to the pathogenesis of colitis and visceral hypersensitivity has regained attention in recent years,12, 13 with nociceptors TRP vanilloid-1 (TRPV1) and TRP ankyrin-1 (TRPA1) as the major factors.14 Both pharmacological and genetic data suggest a deleterious role for TRPV1 in colitis,15, 16 most likely through neuronal pathways. This is associated with the contribution of TRPV1 signaling to visceral hypersensitivity and pain under inflammatory conditions in the colon.17, 18, 19 Indeed, the distal gastrointestinal tract is densely innervated by TRPV1+ sensory neurons.20, 21 These extrinsic TRPV1+ neurons co-express various non-peptidergic (for example, neuronal nitric oxide synthase) and peptidergic markers such as calcitonin gene–related peptide (CGRP), substance P (SP), and neurokinin A.21, 22 Similarly, TRPA1 has been associated with the promotion of colitic responses.23, 24 In addition to their expression by sensory neurons, signaling by TRP channels in non-excitable cells has been shown to have a role in colitis. For example, TRPV4 was found to be expressed by intestinal epithelial cells (IECs) and to promote experimental colitis.25, 26 TRPM2 expression by monocytes enhances their inflammatory capacity; conversely, Trpm2−/− mice were resistant to colitis mediated by innate immune cells.27
Offset against the overwhelming pro-inflammatory effects of various TRP channel family members, a recent report by Ramachandran et al.28 suggested a regulatory role for TRPM8 in experimental colitis. However, in contrast to our previous findings,11 TRPM8 expression was only described in IEC and the myenteric plexus.28 We hypothesized that colonic TRPM8+ primary afferent neurons might have local, anti-inflammatory effects on mucosal inflammatory cells. Indeed, we found that the genetic deletion of TRPM8 increased the susceptibility of mice to acute colitis. Genetic evidence suggested that this was not related to TRPM8 signaling in hematopoietic or IECs. Trpm8−/− mice had increased CGRP contents in mucosal fibers under naive and inflammatory conditions, suggesting deficient release of this neuropeptide in the intestinal microenvironment. Thus, our data suggest that TRPM8+ nerve fibers exert anti-inflammatory effects in the tissue microenvironment and restrict innate immune cell responses under inflammatory conditions via the local release of CGRP.
Results
Genetic deletion of Trpm8 increases the susceptibility to acute experimental colitis
The acute dextran sodium sulfate (DSS)-induced colitis model is representative for the deleterious effects of innate immune responses in the course of mucosal injury. Hence, we evaluated the contribution of TRPM8 signaling to acute colitis by employing Trpm8−/− mice.3 Mice treated with water only were used as controls. After DSS challenge, Trpm8−/− mice showed significantly more weight loss and intestinal bleeding, with a concomitant increase in the mean disease activity index (DAI) compared with wild-type (WT) mice (Figure 1a,b). Histological analyses of distal colon sections showed enhanced mucosal injury and inflammatory cell infiltration in Trpm8−/− mice after DSS treatment (Figure 1c,d). Quantitative analysis of the mucosal integrity in cross-sectioned colons showed significantly increased ulceration in Trpm8−/− mice (Figure 1e), shorter colons (Supplementary Figure S1a,b online), and reduced contents of Goblet cell products Muc2 (secreted MUC2) and Muc3 (membrane-bound MUC3; Supplementary Figure S1c,d). Consistent with these observations, colon explants from Trpm8−/− mice produced significantly higher levels of pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 (Figure 1f). As these data suggested that the lack of TRPM8 signaling confers susceptibility to DSS colitis, we hypothesized that Trpm8−/− mice would also be susceptible to a reduced DSS concentration that is not colitogenic in WT mice (DSSlow). To this end, we treated WT and Trpm8−/− mice with DSS 1% (w/v) for 5 days, followed by normal drinking water. As expected, DSSlow treatment did not result in body weight loss or intestinal bleeding (Figure 1g,h), nor did it result in signs of mucosal injury or inflammation in colons from WT mice (Figure 1i). However, DSSlow treatment in the Trpm8−/− cohort caused significant body weight loss, enhanced DAI (Figure 1g,h), clear histological signs of colitis (Figure 1i), as well as an elevated production of pro-inflammatory cytokines in colon explant cultures (Figure 1j). Together, these data show that TRPM8 deficiency increases the host’s susceptibility to acute mucosal injury in the colon, suggesting that endogenous TRPM8 signaling normally protects against the inflammatory damage associated with colitis.

Trpm8−/− mice are hypersusceptible to experimental colitis. (a) Mice were treated with drinking water with or without dextran sodium sulfate (DSS) 2% (w/v) for 5 days and killed on day 12 for analysis. DSS-treated Trpm8−/− mice showed significantly increased body weight loss and (b) significantly increased disease activity index (DAI) scores compared with wild-type (WT) mice. (c) Increased epithelial damage and inflammatory cell infiltration in the distal colon in DSS-treated Trpm8−/− mice compared with WT mice. Representative cross-sections of the distal colons of WT and Trpm8−/− mice after water or DSS treatment, respectively (hematoxylin and eosine (H&E) staining). No differences were observed between both genotypes in water controls. (d) Significantly increased mean colitis score in DSS-treated Trpm8−/− vs. WT mice, as determined by histological analysis. (e) Significantly increased ulcerated area of colonic mucosa in DSS-treated Trpm8−/− compared with WT mice. (f) Increased production of pro-inflammatory cytokines (tumor necrosis factor (TNF)-α, interleukin (IL)1β, and IL-6) in colonic explants from DSS-treated Trpm8−/− mice compared with WT mice. Of these inflammatory mediators, only IL-6 was detected in colon explant cultures taken from water-treated (control) animals. (g) WT and Trpm8−/− mice were subjected to DSS 1% (DSSlow) for 5 days and killed on day 10. Only Trpm8−/− mice showed significant body weight loss and (h) elevated DAI scores after DSSlow treatment. (i) Trpm8−/− but not WT mice showed mucosal damage and inflammatory activity after DSSlow treatment. (j) Significantly increased pro-inflammatory cytokine production by colonic explants from Trpm8−/− mice. Data are mean±s.e.m. and representative of three independent experiments. *P<0.05 by two-way analysis of variance (ANOVA; a, g) or Mann–Whitney (b, d–f, h, j). Scale bars = 100 μm (c, i).
Trpm8 deficiency does not affect chronic DSS or TNBS-induced colitis
To also evaluate the role of TRPM8 signaling in experimental models of chronic colitis, we subjected WT and Trpm8−/− mice to three cycles of DSS, or 2,4,6-trinitrobenzene sulfonic acid (TNBS).29 These chemically induced colitis models involve activation of pro-inflammatory CD4+ T-helper cell subsets, for example, Th1, Th2, and Th17, in conjunction with innate immune cell-driven inflammation. In the chronic DSS model, we observed significantly increased body weight loss in the Trpm8−/− cohort compared with WT mice during the first DSS challenge. However, these differences were diminished and eventually lost in the second and third DSS cycles, respectively (Supplementary Figure S1e). After three cycles of DSS, there were also no differences in DAI, the extent of mucosal injury or pro-inflammatory cytokine production in colon explant cultures (Supplementary Figure S1f–h). Mesenteric lymph node cells isolated from WT and Trpm8−/− mice after three cycles of DSS showed similar (TNF-α, IL-17A) or even lower (interferon-γ) levels of cytokine production after CD3/CD28 restimulation in the latter (Supplementary Figure S1i). Thus, Trpm8−/− mice did not display a hyperinflammatory phenotype in the chronic DSS colitis model. Similarly, Trpm8−/− mice showed similar or diminished signs of disease severity in the TNBS-mediated colitis model, as measured by body weight loss, mucosal injury, and pro-inflammatory cytokine production (Supplementary Figure S1j–m). Together, these data suggest a protective role for TRPM8 signaling in acute, that is, innate immune cell–mediated colitis, but not chronic colitis.
