
Abstract
The immune mechanisms regulating epithelial cell repair after injury remain poorly defined. We demonstrate here that lymphotoxin beta receptor (LTβR) signaling in intestinal epithelial cells promotes self-repair after mucosal damage. Using a conditional gene-targeted approach, we demonstrate that LTβR signaling in intestinal epithelial cells is essential for epithelial interleukin-23 (IL-23) production and protection against epithelial injury. We further show that epithelial-derived IL-23 promotes mucosal wound healing by inducing the IL-22-mediated proliferation and survival of epithelial cells and mucus production. Additionally, we identified CD4–CCR6+T-bet– RAR-related orphan receptor gamma t (RORγt)+ lymphoid tissue inducer cells as the main producers of protective IL-22 after epithelial damage. Thus, our results reveal a novel role for LTβR signaling in epithelial cells in the regulation of intestinal epithelial cell homeostasis to limit mucosal damage.
Similar content being viewed by others


Introduction
The intricate balance between immune defense and inflammation in the gut is a highly regulated process that requires interactions between the intestinal epithelium and the underlying immune system. A breakdown in this balance is believed to promote the induction and perpetuation of the chronic intestinal inflammation found in patients with intestinal inflammatory diseases.1, 2
Lymphotoxin beta receptor (LTβR), a member of the tumor necrosis factor receptor superfamily of cytokines, has been shown to play a critical role in the regulation of mucosal immune responses.3, 4, 5, 6 Like many other members of the tumor necrosis factor receptor superfamily, LTβR signaling can mediate both protective and pathogenic effects during intestinal inflammation. While inhibition of LTβR signaling has been shown to be beneficial in a T-cell-mediated colitis model,7 studies utilizing other chemically induced and infectious colitis models suggest that LTβR signaling plays a protective role against intestinal injury.4, 5, 8, 9, 10, 11 Additionally, LTβR-dependent production of interleukin-22 (IL-22) by innate lymphoid cells (ILCs) has recently been shown to be essential for protection against the bacterial pathogen Citrobacter rodentium.4, 10 However, the mechanisms whereby LTβR signaling contributes to protection during epithelial damage remain to be fully elucidated.
ILCs are a heterogeneous population of innate lineage-negative lymphoid cells that rapidly produce several cytokines upon stimulation and participate in the regulation of immunity at mucosal surfaces.12, 13, 14 The current classification of ILC populations is based on the expression of specific transcription factors that regulate their development and function, and their cytokine profiles.15 Group 3 ILCs (ILC3s) are dependent on the transcription factors RAR-related orphan receptor gamma t (RORγt) and Gata3 for their development and include CD4+ and CD4– lymphoid tissue inducer cell populations (LTi cells), natural killer (NK)p46+ and colitogenic NKp46– ILC3s.15, 16, 17, 18 A protective role for ILC3s during epithelial injury was suggested by a previous study showing that RORγt-deficient mice, which lack all ILC3 subsets, exhibited increased susceptibility to chemically induced colitis.19 In contrast, a pathogenic role for ILC3s has also been reported in experimental colitis models and in patients suffering from inflammatory bowel disease.16, 20 However, the mechanisms that regulate the function of distinct ILC3s during mucosal damage are still poorly defined.
IL-22, IL-17, and interferon γ (IFNγ), the principal cytokines produced by ILC3s, have major effects on epithelial cells of many tissues and are critical in defense against mucosal bacterial pathogens.15, 21, 22 IL-22 has been identified as an important regulator of inflammation, particularly at the barrier surfaces of the skin, lung, and intestine.14, 23 However, dysregulation of IL-22 signaling may also be associated with disease progression, including tumorigenesis.24, 25, 26 Therefore, understanding the mechanisms controlling IL-22 production by distinct ILC3s populations during mucosal damage is critical for development of effective disease therapies.
In the present study, we investigate the role of LTβR-mediated signaling between intestinal epithelial cells and ILC3s in a model of mucosal wound healing. Our data suggest that during epithelial injury, LTβR engagement in intestinal epithelial cells drives IL-23 production. The IL-23 then induces IL-22 production by CD4− LTi cells thereby promoting epithelial cell repair and homeostasis. These results support a novel mechanism wherein LTβR-mediated cooperation between epithelial cells and LTi cells regulates intestinal homeostasis to limit mucosal damage.
Results
LTβR signaling protects against intestinal epithelial injury by promoting IL-22-dependent epithelial cell repair and homeostasis
To define the role of LTβR signaling in mucosal wound healing, we utilized an acute epithelial injury regeneration model, induced by transient administration of dextran sulfate sodium (DSS) in the drinking water. Since the genetic background of the mouse can significantly influence the outcome of DSS treatments,27 we first generated neo-free LTβR-deficient and LTβR-floxed mice by crossing tri-loxP conditional LTβR-floxed mice4 with ubiquitous MeuCre40 deleter mice,28 (all on C57BL/6 background) and selecting recombinants with a deletion of the neo cassette (Supplementary Figure S1a, b online). We then treated control neo-free LTβR-floxed mice (Ltbrfl/fl) and neo-free LTβR-deficient mice (LtbrΔ/Δ) with drinking water containing DSS for 5 days followed by normal water for 3 days. Compared with Ltbrfl/fl mice, LtbrΔ/Δ mice lost body weight rapidly and displayed reduced survival (Figure 1a, b). Consistently, the disease score, assessed daily as an average of body weight loss and signs of rectal bleeding and diarrhea, was increased in LtbrΔ/Δ mice (Figure 1c). Importantly, these differences were not associated with altered food or water consumption (Supplementary Figure S1c). Macroscopic examination of the colons at day 8 revealed severe pathology and significant reduction of colon length in LtbrΔ/Δ mice compared with control animals (Figure 1d). Additionally, while the colon mucosa and submucosa of Ltbrfl/fl mice exhibited low histological scores reflecting only slight infiltration of inflammatory cells, LtbrΔ/Δ mice exhibited increased histological scores reflecting severe destruction and necrosis of epithelial cells, loss of goblet cells, thickening of the submucosa, and increased infiltration of inflammatory cells (Figure 1e). Together, these data show that LTβR signaling is essential for mucosal wound healing following epithelial injury.

