
Abstract
Invasive infection often begins with asymptomatic colonization of mucosal surfaces. A murine model of bacterial colonization with Streptococcus pneumoniae was used to study the mechanism for mucosal protection by immunoglobulin. In previously colonized immune mice, bacteria were rapidly sequestered within large aggregates in the nasal lumen. To further examine the role of bacterial agglutination in protection by specific antibodies, mice were passively immunized with immunoglobulin G (IgG) purified from antipneumococcal sera or pneumococcal type-specific monoclonal human IgA (hIgA1 or hIgA2). Systemically delivered IgG accessed the mucosal surface and blocked acquisition of colonization and transmission between littermates. Optimal protection by IgG was independent of Fc fragment and complement and, therefore, did not involve an opsonophagocytic mechanism. Enzymatic digestion or reduction of IgG before administration showed that protection required divalent binding that maintained its agglutinating effect. Divalent hIgA1 is cleaved by the pneumococcal member of a family of bacterial proteases that generate monovalent Fabα fragments. Thus, passive immunization with hIgA1 blocked colonization by an IgA1-protease-deficient mutant (agglutinated) but not the protease-producing wild-type parent (not agglutinated), whereas protease-resistant hIgA2 agglutinated and blocked colonization by both. Our findings highlight the importance of agglutinating antibodies in mucosal defense and reveal how successful pathogens evade this effect.
Similar content being viewed by others
INTRODUCTION
Colonization of mucosal surfaces is often the first step in the pathogenesis of disease for many microbial infections. Immunoglobulin has an important role in host defense at mucosal sites and is thought to act by preventing colonization of pathogens. Patients with hypogammaglobulinemia or agammaglobulinemia, for example, typically present in early childhood with recurrent respiratory tract infections, in particular with extracellular, encapsulated bacteria.1 Most mucosal antibodies that are actively transported into the lumen (IgA and IgM) are multivalent with 4 or 10–12 antigen-binding sites per molecule, respectively. Immunoglobulin G (IgG), with two binding sites per molecule, is not secreted by the same mechanism; however, its extravasation from the abundant plasma pool results in effective levels on mucosal surfaces.2 Evidence for the importance of plasma antibody in protection against disease is demonstrated by the effectiveness of systemic immunization against several mucosal pathogens, which correlates with increased specific antibody titers. Polysaccharides that comprise the capsules of common respiratory pathogens (Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis) induce high levels of IgG when conjugated to an immunogenic protein carrier. This approach has been very successful for vaccines licensed to protect against invasive disease.3 Moreover, high plasma titers to these parenterally administered conjugate vaccines correlate with decreased rates of colonization in immunized individuals – an effect attributed to reduced bacterial acquisition.4, 5 Lower rates of colonization in turn diminish the frequency of host-to-host transmission, amplifying the effectiveness of immunization by protecting the unvaccinated population (indirect protection or herd immunity). Extensive surveillance in the United States has revealed that much of the public health benefit of the pneumococcal conjugate vaccine is because of protection of unvaccinated adults and the decreased incidence of pneumonia in this population.6, 7
Several distinct functions of immunoglobulin in supporting host defense against pathogens have been described.8 (1) Antibodies may act as opsonins, leading to enhanced phagocytic recognition or promoting the deposition of complement and subsequent lysis. (2) Antibodies may tag infected cells for destruction through antibody-dependent cell-mediated cytotoxicity. (3) Antibodies may neutralize a pathogen by binding to its surface and inhibiting its growth. (4) Antibodies may coat a pathogen and prevent its adherence to host cells and tissues. (5) Antibodies, because of their di- or multivalent binding properties, may agglutinate their target into larger clusters allowing for more effective recognition and mechanical clearance by the host. However, the specific mechanism(s) by which mucosal antibodies, including systemically generated antibodies derived from plasma, ameliorate colonization by pathogens is poorly understood.
In this study, we used a murine model of S. pneumoniae colonization of the upper respiratory tract to characterize how immunoglobulin affects mucosal colonization. These experiments using passive immunization revealed that multivalency of antibodies was required to protect against colonization in vivo, an outcome that was dependent on their ability to agglutinate the infecting bacteria at the mucosal surface.
RESULTS
Serotype-specific IgG blocks the acquisition of bacterial colonization
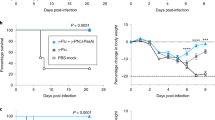
To examine the role of systemic antibody in mucosal protection, adult mice were passively immunized intraperitoneally (IP) with high-titer antipneumococcal rabbit sera and challenged intranasally (IN) with S. pneumoniae. Protection from the acquisition of colonization was determined by quantitative culture of upper respiratory tract lavages 20 h following bacterial challenge. An inoculum of 104 colony-forming unit (CFU) was chosen because this dose was previously established as the 50% colonizing dose for this strain in experimental human carriage studies in healthy adults.9 Mice passively immunized 4 h before pneumococcal challenge with antisera raised to an isolate of the same capsular polysaccharide serotype as the challenge strain (TS, type-specific) were significantly protected from colonization (Figure 1a). Protection against a low-inoculum challenge was dose-dependent, requiring at least 25 μg of specific antibody per animal for significant protection, an amount used in subsequent experiments (Figure 1a). This mucosal protection detected using viable counts was confirmed using an independent non-culture based method—quantitative real-time PCR (qPCR) of nasal lavages amplifying pneumococcal gyrA DNA (Figure 1b). The magnitude of protective effect of TS antiserum was diminished with a higher dose (106 CFU) bacterial challenge (Figure 1a). The timing of antibody exposure was important as the protective effect of TS antiserum was no longer significant when mice were passively immunized 24 h after pneumococcal challenge, when stable colonization on the epithelial surface had already been established.10 These results demonstrate that specific antibody is most effective in blocking primary acquisition.