Lack of epithelial characteristics predisposing for colitis in Trpm8−/− mice
Next, we sought to identify the mechanism by which TRPM8 signaling mediates protective effects in acute colitis. The DSS model is affected by multiple host factors, such as the gut barrier function and host immune responses. With regard to the gut barrier, the mucus layer is an important component of the intestinal lining to physically separate host cells from noxious biochemical and microbial substrates present in the lumen. TRPM8 has been linked to skin barrier function,30 and to the regulation of mucin production in the lungs.31 Whereas reduced expression of mucin transcripts was observed in inflamed colons (Supplementary Figure S1c,d), we found no differences in colonic Muc2 or Muc3 expression between naive WT and Trpm8−/− mice (Supplementary Figure S2a). In addition, there were no differences in intestinal barrier function as measured in vivo (Supplementary Figure S2b,c), or by Ussing chamber experiments ex vivo (Supplementary Figure S2d) between WT and Trpm8−/− mice. There were also no differences in IEC proliferation (Supplementary Figure S2e,f) between the two mouse strains under homeostatic conditions. Furthermore, we found that Trpm8 mRNA was not expressed by IEC (Supplementary Figure S2g). Thus, these data suggest that deficient TRPM8 signaling does not lead to gut barrier dysfunction and does not explain the DSS phenotype of Trpm8−/− mice.
TRPM8 deficiency in nonhematopoietic cells results in a hyperinflammatory phenotype
We then evaluated whether naive Trpm8−/− mice show features of immune dysregulation as a possible explanation for their DSS susceptibility phenotype. We previously reported that conventional CD11chi dendritic cells (DCs) have a crucial role in DSS colitis.32 Thus, we tested whether DCs displayed hyperinflammatory responses in naive Trpm8−/− mice. Given the limited number of DCs that can be isolated from the colonic lamina propria (LP) in naive mice, we compared freshly isolated CD11c+ DCs from the spleens of WT vs. Trpm8−/− mice. Splenic DCs were stimulated with two different toll-like receptor (TLR) ligands, that is, lipopolysaccharide (LPS; TLR4 ligand) or CpG-ODN (TLR9 ligand). We found that splenic CD11c+ DCs from Trpm8−/− mice produced significantly higher levels of TNF-α and IL-6 (Figure 2a). Members of the TRPM family (for example, TRPM1, TRPM2, TRPM4, and TRPM7),33 and TRPM8 in particular,34 may have pivotal roles in innate immune cell functions. Thus, we tested whether our observations could be explained by cell-intrinsic signaling by TRPM8 by using bone marrow–derived DCs (BMDCs) followed by enrichment for CD11c+ cells after ex vivo expansion. However, we did not find any significant differences in TNF-α or IL-6 production after TLR stimulation between WT vs. Trpm8−/− BMDCs (Figure 2b). Consistent with these data, we did not observe expression of Trpm8 in either freshly isolated splenic CD11c+ DCs or cultured CD11c+ BMDCs (Figure 2c).

TRPM8 deficiency in nonhematopoietic cells results in a hyperinflammatory phenotype. (a) Splenic dendritic cells (DCs) from Trpm8−/− mice are hyperinflammatory in response to toll-like receptor (TLR) stimulation. CD11c+ DCs were isolated from the spleens of wild-type (WT) and Trpm8−/− mice, followed by stimulation with 10 ng ml−1 lipopolysaccharide (LPS) or 5 μg ml−1 CpG, respectively. Supernatants were harvested 24 h after stimulation. Tumor necrosis factor (TNF)-α, interleukin (IL)6 levels in supernatants were quantified using enzyme-linked immunosorbent assay (ELISA). *P<0.05 (t-test). (b) Lack of a hyperinflammatory phenotype of Trpm8−/− bone marrow–derived DCs (BMDCs). BMDCs were obtained by expanding bone marrow cells with 10 ng ml−1 granulocyte–monocyte colony–stimulating factor. CD11c+ DCs were obtained by positive selection on day 6. BMDCs were then stimulated with 10 ng ml−1 LPS or 5 μg ml−1 CpG and supernatants were harvested 24 h later for pro-inflammatory cytokine detection. (c) No detection of Trpm8 transcripts in CD11c+ splenic DCs or BMDCs. Positive control: spinal cord homogenate. (d) Non-myeloid expression of TRPM8 determines the susceptible phenotype of Trpm8−/− mice in experimental colitis. Bone marrow chimeras (BMCs) were constructed as described in the Methods section. Six weeks after BM transplantation, mice were treated with dextran sodium sulfate (DSS; 2%) for 5 days and followed for body weight loss. Mice were killed on day 9 for determination of colitis severity. Trpm8−/− recipients showed an increased susceptibility to DSS colitis compared with WT hosts, independent of BM donor genotype, as shown by their body weight loss and disease activity index (DAI). *P<0.05, WT −> Trpm8 or Trpm8 −> Trpm8 vs. WT −> WT (analysis of variance (ANOVA)). (e) Enhanced TNF-α production in colon explant cultures by Trpm8−/− recipients compared to WT mice after DSS treatment. *P<0.05 vs. WT−> WT (ANOVA). (f) Increased mucosal damage and inflammatory cell infiltration in Trpm8−/− recipients compared with WT mice. Scale bar = 100 μm. Data are presented as mean±s.e.m. and are representative of three independent experiments (a–c).
We then hypothesized that the microenvironment in the host, for example, neuronal input, may determine the inflammatory output of CD11c+ DCs. To confirm that nonhematopoietic expression of TRPM8 drives the phenotype of Trpm8−/− mice in the DSS colitis model, we generated bone marrow chimeras (BMCs). WT and Trpm8−/− mouse recipients were reconstituted with bone marrow harvested from either WT or Trpm8−/− donors, yielding four experimental groups (donor −> recipient). After a recovery period of 6 weeks, the recipients were subjected to DSS and colons were harvested for analysis. Importantly, all Trpm8−/− recipients (WT −> Trpm8−/− and Trpm8−/− −> Trpm8−/− groups) showed an increased susceptibility to DSS colitis, independent of their respective bone marrow donors. This was shown by an increased body weight loss, elevated DAI (Figure 2d) and enhanced TNF-α production in colon explant cultures from Trpm8−/− recipients (Figure 2e). These results were consistent with colitis severity represented by the mucosal damage and inflammatory cell infiltration (Figure 2f). In contrast, WT mice grafted with Trpm8−/− bone marrow (Trpm8−/− −> WT) showed comparable body weight loss and similar colitis severity to controls (WT −> WT) (Figure 2d–f). Taken together, these results indicate that TRPM8 signaling in nonhematopoietic cells affects the hyperinflammatory phenotype of Trpm8−/− mice in experimental colitis.
TRPM8+ mucosal fibers in the colon express CGRP
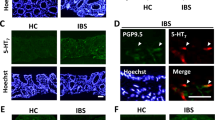
Given our previous observation that TRPM8 is expressed by mucosal fibers in the colon,11 we then addressed the peptidergic features of TRPM8+ neurons during quiescent and inflammatory conditions. The role of neurogenic inflammation in colitis has been established in recent years,12 and most data suggest that TRP channel activation has a deleterious role in this regard. Our previous work identified TRPM8+ afferents in all layers of the colon, including the mucosa, where they co-labeled with CGRP.11 Immunofluorescent staining confirmed expression of both CGRP and TRPM8 throughout the murine colon, with the most dense pattern of positive fibers found in the myenteric plexus (Supplementary Figure S3a, arrowheads) and serosa (Supplementary Figure S3b, double asterisks), respectively. Next, co-labeling of TRPM8/CGRP in mucosal fibers and the myenteric plexus was confirmed in colon sections from naive WT mice (Figure 3a). TRPM8 staining was absent in Trpm8−/− mice in either the mucosa or myenteric plexus (Figure 3b) consistent with the lack of Trpm8 expression in this strain.3 TRPM8+ staining patterns in WT colons that were superimposed to phase-contrast images suggested that the specific TRPM8+ immunoreactivity (IR) observed was located between colonic crypts (Supplementary Figure S3c), consistent with the structural features of mucosal fibers. Importantly, we also observed TRPM8/CGRP co-labeling fibers in the human colon (Figure 3c). In addition, whereas relatively few CGRP+ mucosal fibers were observed in naive WT colons, these were markedly increased after DSS treatment (Figure 3d). Furthermore, enhanced CGRP expression levels were observed in mucosal fibers in Trpm8−/− mice under both homeostatic and inflammatory conditions (Figure 3e). Image quantification of CGRP in colon sections from naive or DSS-treated WT and Trpm8−/− mice confirmed the increased CGRP levels in knockout tissues (Figure 3f). Representative results of CGRP/TRPM8 co-labeling mucosal fibers in DSS-treated WT mice are shown in Figure 3g and Supplementary Figure S3d. The increased expression of CGRP in naive and inflamed Trpm8−/− colons compared with WT was confirmed by measuring neuropeptide levels in colon homogenates (Figure 3h). We then evaluated the intensity of TRPM8-IR in mucosal fibers under both quiescent and inflammatory conditions. Colons from naive and DSS-treated mice were co-labeled for TRPM8 and an epithelial marker, E-cadherin, to visualize the mucosal layer. Analogous to the increased CGRP expression observed under inflammatory conditions, TRPM8-IR+ was more distinct in mucosal fibers in DSS-treated colons compared with controls (Figure 3i). On the basis of these data, we speculate that both TRPM8 and CGRP expressions may increase in colonic fibers during inflammation. Altogether, this suggests a possible role for CGRP release by TRPM8+ sensory neurons in the regulation of colonic inflammation.