Lymphotoxin beta receptor (LTβR) signaling protects against intestinal epithelial injury by promoting interleukin-22 (IL-22)-dependent epithelial cell repair and homeostasis. Ltbrfl/fl and LtbrΔ/Δ mice (n=5) were treated for 5 days with 3.5% dextran sulfate sodium (DSS). (a) Weight loss, (b) survival, (c) disease score, and (d) length and photographs of colons at day 8. (e) Hematoxylin and eosin staining and histological score of colon sections at day 8. (f) Kinetics of colonic Il22 mRNA expression. (g) IL-22 production in the supernatants of colons collected at day 5 and cultured for 24 h. (h) Kinetics of colonic mRNA expression of indicated antimicrobial proteins. (i) Bacterial titers in liver and spleen at day 8. (j) Kinetics of colonic expression of indicated mucin genes. (k) Immunohistochemical staining of Muc2 in sections of colons collected at day 5. (l) Colon sections were stained with terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) (white) to detect apoptotic cells at day 8. Nuclei were stained with Hoechst (blue). The number of apoptotic cells was determined by analyzing 10 crypts per colon section. (m) Colonic expression of antiapoptotic factors at day 5. (n) Intestinal epithelial cell proliferation was analyzed with bromodeoxyuridine (BrdU) pulse injection on day 8. Ten crypts from the colons of five mice per genotype were analyzed. For mRNA expression analysis, Ct values were normalized to Hprt expression. Data are representative of two (g, i, k–n) or three independent (a–f, h, j) experiments.*P<0.05; **P<0.01; ***P<0.001 (Student’s t-test). Error bars represent s.e.m.
To define the mechanism whereby LTβR signaling conferred protection against DSS-induced colitis, we measured the expression of genes known to be involved in the induction or regulation of intestinal inflammation at day 5, when the response of the two groups was beginning to diverge. Surprisingly, Ltbrfl/fl and LtbrΔ/Δ mice displayed similar colonic mRNA expression levels for several chemokines (Cxcl1, Cxcl13, Ccl2, and Ccl20), IL-17 family members (Il17a, Il17c, Il17e, Il17f, and Il17re), key pro-inflammatory cytokines (Il-1b, Il6, ifng, and tnf) and regulatory molecules such as Il10 and Trim30a (Supplementary Figure S1d). However, we found that Il23a and Il22 mRNA expression were significantly reduced in the colons of DSS-treated LtbrΔ/Δ mice (Supplementary Figure S1d). To define the kinetics of IL-22 production, we next measured Il22 mRNA expression during the injury and regeneration stages of colitis. While DSS-treated Ltbrfl/fl mice displayed a marked upregulation of Il22 mRNA expression in the colon between day 5 and day 8 (Figure 1f), LtbrΔ/Δ mice failed to upregulate colonic IL-22 (Figure 1f, g), suggesting that LTβR signaling is required for IL-22 production during intestinal injury.
IL-22 has been shown to exert its antiinflammatory effects via the induction of a wide range of antimicrobial peptides that help to prevent the lethal dissemination of intestinal microbiota, and via reinforcing the mucus barrier and enhancing epithelial cell regeneration.29, 30, 31, 32 Unexpectedly, we found comparable mRNA levels of Reg3b, Reg3g, and Defb4 in the colons of DSS-treated Ltbrfl/fl and LtbrΔ/Δ mice (Figure 1h). Additionally, similar levels of bacterial dissemination were found in the liver and spleen of DSS-treated Ltbrfl/fl and LtbrΔ/Δ mice at day 8 (Figure 1i), suggesting that the sensitivity of LtbrΔ/Δ mice to DSS-induced colitis is not mediated by the pathogenic dissemination of commensal bacteria. We therefore hypothesized that the sensitivity of LtbrΔ/Δ mice is caused by a defect in the promotion of IL-22-dependent tissue protective responses. To test this, we compared the induction of IL-22-associated mucins, which are thought to protect against intestinal inflammation,29, 33 between DSS-treated Ltbrfl/fl and LtbrΔ/Δ mice. Muc2, Muc3, Muc4, and Muc13 mRNA levels were upregulated in the colons of control mice during DSS-induced colitis, whereas the induction of these mucins was impaired in LtbrΔ/Δ mice (Figure 1j). Moreover, Muc2 protein, the most abundant mucin in the large intestine,33, 34 was expressed at a lower level in the colons of LtbrΔ/Δ mice during DSS-induced injury (Figure 1k). Since mucins can protect epithelial cells from damage during intestinal inflammation,29, 33 we next addressed whether there was increased epithelial cell death in the colon of LtbrΔ/Δ mice. We found that, with respect to Ltbrfl/fl mice, LtbrΔ/Δ mice exhibited an increase in the number of apoptotic epithelial cells at day 8 (Figure 1l). Consistent with this observation, expression of the antiapoptotic factors Bcl2 and Bclxl was significantly reduced in the colon of LtbrΔ/Δ mice at day 5 (Figure 1m). Moreover, analysis of epithelial cell proliferation following bromodeoxyuridine pulse injection revealed that intestinal epithelial cell proliferation was markedly reduced in LtbrΔ/Δ mice (Figure 1n). Collectively, these data suggest that LTβR-mediated IL-22 production promotes intestinal wound healing by inducing the regeneration of the epithelial layer.
Exogenous IL-22 expression rescues LTβR-deficient mice from epithelial injury
To determine whether IL-22 production is sufficient to promote tissue repair upon DSS-induced epithelial injury, we intravenously injected control and LtbrΔ/Δ mice with an IL-22-expressing or control plasmid 24 h after initiation of DSS treatment. IL-22-expressing plasmid significantly reduced the severity of DSS-induced colitis in LtbrΔ/Δ mice as indicated by reduced body weight loss, increased survival, decreased disease score, and reduced colon shortening and pathology (Figure 2a–d). These macroscopic observations were corroborated with histological analysis of colon sections which revealed reduced colon inflammation and reduced histological scores in IL-22-expressing plasmid-treated LtbrΔ/Δ mice (Figure 2e). Furthermore, exogenous IL-22 expression also increased Muc2, Muc3, Muc4, and Muc13 mRNA expression in the colons of DSS-treated LtbrΔ/Δ mice (Figure 2f). Taken together, these data suggest that IL-22-mediated induction of mucins is the critical mechanism downstream of LTβR signaling that promotes wound healing after epithelial injury.