Protection against mucosal colonization by systemic antibody. (a) Mice were passively immunized (intraperitoneally, IP) with rabbit antisera raised against S. pneumoniae either 4 h before (Ab→Sp) or 24 h after (Sp→Ab) pneumococcal challenge with a type 23F isolate. Increasing doses of serotype 23F capsular polysaccharide-specific antibody were tested for effects on colonization, which was determined by quantitative culture of upper respiratory tract lavages. Mice were challenged intranasally (IN) with either a low dose (104) or high dose (106) of S. pneumoniae. Colony-forming unit (CFU) per animal was determined 24 h post immunization with type-specific antisera (closed symbols) or control (open symbols). n⩾3 mice per group. ns, not significant. *P<0.05, **P<0.01. (b) Bacterial load was confirmed in the nasal lavages of mice passively immunized 4 h before challenge with low dose (104) S. pneumoniae using quantitative real-time PCR (qPCR). Genomic equivalence per animal±s.e.m. was determined based on a standard curve of DNA of known bacterial quantity. n⩾6 nasal lavage samples per group. **P<0.01.
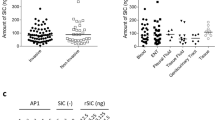
To confirm that this model was broadly applicable, we tested protection by prior IP administration of antisera against low-dose challenge with isolates of three additional common pneumococcal serotypes (Figure 2). In each case, protection was serotype-dependent, as protection was observed with TS antisera but not antisera generated against an isolate of another capsular polysaccharide serotype (HS, heterologous-type).

Type-specific protection against multiple serotypes. Mice were passively immunized with type-specific (TS) or an equivalent amount of antisera generated to a heterologous serotype (HS) and challenged with 104 pneumococci of the serotype indicated below 4 h later. Colony-forming unit (CFU) per animal was determined 24 h post immunization. n⩾6 mice per group. *P<0.05, **P<0.01. The in vitro agglutinating effect of TS and HS antipneumococcal rabbit antiserum was tested for each challenge strain.
Following passive immunization and at the time of bacterial challenge, rabbit IgG was detected on the mucosal surface of the upper respiratory tract (Figure 3a). Passively administered purified serum TS IgG was sufficient for protection against mucosal colonization in adult mice (Figure 3b). The effectiveness of TS IgG in blocking the acquisition of colonization was also demonstrated using infant mice (Figure 3c).