TRPM8+ mucosal fibers in the colon express calcitonin gene–related peptide (CGRP). (a) Co-labeling for CGRP and TRPM8 in colon tissues from naive wild-type (WT) mice. Representative pictures from IF staining showing TRPM8+/CGRP+ fibers in the mucosal layer (asterisk) and myenteric plexus (arrowheads). (b) Absence of TRPM8-IR (immunoreactivity) in colons from Trpm8−/− mice. (c) In normal human ascending colon, CGRP-IR is found in fibers within the lamina propria, and in enteroendocrine (EE) cells lining the epithelium. TRPM8-IR is co-localized with CGRP only in fibers (asterisks). Right panel: interpretation of the merged IF image with TRPM8/CGRP co-labeling fibers in yellow. Representative results are shown (n=4). (d) Representative pictures of CGRP+ mucosal fibers in naive and dextran sodium sulfate (DSS)-treated WT colons. Typically, naive WT mice displayed an absence of CGRP-IR in the mucosa, which was markedly increased in inflamed colons (DSS). (e) Enhanced CGRP-IR was observed in naive Trpm8−/− compared with WT mice, which was further increased after DSS treatment. (f) Quantification of CGRP-IR in colons from naive and DSS-treated WT and Trpm8−/− mice, respectively (n=4 for each group). CGRP expression increased following DSS colitis, with significantly increased expression in Trpm8−/− mice following disease, and when compared with WT DSS. (g) Representative picture of TRPM8/CGRP co-expression in mucosal fibers (asterisks) and myenteric plexus (arrowhead) after DSS treatment of WT mice. (h) Quantification of CGRP levels in colon homogenates (normalized per mg tissue) using enzyme-linked immunosorbent assay (ELISA), showing increased CGRP levels following DSS colitis in WT mice, with significantly elevated levels in naive and DSS-treated Trpm8−/− mice. (i) Co-labeling for TRPM8 and the epithelial marker, E-cadherin, of naive and inflamed colon sections from WT mice. Enhanced TRPM8-IR was observed in the myenteric plexus (indicated by arrowheads) and mucosal fibers (asterisks) in DSS-treated colons. Data are presented as mean±s.e.m. (f, h). *P<0.05 vs. naive WT, or as indicated (analysis of variance (ANOVA)). Imaging was performed at original magnification × 63 (a–e, g) or × 20 (i).
CGRP receptor deficiency in hematopoietic cells confers susceptibility to colitis
On the basis of our assumption that the lack of TRPM8 mimics deficient CGRP signaling, we next addressed the regulatory effects of CGRP in colitis. CGRP and SP are the two predominant neuropeptides in sensory neurons that innervate the colon,12 and they appear to have reciprocal effects on colitis. Whereas CGRP protects against colitis, the effects of SP signaling increase the colitis severity.35, 36, 37 Notably, deletion of either CGRP isoform (α-CGRP and β-CGRP) increases the susceptibility to spontaneous colitis, suggesting a non-redundant immunoregulatory role for CGRP+ colonic afferents.38 Indeed, CGRP has established anti-inflammatory effects on DCs,39 and the CGRP receptor is expressed by CD11c+ DCs.40 However, the mechanism by which CGRP is released during inflammation is unknown. We hypothesized that CGRP released by TRPM8+ sensory neurons could affect the onset and severity of colitis by suppressing local inflammatory responses.
To first evaluate the proximity between CGRP+ mucosal fibers and CD11c+ DCs, we performed double staining on colon sections from DSS-treated WT and Trpm8−/− mice. This showed CGRP+ fibers in close proximity to CD11c+ cells in the inflamed colonic mucosa (Figure 4a), thereby suggesting a possible functional association. In line with the hyperinflammatory phenotype of Trpm8−/− mice, we also observed a significantly increased number of CD11c+ cells per field in the colon early during DSS challenge but not in the spleen (Figure 4b).

Close proximity between calcitonin gene–related peptide+ (CGRP+) mucosal fibers and CD11c+ DCs (dendritic cells). (a) CD11c+ cells were observed in the colonic lamina propria in close proximity to CGRP-IR (immunoreactivity)+ fibers in dextran sodium sulfate (DSS)-treated wild-type (WT) and Trpm8−/− mice, respectively. Imaging was performed at original magnification × 63. (b) CD11c+ cells were significantly increased in numbers in DSS-treated Trpm8−/− compared with WT mice in colons, but not in spleens (n=4 for each group). Data are presented as mean±s.e.m. *P<0.05 (t-test).
Next, we used mice with defective CGRP signaling to test the effect of CGRP signaling on DSS colitis. The CGRP receptor consists of the calcitonin receptor–like receptor and its co-receptor, receptor activity modifying protein-1 (RAMP1). Ramp1−/− mice fail to respond to CGRP stimulation.41 Thus, we subjected Ramp1−/− mice to DSS and compared the outcome to WT mice. The lack of CGRP signaling resulted in increased colitis severity after DSS treatment as shown by significantly increased body weight loss and DAI (Figure 5a,b), mucosal damage (Figure 5c), and enhanced pro-inflammatory cytokine production by colon explants in the Ramp1−/− cohort (Figure 5d). To test the role of CGRP receptor signaling in BM-derived cells in colitis, BMCs were generated with WT or Ramp1−/− mice as donors. After being challenged with DSS, we observed that WT recipients of Ramp1−/− bone marrow displayed more severe colitis (Figure 5e–g). Consistent with these data, we confirmed expression of both calcitonin receptor–like receptor and RAMP1 in CD11c+ splenic DC and BMDC at the mRNA (Figure 5h) and protein levels (Figure 5i). Moreover, we found that BMDC generated from Ramp1−/− mice showed a hyperinflammatory phenotype upon TLR stimulation (Figure 5j). Together, these results suggest that CGRP signaling in BM-derived cells (for example, CD11c+ DCs) protects against experimental colitis. Finally, opposite results were achieved by using SP receptor knockout (Nk1−/−) mice in standard DSS or BMC experiments (Supplementary Figure S4a,b, respectively), confirming the opposing pro-inflammatory role of SP in the colon.

Calcitonin gene–related peptide (CGRP) receptor deficiency in BM-derived cells confers susceptibility to colitis. (a) Wild-type (WT) and Ramp1−/− (receptor activity modifying protein-1) mice were subjected to drinking water with or without dextran sodium sulfate (DSS; 2%) for 5 days, followed by normal drinking water. The mice were killed on day 9 to analyze the colitis severity. DSS-treated Ramp1−/− mice showed significantly increased body weight loss, and (b) mean disease activity index (DAI) compared with WT controls. (c) Ramp1−/− mice showed increased DSS-induced mucosal damage and inflammatory cell infiltration in distal colon sections compared with WT controls (hematoxylin and eosine (H&E)). (d) Significantly increased tumor necrosis factor (TNF)-α, interleukin (IL)1β production in colon explants from DSS-treated Ramp1−/− vs. WT mice. (e) Defective CGRP signaling in bone marrow–derived cells determines the susceptible phenotype of Ramp1−/− mice in DSS colitis. WT mice with reconstituted WT or Ramp1−/− bone marrow were treated with DSS (2%). Mice were killed on day 9. Recipients of Ramp1−/− bone marrow were more sensitive to DSS colitis as demonstrated by increased weight loss. (f) Mice in the Ramp1−/− −> WT cohort showed increased histological features of mucosal injury compared with the WT −> WT control group, as shown by the difference in the mean colitis score. (g) Representative examples of colon cross-sections from recipients of WT and Ramp1−/− BM, respectively. (h) Expression of Clr (calcitonin receptor–like receptor) and Ramp1 transcripts in CD11c+ bone marrow–derived DC (BMDC), as determined using Q-PCR. (i) Flow cytometric analysis of the expression of CRLR and RAMP1 by CD11c+ BMDC. Cells were stained with primary antibodies (Abs), followed by detection with AF488-conjugated secondary Abs (red lines). Isotype controls (blue lines). (j) Ramp1−/− BMDCs display a hyperinflammatory response to lipopolysaccharide (LPS) (10 ng ml−1) or CpG (5 μg ml−1) stimulation compared with WT BMDC. Data are mean±s.e.m. *P<0.05 by two-way analysis of variance (ANOVA) (a, e) or Mann–Whitney (b, d, f, j). Scale bars = 100 μm (c, g).