Exogenous interleukin-22 (IL-22) expression rescues lymphotoxin beta receptor (LTβR)-deficient mice from epithelial injury. Ltbrfl/fl and LtbrΔ/Δ mice (n=5) were treated for 5 days with 3.5% dextran sulfate sodium (DSS). In total, 10 μg of plasmid encoding empty vector (pRK) or IL-22-expressing plasmid were injected intravenously at day 1. (a–c) Weight loss (a), survival (b), and clinical disease score (c). (d) Length and photographs of colons at day 8. (e) Hematoxylin and eosin staining and histological score of colon sections at day 8. (f) Colonic expression of mucin genes at day 8. Data are normalized to Hprt. *P<0.05; **P<0.01; ***P<0.001; NS, not significant (Student’s t-test). Data are representative of two independent experiments. Error bars represent s.e.m.
LTβR signaling promotes epithelial wound healing through IL-22 production by CD4− LTi cells
To identify which cells require LTβR signaling for IL-22 production during intestinal injury, we compared Il22 mRNA expression in distinct innate and adaptive cell populations isolated from the colonic lamina propria (LP) of RORγt–green fluorescent protein (GFP)+/−Ltbr+/Δ and RORγt–GFP+/−LtbrΔ/Δ reporter mice treated with DSS for 5 days. We found that Il22 expression was markedly reduced in CD4− and CD4+ LTi cells isolated from RORγt–GFP+/−LtbrΔ/Δ mice (Figure 3a), indicating that IL-22 production by these cells is LTβR dependent. Interestingly, Il22 expression by NKp46+ ILC3 cells was not significantly impaired in the absence of LTβR signaling (Figure 3a). Importantly, we found comparable numbers and frequencies of CD4− LTi, CD4+ LTi, NKp46+ ILC3, RORγt+ T cells, CD4+ T cells, NK cells, and B cells in the LP of Ltbr+/Δ and LtbrΔ/Δ mice (Figure 3b and Supplementary Figure S2) indicating no defect in the recruitment of IL-22-producing cells to the LP in response to epithelial injury in the absence of LTβR signaling.

Lymphotoxin beta receptor (LTβR)-dependent interleukin-22 (IL-22) production by CD4−CCR6+T-bet− lymphoid tissue inducer (LTi) cells is the primary source of protective IL-22 during epithelial injury. (a–c) Lamina propria (LP) cells were purified from the colon of RAR-related orphan receptor gamma t (RORγt)–green fluorescent protein (GFP)+/– Ltbr+lΔ and RORγt–GFP+/− LtbrΔ/Δ reporter mice treated with 3.5% dextran sulfate sodium (DSS) for 5 days (n=3). (a) Expression of Il22 mRNA in sorted CD4− (CD45+RORγt+CD3−CD4−NKp46−) and CD4+ LTi cells (CD45+RORγt+CD3−CD4+NKp46−), NKp46+ innate lymphoid cell (ILC)3s (CD45+RORγt+CD3−CD4−NKp46+), RORγt+ T cells (CD45+RORγt+CD3+CD4+NKp46−), CD4+ T cells (CD45+RORγt−CD3+CD4+NKp46−) and natural killer (NK) cells (CD45+RORγt−CD3−CD4−NKp46+). (b) Number of indicated LP cell populations per colon. (c) Expression of Il23r mRNA in sorted RORγt+ ILCs and RORγt+ T cells. (d, e) Colonic LP cells from Ltbrfl/fl and LtbrΔ/Δ mice treated with 3.5% DSS for 5 days (n=3 per group) were stimulated ex vivo for 4 h with IL-23 (50 ng ml−1). The number (d) and surface and intracellular marker analysis (e) of colonic LP CD45+Lin−RORγt+ IL-22-producing cells was evaluated by flow cytometry. (f–i) Rag1−/− mice (n=4) were treated for 5 days with 3.5% DSS. Isotype control, αCD4 and αThy1.2 (150 μg per mouse) were injected intraperitoneally (i.p.) at days 0 and 3. (f) Weight loss, (g) survival, and (h) colon length evaluated at day 8. (i) Expression of colonic Il22 mRNA at day 8. For mRNA expression, data are normalized to Hprt. *P<0.05; **P<0.01; NS, not significant (Student’s t-test). Data are representative of two independent experiments. Error bars represent s.e.m.
Production of IL-22 by ILCs has been shown to be triggered by IL-23R stimulation.21, 22 We found that ILCs and RORγt+ T cells isolated from the colons of Ltbr+/Δ and LtbrΔ/Δ mice displayed similar levels of IL-23R expression (Figure 3c). Furthermore, LP ILCs isolated from LtbrΔ/Δ mice were able to produce IL-22 after in vitro stimulation with IL-23 (Figure 3d), indicating that RORγt+ ILCs from LtbrΔ/Δ mice do not have an intrinsic defect in IL-22 production.
To characterize the IL-22-producing ILC subsets, we purified colonic LP cells from DSS-treated Ltbrfl/fl mice and stimulated them with IL-23. The majority of CD45+Lin−RORγt+ IL-22-producing cells expressed IL-7Rα, c-Kit, Thy1.2, and Sca-1, but were poor producers of IL-17A or IFNγ (Figure 3e). Furthermore, the majority of these cells did not express the surface marker NKp46 (4.2% NKp46+ vs. 95.8% NKp46−) confirming them as LTi cells. As T-bet participates in the differentiation of CCR6− LTi cells to IL-22-producing NKp46+ ILCs during mucosal immune responses,15, 35 we sought to determine whether LTi cells and NKp46+ ILCs expressed T-bet during DSS-induced injury. We found that both CD4− and CD4+ LTi cells were CCR6+T-bet− whereas NKp46+ ILC3 cells were CCR6−T-bet+ (Figure 3e). Remarkably, we found that the frequency of IL-22-producing CD4− LTi cells was higher than the frequency of IL-22-producing CD4+ LTi cells (77.2% vs. 22.8%, respectively) (Figure 3e), suggesting that CD4− LTi cells represent a predominant source of IL-22 during epithelial injury.
To further delineate the contribution of CD4− and CD4+ LTi cells to protection against intestinal injury, Rag1−/− mice were treated with anti-CD4 or anti-Thy1 antibodies to deplete CD4+ LTi cells or both CD4− and CD4+ ILCs, respectively. Depletion of Thy1+ ILCs in DSS-treated Rag1−/− mice induced severe pathology and dramatically reduced colonic IL-22 expression (Figure 3f–i). In contrast, anti-CD4 treatment only partially reduced colonic IL-22 expression, yet it had no effect on mortality, body weight loss, or colon shortening in DSS-treated Rag1−/− mice (Figure 3f–i), suggesting that although CD4+ LTi cells may contribute to LTβR-mediated IL-22 production during intestinal injury, they are dispensable for protection. Collectively, these results suggest that LTβR signaling promotes epithelial wound healing through the induction of IL-22 production by CD4− LTi cells.