IgG-mediated protection against colonization. (a) Mice were passively immunized with antipneumococcal rabbit antisera (+) or phosphate-buffered saline (PBS) (−) and killed 4 h later to detect systemic (serum) and mucosal (nasal lavage) rabbit IgG. Rabbit IgG was detected by western analysis with anti-rabbit IgG (raised against whole molecule) using non-reducing conditions. An aliquot of antiserum was used as a positive control (type-specific, TS). Size marker in kilodaltons. (b) Adult mice were passively immunized with equal amounts of heterologous serotype (HS) or TS antisera or an equivalent amount of IgG purified from TS antisera and challenged with 104 pneumococci (type 6A) 4 h later. Colony-forming unit (CFU) per animal was determined 24 h post immunization. n⩾8 mice per group. (c) Two-week-old infant mice were passively immunized with IgG purified from TS or PBS control and 4 h later challenged intranasally (IN) with 103 pneumococci (type 2). CFU per animal was determined 24 h post immunization. The in vitro agglutinating effect of each serum tested was compared. n⩾5 mice per group. ns, not significant. *P<0.05, **P<0.01.
In addition to its role in preventing primary colonization, we investigated the role of antibody in blocking pneumococcal transmission from an infected host. We used a previously described infant mouse model of colonization in the setting of influenza A co-infection.11 At the age of 4 days, one in four mice within a litter (index mice) were colonized with S. pneumoniae, followed by influenza A infection of all mice in the litter at the age of 8 days (Figure 4). The acquisition of pneumococci by day 14 in uncolonized littermates (contact mice) through intralitter transmission was reduced from 5/11 (45%) in unimmunized contact pups to 1/10 (10%) in TS antisera immunized contact pups.

Protection against bacterial transmission. (a) Schematic of inoculation and immunization schedule. S. pneumoniae (Sp) (type 23F; index mice only) and influenza virus (Flu; index and contact mice) were administered intranasally to infant mice. In the ‘immunized’ group, the contact mice were passively immunized intraperitoneally (IP) with type-specific (TS) antisera. (b) Infant mice were infected and sacrificed as described above. Colony-forming unit (CFU) per animal in nasal lavage fluid are shown. Open circles designate unimmunized group, and filled represent TS immunized group. n⩾ 10 mice per group. ***P<0.001. Dotted line indicates limit of detection (10 CFU).
Thus, our model using passive immunization recapitulated many of the clinical observations that suggest that systemic antibody protects against mucosal pneumococcal infection of adults and infants. These included the requirement for TS antibody and the importance of diffusion of IgG from the plasma pool to the mucosal surface in blocking the acquisition of carriage and transmission.
The agglutinating effect of antibody enhances mucosal protection
To determine the mechanism of systemic IgG-mediated mucosal protection, we considered factors required for opsonophagocytosis of pneumococci, as this is the primary mechanism of its clearance from the bloodstream.12 Phagocytes recognize the Fc portion of bound antibody and their recognition is enhanced by the deposition and activation of complement through binding of products derived from complement component 3 (C3). However, TS antiserum was still protective against colonization in complement-deficient (C3−/−) mice (Figure 5a). In addition, we confirmed that TS rabbit antisera promotes the deposition of mouse C3 on S. pneumoniae (Figure 5b), although this process is not required for antibody-dependent protection against colonization. Furthermore, mice passively immunized with TS IgG that had been cleaved to completely remove its Fc domain were also protected, excluding a role for direct Fc binding in the mechanism of antibody-mediated protection (Figure 5c). Together, these findings suggested that the protective effect of mucosal IgG does not require opsonization. The Fc independence of protection also suggested that antibody-dependent cell-mediated cytotoxity did not contribute to protection in this model.