CGRP treatment reverses the hyperinflammatory phenotype of Trpm8−/− mice
On the basis of the findings above, we hypothesized that the increased susceptibility of Trpm8−/− mice to DSS might be because of a lack of CGRP release in the colonic mucosa, which normally acts to suppress local inflammatory responses. Thus, we tested the direct effects of recombinant CGRP on TLR-induced cytokine production by CD11c+ DCs isolated from WT and Trpm8−/− mice. We found that the increased production of TNF-α by splenic DCs isolated from Trpm8−/− mice was reversed by recombinant CGRP (rCGRP) pretreatment (Figure 6a). These results suggest that rCGRP treatment ex vivo recapitulated the normal suppressive environment of the host provided by TRPM8+ sensory neurons. To test whether rCGRP also reversed the increased susceptibility of TRPM8-deficient mice to DSS colitis, we treated WT and Trpm8−/− mice with rCGRP (i.p.) during DSS challenge. Whereas rCGRP treatment did not have marked effects in the WT control group, it successfully reversed the increased body weight loss (Figure 6b), DAI (Figure 6c), mucosal damage, and colonic inflammation (Figure 6d) in the Trpm8−/− cohort. Conversely, treatment with a CGRP receptor antagonist should only affect the course of DSS colitis in WT mice. We found that i.p. treatment with the CGRP antagonist, CGRP8–37, significantly increased the colitis-associated DAI, mucosal damage, and pro-inflammatory cytokine production in colon explants in WT mice, but not in the Trpm8−/− cohort (Supplementary Figure 5a–d). Together, these data suggest that the hyperinflammatory phenotype of Trpm8−/− mice may be explained by deficient CGRP signaling. This led us to propose a working model in which dysfunctional secretion of CGRP in the tissue microenvironment leads to a dysregulation of innate immune responses in Trpm8−/− mice, as summarized in Figure 7.

Calcitonin gene–related peptide (CGRP) treatment reverses the hyperinflammatory phenotype of Trpm8−/− mice. (a) CGRP reverses the hyperinflammatory phenotype of CD11c+ DCs (dendritic cells) from Trpm8−/− mice. CD11c+ DCs were isolated from spleens of wild-type (WT) and Trpm8−/− mice. DCs were pretreated with 100 nM CGRP for 30 min and then stimulated with 5 μg ml−1 CpG. Tumor necrosis factor (TNF)-α levels were determined in the supernatants harvested 24 h after toll-like receptor (TLR) stimulation. (b) Daily administration of CGRP reversed the increased susceptibility of Trpm8−/− mice to dextran sodium sulfate (DSS) colitis. WT and Trpm8−/− mice were treated with i.p. CGRP (2 μg per day) or phosphate-buffered saline (PBS), during and after 2% DSS treatment, until being killed on day 10. CGRP treatment reversed the increased body weight loss in the Trpm8−/− cohort compared with WT mice. (c) Disease activity index (DAI) of WT and Trpm8−/− mice after daily PBS or CGRP administration during DSS treatment, determined on day 10 of follow-up. (d) CGRP treatment reversed the increased histological signs of mucosal injury and inflammation in Trpm8−/− mice during DSS colitis. Scale bar = 100 μm. All data are presented as mean±s.e.m. *P<0.05 vs. WT control or as indicated (analysis of variance (ANOVA)).

Working model of the effects of TRPM8+/CGRP+ (calcitonin gene–related peptide+) mucosal fibers in colitis. (a) Proposed model of the local, anti-inflammatory effects of CGRP released by TRPM8+ sensory nerve fibers in the tissue microenvironment of the colon. In wild-type (WT) mice, the local release of CGRP by TRPM8+ mucosal fibers inhibits pro-inflammatory cytokine production by CD11c+ DCs (dendritic cells) in the course of mucosal damage and inflammation. (b) In Trpm8−/− mice, the lack of TRPM8 triggering results in CGRP accumulation in mucosal fibers. The absence of mucosal CGRP release results in unrestrained production of pro-inflammatory cytokines and enhanced colitis.
Discussion
Our data suggest a regulatory role for TRPM8+ mucosal nerve fibers through the local release of CGRP in an experimental model of acute colitis. This is based on the following observations. First, we found that Trpm8−/− mice are extremely sensitive to DSS colitis (Figure 1). Second, innate immune cells from Trpm8−/− mice displayed hyperinflammatory responses upon stimulation with TLR ligands ex vivo, although this was not related to intrinsic TRPM8 signaling in myeloid cells (Figure 2). Third, we observed CGRP-IR in TRPM8+ fibers in the human and mouse colon, as well as an increase in TRPM8/CGRP expression in mucosal fibers in experimental colitis (Figure 3). These CGRP+ mucosal fibers were observed in close proximity to CD11c+ cells in the inflamed colon (Figure 4). Fourth, we found that the hypersusceptiblity of Trpm8−/− mice to DSS colitis was phenocopied by mice with defective CGRP signaling (Figure 5). Finally, treatment with exogenous CGRP rescued the DSS susceptibility phenotype of Trpm8−/− mice (Figure 6). Thus, we propose a model in which mucosal injury or colitis activates TRPM8 signaling in a subset of mucosal sensory neurons that results in the local release of CGRP. Subsequently, local high concentrations of CGRP suppress pro-inflammatory cytokine production by innate immune cells such as CD11c+ DCs, thereby suppressing colitogenic responses (Figure 7a). This immunoregulatory mechanism is absent in Trpm8-deficient mice, resulting in accumulation of non-released CGRP in mucosal fibers and a hyperinflammatory phenotype (Figure 7b). Alternatively, the surprising and paradoxical observation of CGRP accumulation in Trpm8−/− colons may result from the increased inflammatory activity observed in these mice after DSS challenge. However, the well-established anti-inflammatory effects of CGRP may be more consistent with our proposed model (Figure 7).
In contrast to the skin and oral cavity, the intestines are not subject to temperature changes. Thus, our model implies that endogenous TRPM8 ligands and/or modulators may have an important role in mucosal homeostasis. Furthermore, the production of these substrates may be modulated by inflammatory signaling pathways. Potential candidates are the end products of Ca2+-independent phospholipase A2 that includes lysophosphatidylcholine, lysophosphatidylinositol, and lysophosphatidylserine. These lysophospholipids can shift the gating threshold of TRPM8 to more physiological temperatures.42, 43 As inflammation-associated injury leads to the activation of Ca2+-independent phospholipase A2,44 this putative positive feedback loop may promote TRPM8 gating in the inflamed gut, thereby providing an endogenous pathway for its activation. Similarly, phosphatidylinositol 4,5-bisphosphate (PIP2, a membrane phospholipid) can directly induce gating of the TRPM8 channel independent of temperature or cooling agents.45 This provides an alternative pathway of endogenous TRPM8 activation in the course of inflammatory damage. For example, activation of phosphatidylinositol 4-phosphate 5 kinase in sensory neurons during inflammation, as recently demonstrated,46 may lead to PIP2-mediated TRPM8 activation. Thus, various putative mechanisms could connect the induction of inflammatory injury to TRPM8 activation on mucosal fibers in the intestines. On the other hand, certain components of the ‘inflammatory soup’ are capable of antagonizing TRPM8 signaling, including endovanilloids and endocannabinoids.47 Furthermore, G-protein-coupled receptor ligands such as pro-inflammatory mediators bradykinin and histamine can directly inhibit TRPM8 gating via G-protein subunit Gαq.48 These endogenous signaling pathways often have opposite effects on cold receptor TRPM8 vs. prototypical heat receptor TRPV1 (that is, inhibition of TRPM8 vs. activation of TRPV1, and vice versa).45, 47, 48 Thus, the pleiotropic effects of these endogenous mediators on TRP channel gating imply an important modulatory effect on the regulation of neurogenic inflammation in colitis.