LTβR signaling in epithelial cells protects against intestinal epithelial injury by promoting IL-23-driven IL-22-dependent tissue protective responses
To identify which LTβR-expressing cells are essential for controlling IL-22 production during epithelial injury, we generated reciprocal bone marrow chimeric mice. Following DSS treatment, we observed reduced survival, increased body weight loss, and reduced colonic IL-22 expression in wild type (WT)→LtbrΔ/Δ and to a lesser extent in LtbrΔ/Δ→WT chimeras with respect to WT→WT chimeras (Figure 4), indicating that LTβR expression on both radio-resistant cells and bone marrow-derived cells contributes to protection against epithelial injury. Additionally, mice lacking LTβR in CD11c+ cells (CD11c-Ltbr−/−) displayed increased body weight loss, reduced survival, and reduced colonic IL-22 levels (Supplementary Figure 3), indicating a role for LTβR signaling in dendritic cells (DCs) in protection against epithelial injury, in line with previous studies in an infectious colitis model.10

Expression of lymphotoxin beta receptor (LTβR) on both radio-resistant and hematopoietic cells is required for control of intestinal inflammation. Bone marrow cells from C57BL/6 (wild type (WT)) or LtbrΔ/Δ mice were transferred into lethally irradiated WT or LtbrΔ/Δ mice. Six weeks later, WT→WT (n=16), LtbrΔ/Δ→LtbrΔ/Δ (n=9), WT→LtbrΔ/Δ (n=12) and LtbrΔ/Δ→WT (n=9) chimeric mice were treated for 5 days with 5% dextran sulfate sodium (DSS). (a–d) Weight loss (a), survival (b), clinical disease score (c), and length and photographs of colons at day 10 (d). (e) Hematoxylin and eosin staining and histological score of colon sections from chimeric mice at day 10. (f) Il22 mRNA expression in the colon of chimeric mice (n=5) at day 10. Data are normalized to Hprt. *P<0.05; **P<0.01; ***P<0.001 (Student’s t-test). Data are representative (e, f) or a combination (a–d) of two independent experiments. Error bars represent s.e.m.
Since WT→LtbrΔ/Δ chimeras displayed increased morbidity and colon pathology compared with WT→WT chimeras (Figure 4), we hypothesized that LTβR signaling in intestinal epithelial cells maybe important for IL-22-mediated wound healing after epithelial injury. To test this, we generated mice with specific inactivation of LTβR in epithelial cells (Vil-Ltbr−/− mice). Compared with Ltbrfl/fl mice, DSS-treated Vil-Ltbr−/− mice displayed increased body weight loss, reduced survival, reduced colon length, and severe colon inflammation (Figure 5a–d) indicating that LTβR signaling in epithelial cells is essential for protection against epithelial injury. Comparable numbers of CD4+ and CD8+ T cells, CD11c+ cells, neutrophils and RORγt+ ILCs were found in the colons of Ltbrfl/fl and Vil-Ltbr−/− mice (Supplementary Figure S4), suggesting that the migration or expansion of these cells to the LP during DSS-induced injury is not impaired in the absence of LTβR signaling in epithelial cells. However, epithelial cell proliferation and the expression of the antiapoptotic factors Bcl2 and Bclxl were significantly reduced in the colons of Vil-Ltbr−/− mice (Figure 5e, f). Accordingly, Il22, Muc2, Muc4, and Muc13 expression was reduced in the colons of Vil-Ltbr−/− mice (Figure 5g, h). These results indicate that LTβR signaling in epithelial cells is critical for the induction of IL-22-dependent tissue protective responses.

Lymphotoxin beta receptor (LTβR) signaling in epithelial cells inhibits intestinal epithelial injury by promoting interleukin-23 (IL-23)-driven IL-22-dependent tissue protective responses. Ltbrfl/fl and Vil-Ltbr−/− mice (n=5) were treated for 5 days with 3.5% dextran sulfate sodium (DSS). (a) Weight loss and (b) survival. (c) Colon length, and (d) hematoxylin and eosin staining and histological score of colon sections at day 11. (e) Intestinal epithelial cell proliferation was analyzed after bromodeoxyuridine (BrdU) pulse injection on day 8. Ten crypts from the colons of five mice per genotype were analyzed. (f–j) Expression of the indicated genes (f–i) and IL-23 production in culture supernatants of colons (j) was evaluated at day 5. (k) Ltbrfl/fl and Vil-Ltbr−/− mice (n=3–4) were treated for 5 days with 5% DSS. 10 μg of plasmid encoding empty vector (pRK) or IL-23-expressing plasmid (IL-23Fc) was injected intravenously at day 1. Colonic expression of Il22 mRNA was evaluated at day 5. For mRNA expression, data are normalized to Hprt. *P<0.05; **P<0.01; ***P<0.001 (Student’s t-test). Data are representative of two (e, j, k) or three (a–d, f–i) independent experiments. Error bars represent s.e.m.
IL-23 is the principal cytokine required for the induction of IL-22 in the gut.21, 22 Based on our findings that LTβR signaling in epithelial cells is essential for colonic IL-22 production and that Il23a mRNA expression was significantly reduced in the colons of LtbrΔ/Δ mice (Figure 5g and Supplementary Figure S1d), we hypothesized that LTβR expression in epithelial cells may be necessary for IL-23 production. We found that with respect to Ltbrfl/fl mice, Il23a mRNA expression and IL-23 protein production were significantly reduced in DSS-treated Vil-Ltbr−/− mice (Figure 5i, j), suggesting that LTβR signaling in epithelial cells is indeed required for IL-23 production during DSS-induced injury. Furthermore, hydrodynamic injection of a construct encoding both p19 and p40 IL-23 subunits, restored Il22 mRNA expression in Vil-Ltbr−/− mice (Figure 5k), providing additional evidence that LTβR expression by epithelial cells regulates the IL-23-IL-22 axis.