Early reports suggested that the expression of neuronal TRPM8 did not overlap with prototypical nociceptive markers, that is, CGRP and TRPV1, based on immunostaining of rodent dorsal root ganglions.5 Co-expression of TRPM8 and CGRP was also not observed in nerve fibers that innervate the oral mucosa.6 However, careful dissection of the neurochemical properties of TRPM8+ fibers with a reporter mouse strain (Trpm8GFP) showed a more diverse and heterogeneous picture. A mixture of TRPM8+ putative nociceptors and non-nociceptors was observed in various neuronal structures, including TRPM8/CGRP double-positive neurons in the trigeminal ganglion, skin, and oral cavity.49 We and others confirmed the expression of CGRP in TRPM8+ extrinsic fibers that penetrate the skin,50 bladder,7 colon (Harrington et al.11 and Figure 3), as well as the trigeminal ganglion.51 These peptidergic properties of TRPM8+ fibers are relevant in the context of their proposed physiological functions. CGRP is a classic nociceptive marker and its role in pain perception is well established, in addition to its contribution to neurogenic inflammation. The interplay between CGRP and SP on the activation of infiltrating inflammatory cells in the colon is particularly important in this regard.12 Whereas experimental colitis models show that SP mainly displays colitogenic properties,24, 35, 36 CGRP is protective.36, 37, 38 Our data confirmed the opposite roles of these two neuropeptides in colitis (Figure 5a–g, Supplementary Figure S4). In addition, our results suggest that TRPM8+ mucosal fibers are crucial for local CGRP release in the colonic mucosa upon acute mucosal injury (Figure 3d–g). Notably, the susceptibility phenotype of Trpm8−/− mice was not observed in chronic colitis induced by three cycles of DSS, or TNBS administration (Supplementary Figure S1e–m), which may be related to differences in the type of infiltrating inflammatory cells between these models. Finally, the anticolitogenic role of CGRP seems at odds with the reported pro-inflammatory properties of TRPV1 signaling,12, 15 as TRPV1 is usually co-expressed with CGRP in mucosal fibers in the intestines.20, 21, 22 However, this may be explained by the co-expression of SP in TRPV1+ fibers,21, 22 which could shift the balance to a predominantly pro-inflammatory output.52 In contrast, TRPM8+ fibers show no detectable,6 or very low,50 levels of SP.
To further dissect the features of TRPM8/CGRP signaling in neurogenic inflammation, we applied BMC to show that it is the expression of neuropeptide receptors on BM-derived cells that determines the outcome of colitis. The transfer of CGRP receptor–deficient (Ramp1−/−) BM conferred enhanced DSS susceptibility to WT mice (Figure 5e–g). Conversely, the transfer of SP receptor–deficient (Nk1−/−) bone marrow protected WT mice from DSS colitis (Supplementary Figure S4b). We also confirmed the expression of the receptor and co-receptor for CGRP (that is, calcitonin receptor–like receptor and RAMP1, respectively) in CD11c+ cells at the mRNA and protein levels (Figure 5h,i). The immunoregulatory effects of CGRP on CD11c+ DCs have been previously demonstrated in the context of sepsis,41 allergic airway responses,40 and skin inflammation.53 Mechanistically, the inhibition of TLR pathways by CGRP was shown to be dependent on transcriptional inducible cAMP early repressor in DCs.54 Inducible cAMP early repressor is selectively induced in DCs by CGRP through intracellular (cAMP) elevations. Inducible cAMP early repressor then interferes with the recruitment of ATF-2 to the Tnfa promoter.39 Given the central role of TNF-α in the pathogenesis of colitis, these data are consistent with our proposed model in which TRPM8+/CGRP+ neuronal fibers provide local, anti-inflammatory signals upon the perturbation of mucosal homeostasis in the colon. A practical limitation of our study is the use of CD11c+ splenic DCs or BMDCs to demonstrate the effects of the TRPM8/CGRP axis on innate immune cells in the gut. CD11c is generally regarded as a marker for ‘conventional DCs’ (cDCs), which co-express major histocompatibility complex class II but are negative for the high-affinity IgG receptor FcγR1 (CD64). In the intestinal LP, DCs can be derived from blood-derived committed precursors, which also form splenic CD11c+ cDCs, whereas other subsets are thought to derive from gut-associated lymphoid tissue.55, 56 Given the limited number of CD11c+ cDCs in the colonic LP,57 we have used CD11c+ cDCs isolated from the spleen or ex vivo cultured CD11c+ BMDC in our assays, with the assumption that their responses to CGRP are similar to those from CD11c+ LP cDCs. Thus, the regulatory effects of CGRP on the response of various intestinal LP cDCs subsets to TLR stimulation should be studied in more detail.
A recent report by Ramachandran et al.28 confirmed the regulatory role of TRPM8 signaling in two different models of chemically induced colitis (DSS, TNBS) through pharmacological studies (that is, with menthol or icilin) and data generated with the TRPM8GFP reporter strain. They suggest a model in which agonist-mediated TRPM8 receptor activation inhibits the pro-inflammatory effects of TRPV1. Our data, based on genetic evidence, BMC, and protein expression data, are conceptually different in some aspects. Although we also confirmed TRPM8 expression in the myenteric plexus (Harrington et al.Ref.11 Figure 3a), we could not find clear evidence for Trpm8 expression in IEC (Figure 3i, Supplementary Figures S2g–S3). We also observed that CGRP is co-expressed with TRPM8 in colonic mucosal fibers in mouse and human (Harrington et al.11 Figure 3a,c), and that CGRP levels in mucosal fibers are increased in naive and DSS-treated Trpm8−/− mice compared with WT controls (Figure 3d–g), whereas their data suggest that TRPM8+ sensory neurons do not express CGRP.28 Thus, our findings led us to propose a different model in which TRPM8+ mucosal fibers directly suppress innate immune cells through CGRP (Figure 7). This model is also consistent with the established anti-inflammatory effects of CGRP on DCs and colitis. Of note, a recent publication by Hosoya et al.58 confirmed positive TRPM8 staining in fiber-like structures in the colon, in addition to TRPM8-IR+ in IEC. Thus, the expression patterns of TRPM8 in the gut, that is, in both neuronal and non-neuronal cells, might account for multiple mechanisms of immune modulation. Furthermore, Hosoya et al.58 also confirmed TRPM8/CGRP co-labeling in colonic mucosal fibers, in addition to increased TRPM8-IR+ in these fibers under inflammatory conditions. Together with these data, our findings contribute to a fundamental understanding of the role of TRP channel and neuropeptide signaling in the regulation of mucosal inflammation.
Methods
Reagents. DSS was purchased from MP Biomedicals (Solon, OH). DL-dithiothreitol, EDTA, fluorescein isothiocyanate (FITC)-Dextran, LPS (Escherichia coli O111:B4) and TNBS were obtained from Sigma (St. Louis, MO). Recombinant murine GM-CSF was purchased from eBioscience (San Diego, CA). LPS-free 1018 CpG-ODN (5-TGACTGTGAACGTTCGAGATGA-3) was obtained from TriLink Biotechnologies (San Diego, CA). Recombinant CGRP (Human) and CGRP8–37 were obtained from Phoenix Pharmaceuticals (Burlingame, CA).
Mice. Eight-to-twelve-week-old, sex-matched mice were used for all experiments. Specific pathogen-free C57BL/6 mice were acquired from Harlan Sprague (Hayward, CA) and bred in our vivarium before colitis experiments were performed to minimize differences in intestinal flora. Trpm8−/− mice3 were kindly provided by Dr Patapoutian (TSRI, La Jolla). Ramp1−/− mice41 were kindly provided by Dr Tsujikawa (Osaka University, Osaka, Japan). Nk1−/− mice59 were kindly provided by Dr Weinstock (Tufts Medical Center, Boston). Experimental procedures were conducted in accordance with the University of California, San Diego institutional guidelines for Animal Care and Use.
DSS and TNBS colitis. On the basis of previous experience with the acute DSS model in our vivarium,32 we used DSS dissolved at 1% (DSSlow) or 2% (w/v) in sterile drinking water during 5 days before switching to water. For some experiments, control groups were treated with drinking water only. In the chronic DSS model, three consecutive cycles of 2% (w/v) DSS were applied during 5 days, followed by 2 weeks of water. Mice were killed on day 10 of the final DSS cycle. For the TNBS colitis model, mice were sensitized on day −7 by cutaneous application of 1% (w/v) TNBS or EtOH (negative control) in a mixture of acetone/olive oil (4:1). All mice were re-challenged on day 0 with 2.5% (w/v) TNBS by rectal administration. Cohorts of TNBS-treated mice were killed on day 3 or day 5 of follow-up for analysis of colitis severity and inflammatory cytokine production. For intervention studies in the acute DSS model, CGRP (agonist), CGRP8–37 (antagonist), or phosphate-buffered saline (PBS) was administered to mice at 2 μg per day (i.p.). Bodyweights of mice were recorded daily. Mice were killed at the time of maximal body weight differences between groups, as indicated in the figure legends. Intestinal bleeding was monitored with the hemoccult test (Beckman Coulter) to calculate the DAI. Body weight loss was scored as follows: 0=0–4.9%; 1=5.0–9.9%; 2=10.0–14.9%; 3=15.0–19.9%; 4=>20.0% body weight loss. Intestinal bleeding was scored as follows: 0=negative hemoccult test; 2=positive hemoccult test; 4=gross rectal bleeding. Body weight loss and intestinal bleeding scores were added, resulting in a total score ranging from 0 to 8.
Functional intestinal barrier tests. FITC-Dextran was dissolved at 60 mg/ml in water and orally delivered to mice. Blood serum was obtained by retro-orbital bleeding 2 h later. The fluorescence (FU) in serum samples was compared to a standard range of FITC-Dextran to calculate FITC-Dextran values in experimental samples. Fecal albumin measurements were performed with dried fecal pellets using the Mouse Albumin ELISA (enzyme-linked immunosorbent assay) Quantitation Set obtained from Bethyl Laboratories (Montgomery, TX) according to manufacturer’s instructions.
Ussing chamber experiments. Segments of the distal colon were cut along the mesenteric border and mounted in Ussing chambers (Physiological instruments, San Diego, CA), with a window area of 0.09 cm2, according to the protocol.60, 61 Baseline short-circuit current (Isc) values were obtained at equilibrium, 15 min after the tissues were mounted and expressed as μA cm−2. Conductance (G) was also determined at baseline as an indicator of ion flux, or paracellular permeability, and expressed as mS cm−2. FITC-labeled dextran (4 kDa, Sigma) was used as a probe to assess macromolecular permeability and was added to the luminal buffer at a final concentration of 2.2 mg ml−1 once equilibrium was reached. Serosal samples were taken at 30-min intervals for 2 h and replaced with fresh buffer to maintain constant volumes. Fluorescence was measured by end point assay (Victor4X, Perkin Elmer, Waltham, MA) and the flux of FITC-dextran from the mucosa to the serosa was calculated as the average value of three consecutive stable flux periods (30–60, 60–90, and 90–120 min) and expressed as μg ml−1cm−2h−1.
Histological analysis of colitis. The excised colon was opened longitudinally with surgical scissors and rolled from proximal to distal. Colon rolls were fixed with 10% buffered formalin for 24 h. Paraffin cross-sections (5 μm) were stained with hematoxylin and eosine and the severity of colitis was determined by evaluating epithelial damage and inflammatory cell infiltration, respectively. Colonic epithelial damage was scored as follows: 0=normal; 1=hyperproliferation, irregular crypts, and goblet cell loss; 2=mild to moderate crypt loss (10–50%); 3=severe crypt loss (50–90%); 4=complete crypt loss, surface epithelium intact; 5=small to medium sized ulcer (<10 crypt widths); 6=large ulcer (>10 crypt widths). Infiltration with inflammatory cells was scored separately for mucosa (0=normal; 1=mild; 2=modest; 3=severe), submucosa (0=normal; 1=mild to modest; 2=severe), and muscle/serosa (0=normal; 1=moderate to severe). Scores for epithelial damage and inflammatory cell infiltration were added, resulting in a total colitis score ranging from 0 to 12.
Colonic explants and mesenteric lymph node T-cell stimulation. After extraction and longitudinal opening of the colon, three rectangular tissue specimens (2 mm × 8–10 mm) from the proximal, middle, and distal colon were excised. The tissues were weighed, then washed in RPMI-1640 medium (Gibco, Grand Island, NY) containing streptomycin (100 μg ml−1) and penicillin (100 U ml−1). Tissue explants were incubated in 24-well plates in 500 μl supplemented RPMI-1640 culture medium for 18 h (at 37 °C, with 5% CO2 humidified air). After incubation, supernatants were harvested and stored at −80°C until cytokine quantification using ELISA. Cytokine levels were normalized per mg colon tissue. For T-cell stimulation assays, mesenteric lymph node cells were stimulated with 10 μg ml−1 plate-bound anti-CD3 and 1 μg ml−1 soluble anti-CD28 Abs in complete RPMI-1640 medium. Supernatants were collected 24 h after stimulation for evaluation of cytokine levels by ELISA.
ELISA and EIA. Quantitative analyses for TNF-α, IL-1β, IL-6, IL-17A, and interferon-γ were performed with commercial ELISA kits (eBioscience) according to the manufacturer’s instructions. CGRP levels in colon homogenates were quantified with the CGRP (rat) EIA kit obtained from Cayman (Ann Arbor, MI) following the manufacturer’s instructions.
Intestinal epithelial cell isolation. Intestinal epithelial cell isolation was performed as previously described.62 In short, colons were washed three times in PBS (1 mM dithiothreitol), followed by incubation in HBSS (5 mM EDTA, 0.5 mM dithiothreitol) for 1 h at 37 °C. Detached single cells and crypts (supernatants) were centrifuged at 800 g for 5 min in a table-top centrifuge, and washed three times in PBS before RNA extraction.
Bone marrow chimeras. Groups of 8- to 10-week-old, sex-matched recipients (WT or knockout mice) were γ-irradiated (9 Gy). Recipient mice were then intravenously injected with 107 bone marrow cells from sex-matched donor mice within 24 h after irradiation. Recipients received sulfamethoxazole/trimethoprim in drinking water for 2 weeks at the beginning of the protocol. After 6 weeks, mice were subjected to DSS. Chimerism was quantified using Q-PCR of peripheral blood cells at the time of killing, as previously described.63
Flow cytometry. Flow cytometry was performed as previously described.64 Briefly, enriched CD11c+ BMDCs were pretreated with Fc-Block (Anti-Mouse CD16/CD32 Purified; eBioscience). Cells were then stained with anti-CRLR (H-42; Santa Cruz, Dallas, TX), anti-RAMP1 (FL-148; Santa Cruz) Abs or control serum, followed by detection with AF488-conjugated secondary Abs. Data were acquired with the C6 Accuri flow cytometer (BD Biosciences, San Jose, CA).
Immunohistochemistry and BrdU labeling. Immunohistochemistry and BrdU labeling was performed as previously reported.62, 63 Briefly, formalin-fixed colons were paraffin-embedded and sectioned (5 μm). Tissue sections were incubated overnight at 4 °C with optimized dilutions of anti-Ki67 (Abcam, Cambridge, MA) antibodies (Abs), incubated with horseradish peroxidase-conjugated secondary Abs for 1 h, followed by visualization with the DAB peroxidase substrate kit (Vector Laboratories, Burlingame, CA). For BrdU stainings, mice were injected i.p. with 2 mg of BrdU solution at 2, 48, or 72 h before being killed. The BrdU In-Situ Detection Kit (BD) was used according to the manufacturer’s instructions. Slides were counterstained with Hematoxylin 560 (Surgipath, Richmond, IL).
Immunofluorescent stainings and tissue analysis. Mouse spleen and colon were removed, washed, and fixed overnight (at 4 °C) in 4% paraformaldehyde. Human colon was obtained from consented patients undergoing right hemicolectomies for cancer, according to ethical regulations (NREC 09/H0704/02). Ascending colon that was a minimum of 10 cm away from the tumor was used for IF stainings. The specimens were macroscopically uninflamed. The image shown is representative of the ascending colon samples from four patients. Age range was 59–83 years old, two females and two males. These colon tissues were fixed overnight in 4% paraformaldehyde. Briefly, 10-μm sections of cryoprotected tissue were cut, incubated with blocking buffer (Dako, Carpinteria, CA) for 1 h, before primary Abs were applied overnight (4 °C); TRPM8 (1:200, Abcam: ab104569—for mouse tissue; 1:200, Alomone: ACC-049—for human colon), CGRP (1:400, Abcam: ab36001), CD11c (1:100, Abcam: ab33483), and E-cadherin (1:50, Millipore, MABT26). Tissues were then washed and species-specific AF secondary Ab (1:400, Invitrogen, Carlsbad, CA) applied for 1 h. A Leica DM4000 epifluorescence microscope was used to visualize IR. Five fields of view (1.44 megapixel) were randomly obtained with a QImaging camera and analyzed with ImageJ for total number of IR pixels in the region of interest within the mucosal layer.