Epithelial cells can produce IL-23 in response to mucosal damage in a LTβR-dependent manner
To determine which cells produce IL-23 during DSS-induced epithelial injury, we generated bone marrow chimeric mice using WT and Il23a−/− mice36 and treated them for 5 days with DSS. Unexpectedly, we found that colonic Il23a mRNA expression was comparable in WT→WT and Il23a→WT mice whereas it was greatly reduced in WT→Il23a−/− mice (Figure 6a). These results suggest that Il23a is mainly produced by radio-resistant cells in WT mice during DSS-induced colitis. We also observed that WT→Il23a−/− mice displayed increased pathology with significant reduction of colon length and increased disease score after DSS treatment, compared with WT→WT and Il23a→WT chimeras (Figure 6b), suggesting that Il23a expression by radio-resistant cells contributes to protection against intestinal inflammation.

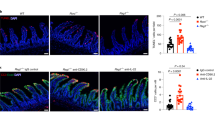
Epithelial cells produce interleukin-23 (IL-23) in response to mucosal damage in a lymphotoxin beta receptor (LTβR)-dependent manner. (a, b) Bone marrow cells from C57BL/6 (wild type (WT)) or Il23a−/− mice were transferred into lethally irradiated WT or Il23a−/− mice. Six weeks later, WT→WT, Il23a−/−→WT, and WT→Il23a−/− chimeric mice were treated for 5 days with 5% dextran sulfate sodium (DSS). (a) Il23a mRNA expression in the colon of chimeric mice at day 5. (b) Colon length and clinical disease score at day 5. (c, d) Mice were treated for 5 days with 3.5% DSS or left untreated. LTβR–Ig fusion protein or control IgG was administered intraperitoneally (i.p.) at days 0 and 3 (150 μg per mouse). (c) IL-23-driven green fluorescent protein (GFP) expression was evaluated at day 5 in the colon of IL-23-GFP+/− mice by GFP staining (green). Nuclei were stained with Hoechst (blue). (d) Il23a and Il22 expression was evaluated in the colon of C57BL/6 mice at day 5. (e) C57BL/6 mice were treated for 5 days with 3.5% DSS. Agonistic αLTβR or control antibody was injected i.p. at days 0 and 3 (100 μg per mouse). Colonic Il23a and Il22 mRNA expression was evaluated at day 5. (f) CMT-93 epithelial cells were stimulated with agonistic αLTβR antibody (5 μg ml−1) or lipopolysaccharide (LPS) (1 μg ml−1). Il23a mRNA expression and IL-23 protein production in culture supernatant was measured. (g) Il23a mRNA expression in sorted EpCAM+CD45− colon epithelial cells from wild-type (WT) mice, stimulated in vitro with agonistic αLTβR antibody (5 μg ml−1) or LPS (1 μg ml−1) for 12 h. (h) Il23a mRNA expression in sorted EpCAM+CD45− colon epithelial cells at day 5 after DSS treatment. For mRNA expression, data are normalized to Hprt. *P<0.05; **P<0.01; ***P<0.001 (Student’s t-test (a, b, d, e, f left panel, g, h) or two-tailed Mann–Whitney test (f right panel)). Data are representative of two independent experiments with 3–7 mice per group. Error bars represent s.e.m.
To further define the cell types responsible for IL-23 production following epithelial injury, we induced colitis in IL-23-GFP+/− reporter mice.36 Surprisingly, we found that IL-23-driven GFP expression was upregulated in epithelial cells, as well as DCs and macrophages (Figure 6c and Supplementary Figure 5) at day 5 post DSS treatment, indicating that epithelial cells are capable of producing IL-23 during epithelial injury. Furthermore, inhibition of LTβR signaling using an LTβR–Ig fusion protein reduced both IL-23-driven GFP and colonic Il23a and Il22 mRNA expression (Figure 6c, d), suggesting that LTβR signaling promotes IL-23 production by epithelial cells during mucosal damage. In contrast to LTβR–Ig treatment, activation of the LTβR pathway using agonistic αLTβR antibody was able to upregulate colonic Il23a and Il22 expression during epithelial injury (Figure 6e).
To confirm the ability of LTβR signaling to directly induce IL-23 production by epithelial cells, we stimulated both transformed colonic epithelial cells (CMT-93 cells) as well as primary intestinal epithelial cells with agonistic αLTβR antibody. Agonistic αLTβR antibody treatment significantly induced Il23a mRNA expression and IL-23 protein production (Figure 6f, g), suggesting that direct activation of LTβR on epithelial cells can induce IL-23. Furthermore, IL-23 production was also induced after stimulation with lipopolysaccharide (Figure 6f, g), indicating that epithelial cells can produce IL-23 in response to toll like receptor 4 agonists. Consistently, Il23a expression was reduced in purified colon epithelial cells of Vil-Ltbr−/− mice during DSS-induced injury (Figure 6h). Together, these data indicate that LTβR signaling in intestinal epithelial cells promotes IL-23 production during mucosal damage.
Given that IL-23 is composed of p19 (Il23a) and p40 (Il12b) subunits, we then investigated the ability of epithelial cells to induce IL12b expression during DSS-induced injury.
IL12b expression was upregulated in both purified colonic epithelial cell fraction and sorted colonic epithelial cells (Supplementary Figure 6a, b). To further define the role of Il12b production by radio-resistant cells in mucosal protection, we generated reciprocal bone marrow chimeric mice. Although both radio-resistant and bone marrow-derived cells contributed to Il12b expression during DSS-induced injury, mice lacking Il12b in radio-resistant cells displayed increased disease score, reduced colon length, and reduced colonic IL-22 production during DSS-induced injury (Supplementary Figure 6c–f). These results indicate that Il12b can be expressed by epithelial cells and participate in epithelial repair after injury. Taken together, these data suggest that LTβR signaling in intestinal epithelial cells is critical for IL-23 production for intestinal epithelial cell repair and intestinal homeostasis after injury.
Discussion
Interactions between epithelial cells and ILCs during epithelial damage and how these cells contribute to disease pathogenesis remains unclear. Our results suggest that during epithelial injury, activation of LTβR triggers IL-23 production by intestinal epithelial cells. IL-23 in turn promotes IL-22 production by CD4–CCR6+T-bet– LTi cells. The IL-22 then induces intestinal epithelial cell proliferation and survival, and the production of mucus thereby promoting mucosal wound healing. These results provide evidence that support a novel mechanism of protection against intestinal injury in which the LTβR-dependent production of IL-23 by epithelial cells regulates mucosal wound healing.