CD11c+ splenic DC isolation and BMDC culture. Spleens were harvested and placed in 35-mm petri dishes containing RPMI-1640 (2% fetal bovine serum), followed by homogenization. Splenocytes were filtered (100 μm) and erythrocytes were lysed by adding ACK Lysing Buffer (Gibco). CD11c+ cells were then isolated by using the EasySep mouse CD11c positive selection kit (StemCell, Vancouver, British Columbia, Canada), according to the manufacturer’s instructions. CD11c+ BMDCs were cultured and harvested as previously described.65 Briefly, BM cells from femurs and tibias were aseptically flushed with RPMI-1640 (2% fetal bovine serum). BM cells were then passed through a 100-μm nylon mesh. BM cells were seeded at a density of 1 × 109 cells in complete RPMI medium containing 10 ng ml−1 rmGM-CSF. Medium was replenished (50% v/v) on day 3 and day 6, respectively. CD11c+ DC cell selection was performed on day 7 by positive selection.
CD11c+ DC stimulation with TLR ligands. Enriched CD11c+ DCs were seeded at a density of 2 × 105 per well in 96-well plates in complete RPMI medium in the presence of 10 ng ml−1 LPS or 5 μg ml−1 CpG. For some experiments, DCs were pretreated with 100 nM CGRP for 30 min before TLR stimulation. Supernatants were harvested after 24 h and cytokine concentrations in the supernatant were measured with ELISA.
RNA isolation and Q-PCR. Colonic tissue samples from proximal, middle, and distal colon were snap-frozen and kept at −80 °C until RNA isolation. IEC and CD11c+ DC cell suspensions were directly lysed in RLT buffer after isolation and enrichment procedures and stored at −80 °C. RNA isolations were performed with the RNeasy Mini Kit (Qiagen, Hilden, Germany), cDNA synthesis with the qScript cDNA superMix kit (Quanta Biosciences, Gaithersburg, MD). Q-PCR was performed on the AB7300 Real-Time PCR System (Applied Biosystems) using the PerfeCta SYBR Green FastMix (Quanta Biosciences). Custom-designed oligonucleotide sequences (IDT Technologies, Coralville, IA) were used for Q-PCR (see Supplementary Table 1). To validate the specificity of the primer pairs, PCR products were run on 2% agarose gels and visualized with SYBR Safe DNA (Invitrogen).
Statistical analysis. Data are presented as mean±s.d. or mean±s.e.m., as indicated. P-values are stated in figure legends. Unpaired Student’s t-test was used for statistical analyses to compare two data sets with normal distribution, Mann–Whitney U-test was used for nonparametric data, analysis of variance was used to compare multiple data sets (GraphPad Prism 5.0, La Jolla, CA).
References
Nilius, B., Owsianik, G., Voets, T. & Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 87, 165–217 (2007).
Holzer, P. TRP channels in the digestive system. Curr. Pharm. Biotechnol. 12, 24–34 (2011).
Dhaka, A., Murray, A.N., Mathur, J., Earley, T.J., Petrus, M.J. & Patapoutian, A. TRPM8 is required for cold sensation in mice. Neuron 54, 371–378 (2007).
McKemy, D.D., Neuhausser, W.M. & Julius, D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58 (2002).
Peier, A.M. et al. A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715 (2002).
Dhaka, A., Earley, T.J., Watson, J. & Patapoutian, A. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J. Neurosci. 28, 566–575 (2008).
Hayashi, T. et al. Expression of the TRPM8-immunoreactivity in dorsal root ganglion neurons innervating the rat urinary bladder. Neurosci. Res. 65, 245–251 (2009).
Fisher, J.T. TRPM8 and dyspnea: from the frigid and fascinating past to the cool future? Curr. Opin. Pharmacol 11, 218–223 (2011).
Almeida, M.C. et al. Pharmacological blockade of the cold receptor TRPM8 attenuates autonomic and behavioral cold defenses and decreases deep body temperature. J. Neurosci. 32, 2086–2099 (2012).
Zhang, L., Jones, S., Brody, K., Costa, M. & Brookes, S.J. Thermosensitive transient receptor potential channels in vagal afferent neurons of the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G983–G991 (2004).
Harrington, A.M. et al. A novel role for TRPM8 in visceral afferent function. Pain 152, 1459–1468 (2011).
Engel, M.A., Becker, C., Reeh, P.W. & Neurath, M.F. Role of sensory neurons in colitis: increasing evidence for a neuroimmune link in the gut. Inflamm. Bowel Dis. 17, 1030–1033 (2010).
Holzer, P. Vanilloid receptor TRPV1: hot on the tongue and inflaming the colon. Neurogastroenterol. Motil. 16, 697–699 (2004).
Blackshaw, L.A., Brierley, S.M. & Hughes, P.A. TRP channels: new targets for visceral pain. Gut 59, 126–135 (2010).
Kimball, E.S., Wallace, N.H., Schneider, C.R., D’Andrea, M.R. & Hornby, P.J. Vanilloid receptor 1 antagonists attenuate disease severity in dextran sulphate sodium-induced colitis in mice. Neurogastroenterol. Motil. 16, 811–818 (2004).
Szitter, I. et al. The role of transient receptor potential vanilloid 1 (TRPV1) receptors in dextran sulfate-induced colitis in mice. J. Mol. Neurosci. 42, 80–88 (2010).
Eijkelkamp, N., Kavelaars, A., Elsenbruch, S., Schedlowski, M., Holtmann, G. & Heijnen, C.J. Increased visceral sensitivity to capsaicin after DSS-induced colitis in mice: spinal cord c-Fos expression and behavior. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G749–G757 (2007).
Miranda, A., Nordstrom, E., Mannem, A., Smith, C., Banerjee, B. & Sengupta, J.N. The role of transient receptor potential vanilloid 1 in mechanical and chemical visceral hyperalgesia following experimental colitis. Neuroscience 148, 1021–1032 (2007).
Phillis, B.D. et al. Role of TRPV1 in high-threshold rat colonic splanchnic afferents is revealed by inflammation. Neurosci. Lett. 459, 57–61 (2009).
Christianson, J.A., McIlwrath, S.L., Koerber, H.R. & Davis, B.M. Transient receptor potential vanilloid 1-immunopositive neurons in the mouse are more prevalent within colon afferents compared to skin and muscle afferents. Neuroscience 140, 247–257 (2006).
Matsumoto, K., Hosoya, T., Tashima, K., Namiki, T., Murayama, T. & Horie, S. Distribution of transient receptor potential vanilloid 1 channel-expressing nerve fibers in mouse rectal and colonic enteric nervous system: relationship to peptidergic and nitrergic neurons. Neuroscience 172, 518–534 (2011).
Tan, L.L., Bornstein, J.C. & Anderson, C.R. Distinct chemical classes of medium-sized transient receptor potential channel vanilloid 1-immunoreactive dorsal root ganglion neurons innervate the adult mouse jejunum and colon. Neuroscience 156, 334–343 (2008).
Kimball, E.S., Prouty, S.P., Pavlick, K.P., Wallace, N.H., Schneider, C.R. & Hornby, P.J. Stimulation of neuronal receptors, neuropeptides and cytokines during experimental oil of mustard colitis. Neurogastroenterol. Motil. 19, 390–400 (2007).
Engel, M.A. et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology 141, 1346–1358 (2011).
D’Aldebert, E. et al. Transient receptor potential vanilloid 4 activated inflammatory signals by intestinal epithelial cells and colitis in mice. Gastroenterology 140, 275–285 (2011).
Fichna, J. et al. Transient receptor potential vanilloid 4 blockade protects against experimental colitis in mice: a new strategy for inflammatory bowel diseases treatment? Neurogastroenterol. Motil. 24, e557–e560 (2012).
Yamamoto, S. et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747 (2008).
Ramachandran, R. et al. TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc. Natl Acad. Sci. USA 110, 7476–7481 (2013).
Wirtz, S., Neufert, C., Weigmann, B. & Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2, 541–546 (2007).
Denda, M., Tsutsumi, M. & Denda, S. Topical application of TRPM8 agonists accelerates skin permeability barrier recovery and reduces epidermal proliferation induced by barrier insult: role of cold-sensitive TRP receptors in epidermal permeability barrier homoeostasis. Exp. Dermatol. 19, 791–795 (2010).
Li, M., Li, Q., Yang, G., Kolosov, V.P., Perelman, J.M. & Zhou, X.D. Cold temperature induces mucin hypersecretion from normal human bronchial epithelial cells in vitro through a transient receptor potential melastatin 8 (TRPM8)-mediated mechanism. J. Allergy Clin. Immunol. 128, 626–634 (2011).
Abe, K. et al. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc. Natl Acad. Sci. USA 104, 17022–17027 (2007).
Perraud, A.L., Knowles, H.M. & Schmitz, C. Novel aspects of signaling and ion-homeostasis regulation in immunocytes. The TRPM ion channels and their potential role in modulating the immune response. Mol. Immunol. 41, 657–673 (2004).