IL-23 is critical for IL-23R-mediated production of IL-22 by ILCs.21, 22 A dichotomy between beneficial and pathogenic roles for IL-23R-responsive innate and adaptive cell populations has been described in the DSS-induced colitis model.37, 38 DCs and macrophages are thought to be the major IL-23 producers during infection, inflammation, and cancer progression in the colon, although epithelial cells can also contribute to IL-23 production.39, 40, 41, 42 Accordingly, we have previously demonstrated that LTβR-mediated production of IL-23 by DCs promotes IL-22 production by RORγt+ ILCs during mucosal bacterial infection.10 Additionally, our results using CD11c-Ltbr−/− mice suggest that LTβR signaling in DCs contributes to protection against DSS-induced injury. Unexpectedly, in the current study, we find that IL-23 and IL-22 production during epithelial injury are impaired in the absence of LTβR signaling in intestinal epithelial cells. We demonstrate that activation of LTβR signaling can promote IL-23 production by epithelial cells, identifying a previously unrecognized mechanism of LTβR-dependent regulation of intestinal wound healing. Our results indicate that both p19 and p40 subunits of IL-23 can be expressed by epithelial cells during DSS-induced injury. These results suggest that following epithelial damage, epithelial cells represent an important source of IL-23 for mucosal repair.
LTβR signaling is known as a key regulator of lymphoid organ development and maintenance; however, its function outside of lymphoid organs is still elusive. In the present study, we reveal that LTβR signaling in intestinal epithelial cells is essential for IL-22 production by RORγt+ ILC3s. Although mice with specific inactivation of LTβR in epithelial cells (Vil-Ltbr−/− mice) were highly sensitive to DSS-induced injury, LTβR signaling in other cells could also contribute to protection. Indeed, a protective role for LTβR activation in DCs and macrophages was recently described in infectious and chemically induced colitis models.4, 9, 10
Recent studies have implicated the role of the gut microbiota in the pathogenesis of intestinal inflammation.2, 43 LTβR signaling, the IL-22 pathway and RORγt+ ILC3s have been identified as regulators of commensal microbiota composition.31, 44, 45 The comparable colonic expression of antimicrobial peptides and bacterial dissemination observed between Ltbrfl/fl and LtbrΔ/Δ mice suggested that the sensitivity of LtbrΔ/Δ mice to DSS-induced colitis was not primarily mediated by the dissemination of commensal bacteria. Instead, we found that, during epithelial injury, LTβR signaling regulates epithelial cell homeostasis via the IL-22-dependent promotion of mucins expression and proliferation/survival of intestinal epithelial cells.
While RORγt+ ILC3s have been shown to be essential for protection against intestinal inflammation,4, 19, 43 accumulating evidence suggests that, in response to IL-23, the production of IL-22 by distinct RORγt+ ILC3 subsets may differentially impact the outcome of intestinal inflammation. Indeed, exuberant IL-22 responses together with IL-17 and IFNγ production by colitogenic NKp46– ILC3s has been shown to promote intestinal immunopathology.16, 26 These observations were corroborated by the presence of IL-23-responsive ILCs in the inflamed tissue of inflammatory bowel disease patients.20 In contrast, IL-22 production by NKp46+ ILC3s and LTi cells has been shown to inhibit intestinal inflammation in the C. rodentium-induced colitis model.10, 21 Our results show that CCR6+Tbet– CD4– and CD4+ LTi cells but not NKp46+ ILC3s, colitogenic NKp46– ILC3s or T cells were the primary source of IL-22 during DSS-induced injury. NKp46+ ILC3s represented less than 5% of IL-22-producing ILC3s, and IL-22 production by those cells was LTβR independent, in agreement with previous results.46 A recent study suggested a critical role for CD4+ LTi cells in IL-22-mediated protection against infectious colitis,47 whereas the function of CD4− LTi cells in mucosal homeostasis remained obscure. Unexpectedly, we found that depletion of CD4+ LTi cells in Rag1−/− mice only partially reduced colonic IL-22 levels but had no effect on colon immunopathology and mice survival, indicating that CD4+ LTi cells are not critical for protection against DSS-induced intestinal injury. This observation might be explained by the fact that the vast majority (approximately 75%) of IL-22-producing LTi cells did not express CD4. Therefore, our results suggest that CD4– LTi cells are the main producers of IL-22 during epithelial injury. Since the developmental plasticity between RORγt+ ILC subsets remains controversial,15, 17 it is important to determine the role of LT in the regulation, and function of CD4– and CD4+ LTi subsets in future studies.
Increased LT expression has previously been found in the mucosa of inflammatory bowel disease patients.48 Additionally, chronic overexpression of LT, as a transgene, has been connected with the formation of tertiary lymphoid tissue, chronic inflammation, immunopathology, and tumor progression.49 Therefore, future studies will be required to better define whether LT expression by distinct ILC populations correlates with disease severity in human inflammatory bowel disease patients. In summary, our findings suggest that the LTβR-dependent IL-23 production by epithelial cells limits mucosal damage via induction of IL-22 by CD4– LTi cells and that the manipulation of LTβR signaling may represent a novel therapeutic avenue in inflammatory diseases of the intestine.
Methods
Mice. C57BL/6 and Rag1−/− were originally obtained from The Jackson Laboratory (Bar Harbor ME) and bred at Trudeau Institute. Neo-free LtbrΔ/Δ and Ltbrfl/fl mice were generated from LTβR tri-loxP-floxed mice4 by crossing with MeuCre40 mice,28 all on C57BL/6 background. Vil-Ltbr−/− mice were generated by crossing neo-free Ltbrfl/fl mice with Vil-Cre mice. RORγt–GFP+/−, IL-23-GFP+/−, and Il12b−/− mice were described previously.18, 36, 50 All mice used in this study were on C57BL/6 background and maintained under specific pathogen-free conditions. When possible, age/sex-matched controls or littermate controls were co-housed with experimental mice. When this was not possible, bedding exchange was performed for 2 weeks before initiation of the experiment. All animal studies were performed in accordance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the Trudeau Institute.
Generation of bone marrow chimeras. Recipient mice were irradiated twice for a combined dose of 950 rad and reconstituted with 4 × 106 total bone marrow cells. Recipient mice were kept on antibiotic-containing food for 4 weeks after the bone marrow transfer and used for experiments 6–8 weeks after the transfer.