Wu, S.N., Wu, P.Y. & Tsai, M.L. Characterization of TRPM8-like channels activated by the cooling agent icilin in the macrophage cell line RAW 264.7. J. Membr. Biol. 241, 11–20 (2011).
Weinstock, J.V., Blum, A., Metwali, A., Elliott, D., Bunnett, N. & Arsenescu, R. Substance P regulates Th1-type colitis in IL-10 knockout mice. J. Immunol. 171, 3762–3767 (2003).
Engel, M.A. et al. Opposite effects of substance P and calcitonin gene-related peptide in oxazolone colitis. Dig. Liver Dis. 44, 24–29 (2012).
Reinshagen, M. et al. Calcitonin gene-related peptide mediates the protective effect of sensory nerves in a model of colonic injury. J. Pharmacol. Exp. Ther. 286, 657–661 (1998).
Thompson, B.J., Washington, M.K., Kurre, U., Singh, M., Rula, E.Y. & Emeson, R.B. Protective roles of alpha-calcitonin and beta-calcitonin gene-related peptide in spontaneous and experimentally induced colitis. Dig. Dis. Sci. 53, 229–241 (2008).
Altmayr, F., Jusek, G. & Holzmann, B. The neuropeptide calcitonin gene-related peptide causes repression of tumor necrosis factor-alpha transcription and suppression of ATF-2 promoter recruitment in Toll-like receptor-stimulated dendritic cells. J. Biol. Chem. 285, 3525–3531 (2010).
Rochlitzer, S. et al. The neuropeptide calcitonin gene-related peptide affects allergic airway inflammation by modulating dendritic cell function. Clin. Exp. Allergy 41, 1609–1621 (2011).
Tsujikawa, K. et al. Hypertension and dysregulated proinflammatory cytokine production in receptor activity-modifying protein 1-deficient mice. Proc. Natl Acad. Sci. USA 104, 16702–16707 (2007).
Vanden Abeele, F. et al. Ca2+-independent phospholipase A2-dependent gating of TRPM8 by lysophospholipids. J. Biol. Chem. 281, 40174–40182 (2006).
Andersson, D.A., Nash, M. & Bevan, S. Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J. Neurosci. 27, 3347–3355 (2007).
Meyer, M.C., Creer, M.H. & McHowat, J. Potential role for mast cell tryptase in recruitment of inflammatory cells to endothelium. Am. J. Physiol. Cell Physiol. 289, C1485–C1491 (2005).
Rohacs, T., Lopes, C.M., Michailidis, I. & Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8, 626–634 (2005).
Wright, B.D. et al. The lipid kinase PIP5K1C regulates pain signaling and sensitization. Neuron 82, 836–847 (2014).
De Petrocellis, L., Starowicz, K., Moriello, A.S., Vivese, M., Orlando, P. & Di Marzo, V. Regulation of transient receptor potential channels of melastatin type 8 (TRPM8): effect of cAMP, cannabinoid CB(1) receptors and endovanilloids. Exp. Cell Res. 313, 1911–1920 (2007).
Zhang, X. et al. Direct inhibition of the cold-activated TRPM8 ion channel by Galphaq. Nat. Cell Biol. 14, 851–858 (2012).
Takashima, Y., Daniels, R.L., Knowlton, W., Teng, J., Liman, E.R. & McKemy, D.D. Diversity in the neural circuitry of cold sensing revealed by genetic axonal labeling of transient receptor potential melastatin 8 neurons. J. Neurosci. 27, 14147–14157 (2007).
Axelsson, H.E., Minde, J.K., Sonesson, A., Toolanen, G., Hogestatt, E.D. & Zygmunt, P.M. Transient receptor potential vanilloid 1, vanilloid 2 and melastatin 8 immunoreactive nerve fibers in human skin from individuals with and without Norrbottnian congenital insensitivity to pain. Neuroscience 162, 1322–1332 (2009).
Abe, J. et al. TRPM8 protein localization in trigeminal ganglion and taste papillae. Brain Res. Mol. Brain Res. 136, 91–98 (2005).
Engel, M.A. et al. The proximodistal aggravation of colitis depends on substance P released from TRPV1-expressing sensory neurons. J. Gastroenterol. 47, 256–265 (2012).
Mikami, N. et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions. J. Immunol. 186, 6886–6893 (2011).
Harzenetter, M.D., Novotny, A.R., Gais, P., Molina, C.A., Altmayr, F. & Holzmann, B. Negative regulation of TLR responses by the neuropeptide CGRP is mediated by the transcriptional repressor ICER. J. Immunol. 179, 607–615 (2007).
Persson, E.K., Scott, C.L., Mowat, A.M. & Agace, W.W. Dendritic cell subsets in the intestinal lamina propria: ontogeny and function. Eur. J. Immunol. 43, 3098–3107 (2013).
Milling, S., Yrlid, U., Cerovic, V. & MacPherson, G. Subsets of migrating intestinal dendritic cells. Immunol. Rev. 234, 259–267 (2010).
Cerovic, V., Bain, C.C., Mowat, A.M. & Milling, S.W. Intestinal macrophages and dendritic cells: what's the difference? Trends Immunol. 35, 270–277 (2014).
Hosoya, T. et al. TRPM8 has a key role in experimental colitis-induced visceral hyperalgesia in mice. Neurogastroenterol. Motil. 26, 1112–1121 (2014).
Garza, A., Weinstock, J. & Robinson, P. Absence of the SP/SP receptor circuitry in the substance P-precursor knockout mice or SP receptor, neurokinin (NK)1 knockout mice leads to an inhibited cytokine response in granulomas associated with murine Taenia crassiceps infection. J. Parasitol. 94, 1253–1258 (2008).
Gareau, M.G., Jury, J., MacQueen, G., Sherman, P.M. & Perdue, M.H. Probiotic treatment of rat pups normalises corticosterone release and ameliorates colonic dysfunction induced by maternal separation. Gut 56, 1522–1528 (2007).
Marchelletta, R.R. et al. Altered expression and localization of ion transporters contribute to diarrhea in mice with Salmonella-induced enteritis. Gastroenterology 145, 1358–1368 (2013).
de Jong, P.R. et al. Ion channel TRPV1-dependent activation of PTP1B suppresses EGFR-associated intestinal tumorigenesis. J. Clin. Invest. 124, 3793–3806 (2014).
Lee, S.H. et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat. Med. 16, 665–670 (2010).
Li, X. et al. Divergent requirement for Galphas and cAMP in the differentiation and inflammatory profile of distinct mouse Th subsets. J. Clin. Invest. 122, 963–973 (2012).
Gonzalez-Navajas, J.M. et al. Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9-driven antiinflammatory cytokine production. J. Exp. Med. 207, 2799–2807 (2010).
Acknowledgements
We thank Melissa Scholl for animal breeding. Liwen Deng and Steve Shenouda performed the tissue processing and Professor Nissi Varki from the Department of Pathology (UCSD) assisted with histological evaluations. Richard Blackshaw and James Blackshaw assisted with image analysis. We thank Jennifer Santini for assistance in confocal imaging at the University of California at San Diego Neuroscience Microscopy Shared Facility, funded by NIH Grant P30 NS047101 through the NINDS. LAB was the principal investigator on the immunohistochemical studies, funded by the Wellcome Trust. This study was supported by grants from the Crohn’s and Colitis Foundation of America (CCFA) to PRJ (Research Fellowship Award no. 2927), SB (RFA no. 3574), and ER (Senior Research Award no. 3038); the Japan Society for the Promotion of Science (JSPS) to NT; and the NIH (AI095623, DK35108, and AI077989-05).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
de Jong, P., Takahashi, N., Peiris, M. et al. TRPM8 on mucosal sensory nerves regulates colitogenic responses by innate immune cells via CGRP. Mucosal Immunol 8, 491–504 (2015). https://doi.org/10.1038/mi.2014.82
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.82
This article is cited by
-
Genetic variants in migraine: a field synopsis and systematic re-analysis of meta-analyses
The Journal of Headache and Pain (2020)
-
Pain and immunity: implications for host defence
Nature Reviews Immunology (2019)
-
Sensory TRP channels contribute differentially to skin inflammation and persistent itch
Nature Communications (2017)
-
TRPM8 in the negative regulation of TNFα expression during cold stress
Scientific Reports (2017)
-
ERK5 signalling rescues intestinal epithelial turnover and tumour cell proliferation upon ERK1/2 abrogation
Nature Communications (2016)