DSS-induced colitis and study design. Mice were given 3.5–5% (w/v) DSS (molecular weight=36,000–50,000 Da; MP Biomedicals, Solon, OH) in their drinking water for 5 days replaced then by regular water for an additional 3–6 days. For Thy1.2- and CD4-depletion experiments, mice were intraperitoneally injected with 150 μg of rat αCD4 (GK1.5), αThy1.2 (30H12), or IgG2b isotype control at days 0 and 3. To inhibit LTβR signaling in WT mice and IL-23-GFP+/− mice, 150 μg of mouse LTβR–Ig fusion protein or control IgG antibody (Biogen Idec, Cambridge, MA) was intraperitoneally injected at days 0 and 3. To activate LTβR signaling, 100 μg of agonistic αLTβR antibody (ACH6, Biogen Idec) or control antibody (MOPC) was intraperitoneally injected at days 3 and 5.
Assessment of DSS-induced colitis. Mice were weighed daily. The disease score was determined as an average of body weight loss (0 points, no weight loss; 1 point, weight loss of 1–5%; 2 points, weight loss of 5–10%; 3 points, weight loss of 10–20%; 4 points, weight loss >20%), signs of rectal bleeding (0 points, no blood in feces; 2 points, presence of blood in feces; 4 points, gross bleeding) and stool consistency (0 points, well-formed pellet; 1 point, soft pellet; 2 points, loose stool; 4 points, diarrhea). The scores were added to obtain a disease score ranging from 0 (healthy) to 12 (maximal activity of the disease). Post mortem, colons were removed and their length was measured from rectum to cecum. Swiss rolls of the colon were fixed in 10% buffered formalin, and 4–5 μm paraffin-embedded sections were stained with hematoxylin and eosin. The severity of colitis was blindly determined as an average of colonic epithelial damage (0 points, normal epithelium; 1 point, hyperproliferation, irregular crypts, and goblet cell loss; 2 points, mild-to-moderate crypt loss (10–50%); 3 points, severe crypt loss (50–90%); 4 points complete crypt loss with the surface of the epithelium intact; 5 points, small-to-medium size ulcer (<10 crypts width); 6 points large ulcers (>10 crypts width)), and the degree of inflammatory cell infiltration in the mucosa (0 points, normal infiltration; 2 points, modest infiltration; 3 points, severe infiltration), the submucosa (0 points, normal infiltration; 1 point, modest infiltration; 2 points, severe infiltration) and the muscularis mucosae (0 points, normal infiltration; 1 point, moderate-to-severe infiltration). Scores for epithelial damage and inflammatory cell infiltration were added, resulting in a total scoring range of 0–12.
Hydrodynamic IL-22 and IL-23 plasmid injection. 10 μg of IL-22-expressing plasmid (pRK-mIL-22, Genentech, San Francisco, CA), IL-23 p19-p40-expressing plasmid (IL-23Fc)44 or control plasmid (pRK) in 1.7 ml of TransIT-EE Hydrodynamic Delivery Solution (Mirus Bio, Madison, WI) were intravenously injected in the tail vein one day after start of DSS treatment.
RNA isolation, reverse transcription and real-time PCR. Total RNA from cells or frozen tissues was isolated using the RNeasy Mini Kits (Qiagen, Hilden, Germany). cDNA was synthesized using MLV Reverse Transcriptase (Promega, Madison, WI) and random primers (Promega). Quantitative real-time PCR reactions were performed using Power Sybr Green PCR master mix and the 7500 cycler (Applied Biosystems, Foster City, CA). Relative mRNA expression of the target gene was determined using the comparative 2−ΔΔCt method.
Flow cytometry. Fluorochrome-conjugated antibodies against mouse CD45 (30-F11), CD3 (145-2C11), CD8 (53-6.7), B220 (RA3-6B2), CD11b (M1/70), CD11c (N418), Gr1 (RB6-8C5), TER119 (TER119), NKp46 (29A1.4), CD4 (RM4-5), Thy1.2 (53-2.1), SCA-1 (D7), CCR6 (29-2L17), c-Kit (2B8), IL-7Rα (A7R34), IL-22 (1H8PWSR), IL-17A (TC11-18H10.1), IFNγ (XMG1.2), T-bet (4B10), RORγt (2B2), EpCAM (G8.8) were purchased from eBioscience (San Diego, CA), BD Biosciences or Biolegend (San Diego, CA). For intracellular cytokine staining, cells were stimulated with IL-23 (50 ng ml−1) in the presence of brefeldin A (10 μg ml−1) for 4 h at 37 °C. Cell sorting and flow cytometry were performed using an Influx cell sorter and a LSRII flow cytometer (BD Biosciences), respectively. Data were analyzed using FlowJo vX software (TreeStar, Ashland, OR).
Determination of bacterial dissemination. Spleens and livers were aseptically removed and homogenized in sterile phosphate-buffered saline. Viable counts were determined by plating serial dilutions on sheep blood agar plates.
Enzyme-linked immunosorbent assay analysis. IL-22 and IL-23 concentrations were measured in culture supernatants or colon extracts using specific R&D Duoset (Minneapolis, MN) and eBioscience enzyme-linked immunosorbent assay kits, respectively, according to the manufacturer’s instructions.
Epithelial cell line CMT-93. Mouse colonic CMT-93 epithelial cells were obtained from American Type Culture Collection. CMT-93 cells were maintained in DMEM (Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum, penicillin and streptomycin. Cells were serum starved for 24 h before in vitro stimulation with lipopolysaccharide from Escherichia coli O111:B4 (Sigma-Aldrich, St Louis, MO) or agonistic αLTβR antibody (ACH6, Biogen Idec) for 6 h (gene expression) or 24 h (enzyme-linked immunosorbent assay).
Statistical analysis. Statistical analysis was performed using the two-tailed Student’s t-test or the Mann–Whitney test when appropriate in Prism V5 (GraphPad Software, La Jolla, CA). P-values <0.05 were considered significant.
References
Kaser, A., Zeissig, S. & Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 28, 573–621 (2010).
Hooper, L.V., Littman, D.R. & Macpherson, A.J. Interactions between the microbiota and the immune system. Science 336, 1268–1273 (2012).
Upadhyay, V. & Fu, Y.X. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat. Rev. Immunol. 13, 270–279 (2013).
Wang, Y. et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 32, 403–413 (2010).
Ota, N. et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat. Immunol. 12, 941–948 (2011).
Boulianne, B., Porfilio, E.A., Pikor, N. & Gommerman, J.L. Lymphotoxin-sensitive microenvironments in homeostasis and inflammation. Front. Immunol. 3, 243 (2012).
Mackay, F. et al. Both the lymphotoxin and tumor necrosis factor pathways are involved in experimental murine models of colitis. Gastroenterology 115, 1464–1475 (1998).
Jungbeck, M., Stopfer, P., Bataille, F., Nedospasov, S.A., Mannel, D.N. & Hehlgans, T. Blocking lymphotoxin beta receptor signalling exacerbates acute DSS-induced intestinal inflammation-Opposite functions for surface lymphotoxin expressed by T and B lymphocytes. Mol. Immunol. 45, 34–41 (2008).
Wimmer, N. et al. Lymphotoxin-beta receptor activation on macrophages ameliorates acute DSS-induced intestinal inflammation in a TRIM30alpha-dependent manner. Mol. Immunol. 51, 128–135 (2012).
Tumanov, A.V. et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 10, 44–53 (2011).
Krause, P., Zahner, S.P., Kim, G., Shaikh, R.B., Steinberg, M.W. & Kronenberg, M. The tumor necrosis factor family member TNFSF14 (LIGHT) is required for resolution of intestinal inflammation in mice. Gastroenterology 146, 1752–1762 e1754 (2014).
Spits, H. & Cupedo, T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 30, 647–675 (2012).
Walker, J.A., Barlow, J.L. & McKenzie, A.N. Innate lymphoid cells—how did we miss them? Nat. Rev. Immunol. 13, 75–87 (2013).
Sonnenberg, G.F., Fouser, L.A. & Artis, D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390 (2011).
Spits, H. et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Serafini, N. et al. Gata3 drives development of RORgammat+ group 3 innate lymphoid cells. J. Exp. Med. 211, 199–208 (2014).
Eberl, G., Marmon, S., Sunshine, M.J., Rennert, P.D., Choi, Y. & Littman, D.R. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73 (2004).
Sawa, S. et al. RORgammat(+) innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326 (2011).
Geremia, A. et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 208, 1127–1133 (2011).
Cella, M. et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457, 722–725 (2009).
Sanos, S.L. et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 10, 83–91 (2009).
Ouyang, W., Rutz, S., Crellin, N.K., Valdez, P.A. & Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 29, 71–109 (2011).
Leung, J.M. et al. IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol. 7, 124–133 (2013).
Huber, S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012).
Eken, A., Singh, A.K., Treuting, P.M. & Oukka, M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 7, 143–154 (2014).
Perse, M. & Cerar, A. Dextran sodium sulphate colitis mouse model: traps and tricks. J. Biomed. Biotechnol. 2012, 718617 (2012).
Leneuve, P. et al. Cre-mediated germline mosaicism: a new transgenic mouse for the selective removal of residual markers from tri-lox conditional alleles. Nucleic Acids Res. 31, e21 (2003).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Zenewicz, L.A., Yancopoulos, G.D., Valenzuela, D.M., Murphy, A.J., Stevens, S. & Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957 (2008).
Sonnenberg, G.F. et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336, 1321–1325 (2012).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
McGuckin, M.A., Linden, S.K., Sutton, P. & Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9, 265–278 (2011).
Van der Sluis, M. et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129 (2006).
Klose, C.S. et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494, 261–265 (2013).
Ghilardi, N., Kljavin, N., Chen, Q., Lucas, S., Gurney, A.L. & De Sauvage, F.J. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J. Immunol. 172, 2827–2833 (2004).
Cox, J.H. et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 5, 99–109 (2012).
Becker, C. et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 177, 2760–2764 (2006).
Vignali, D.A. & Kuchroo, V.K. IL-12 family cytokines: immunological playmakers. Nat. Immunol. 13, 722–728 (2012).
Piskin, G., Sylva-Steenland, R.M., Bos, J.D. & Teunissen, M.B. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J. Immunol. 176, 1908–1915 (2006).
Grivennikov, S.I. et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258 (2012).
Hino, A., Kweon, M.N., Fujihashi, K., McGhee, J.R. & Kiyono, H. Pathological role of large intestinal IL-12p40 for the induction of Th2-type allergic diarrhea. Am. J. Pathol. 164, 1327–1335 (2004).
Lochner, M. et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J. Exp. Med. 208, 125–134 (2011).
Upadhyay, V. et al. Lymphotoxin regulates commensal responses to enable diet-induced obesity. Nat. Immunol. 13, 947–953 (2012).
Kruglov, A.A. et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science 342, 1243–1246 (2013).
Satoh-Takayama, N., Lesjean-Pottier, S., Sawa, S., Vosshenrich, C.A., Eberl, G. & Di Santo, J.P. Lymphotoxin-beta receptor-independent development of intestinal IL-22-producing NKp46+ innate lymphoid cells. Eur. J. Immunol. 41, 780–786 (2011).
Sonnenberg, G.F., Monticelli, L.A., Elloso, M.M., Fouser, L.A. & Artis, D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011).
Agyekum, S. et al. Expression of lymphotoxin-beta (LT-beta) in chronic inflammatory conditions. J. Pathol. 199, 115–121 (2003).
Pitzalis, C., Jones, G.W., Bombardieri, M. & Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 14, 447–462 (2014).
Magram, J. et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity 4, 471–481 (1996).
Acknowledgements
This work was supported by the National Institutes of Health (CA141975 to Y.-X.F.), Crohn's and Colitis Foundation of America (Senior Research Award #294083) to A.V.T., and by Trudeau Institute. We are grateful to N. Ghilardi and W. Ouyang (Genentech, CA) for providing IL-23-GFP+/− mice and IL-22-expressing plasmid, M. Holzenberger (INSERM, France) for providing MeuCre40 mice. We thank J. Browning and M. Ols (Biogen Idec) for providing reagents.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary information
Rights and permissions
About this article

Cite this article
Macho-Fernandez, E., Koroleva, E., Spencer, C. et al. Lymphotoxin beta receptor signaling limits mucosal damage through driving IL-23 production by epithelial cells. Mucosal Immunol 8, 403–413 (2015). https://doi.org/10.1038/mi.2014.78
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.78
This article is cited by
-
Dimethyl itaconate ameliorates cognitive impairment induced by a high-fat diet via the gut-brain axis in mice
Microbiome (2023)
-
A20 undermines alternative NF-κB activity and expression of anti-apoptotic genes in Helicobacter pylori infection
Cellular and Molecular Life Sciences (2022)
-
Metabolic activation and colitis pathogenesis is prevented by lymphotoxin β receptor expression in neutrophils
Mucosal Immunology (2021)
-
Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair
Nature Communications (2020)
-
Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation
Nature Communications (2019)