Complement, and Fc-fragment, -independent mucosal protection. (a) C3−/− mice were immunized with heterologous serotype (HS) or type-specific (TS) antipneumococcal rabbit antisera and challenged with 104 pneumococci (type 23F) 4 h later. Colony-forming unit (CFU) per animal was determined 24 h post immunization. n⩾ 7 mice per group. (b) Serotype 14 or 23F pneumococci were incubated with fresh mouse sera as a complement source and TS antipneumococcal rabbit antisera (black bars) or buffer control (white bars). Deposition of murine C3 was detected using fluorescein isothiocyanate-conjugated anti-mouse C3 by flow cytometry to quantify the percent of events above background±s.e.m. (c) Mice were immunized with HS, TS, or an equivalent amount of TS antipneumococcal rabbit antisera treated with pepsin under conditions that completely digested the Fc fragment and challenged with 104 pneumococci (type 14) 4 h later. CFU per animal was determined 24 h post immunization. n⩾ 7 mice per group. **P<0.01.
Because of the low inoculum required for protection from passively administered antibody, we were unable to detect bacteria in the upper respiratory tract in tissue sections using this model. However, we visualized the effect of antibody during the early events in colonization following a larger inoculum (107 CFU) by comparing previously colonized (immune) and sham-colonized (naive) mice using immunofluorescence on nasal tissue sections obtained during the initial 24-h post challenge. Immune mice expressed higher antibody levels at the nasal mucosa (IgA) and in serum (IgG) (Figure 6a) and have previously been shown to be less susceptible to secondary high-dose IN challenge in an antibody-dependent manner.13 As early as 30 min post challenge, S. pneumoniae was found in the lumen predominantly in large aggregates in immune mice, whereas in naive mice the bacteria in these spaces were mostly single or in pairs (Figure 6b). Consistent with results in naive immunocompetent animals, large aggregates of pneumococci were not seen in previously colonized μMT mice (Figure 6b Insert), which do not generate specific antibody and remain susceptible to secondary challenge. The observation that the presence of local and systemic antibody was associated with agglutination in vivo led us to investigate whether the agglutinating effect of antibody was responsible for mucosal protection.

Agglutination of S. pneumoniae in mice with mucosal immunity. Wild-type (WT) C57Bl6/J or μMT mice were serially immunized with three doses of colonizing pneumococci (immune) or phosphate-buffered saline (PBS; naive) and rechallenged intranasally (IN) with 107 Colony-forming unit (CFU) of the same isolate (type 4) 2 weeks after final immunization. Mice were killed at 30 min post challenge to visualize early events during colonization. (a) Pneumococcal-specific IgA in lavage fluid and IgG in serum from naive (white bars) and immune (black bars) mice was quantified using enzyme-linked immunosorbent assay (ELISA) and expressed as geometric mean titer (GMT)±s.e.m. n=4 for WT mice, and a representative sample for μMT mice. *P<0.05. (b) Immunofluorescent images of nasal tissue sections from naive and immune WT mice 30 min post challenge stained with anticapsular antibodies for pneumococci (red) and host cells with 4′,6-diamidino-2-phenylindole (DAPI; blue). Tissue autofluorescence was also detected (green). Representative images from mice in triplicate. Images taken at × 400 magnification, with 10 μm scale bars. Insert: immunized μMT mice at 24 h post pneumococcal challenge at × 400 magnification, with 10 μm scale bar.
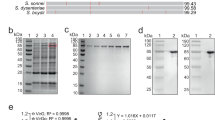
Each of the TS antiserum tested agglutinated the corresponding protected challenge strain, whereas the heterologous-type antiserum did not protect or agglutinate this strain (Figures 2 and 3b). Importantly, agglutination did not affect our ability to accurately assess colonization density on the mucosal surface as qPCR data correlated with CFU counts (Figure 1b). To further examine the contribution of agglutination, TS IgG was cleaved into Fab (monovalent) and F(ab′)2 (divalent) fragments with papain or pepsin, respectively (Figure 7a). Pneumococci incubated with divalent F(ab′)2, but not monovalent Fab, were heavily agglutinated in vitro (Figure 7b), although both bound to homologous capsular polysaccharide with similar titers using an enzyme-linked immunosorbent assay detecting rabbit light chains (Figure 7c). Mice passively immunized with these fragments showed a difference in protection, with the agglutinating F(ab′)2 significantly more effective than equivalent amounts of non-agglutinating Fab (Figure 7d). Only two groups, Fab versus F(ab′)2, were compared in this experiment as whole IgG bound approximately threefold more efficiently to bacteria than fragments (Figure 7c). This difference in protection was not due to any differences in the ability of fragments to access the nasal mucosa as both were detected in nasal lavages (Figure 7e).

Agglutination enhances protection by antibody. Type-specific (TS) IgG was cleaved into either F(ab′)2 or Fab fragments by pepsin or papain, respectively. (a) IgG, Fab, and F(ab′)2 fragments were separated on a reducing SDS-PAGE gel and detected with coomassie to determine the completeness of digestion. Size markers in kilodaltons. Schematic of IgG digestion is shown, with dashed lines indicating enzymatic cleavage sites relative to disulfide bonds (thin lines). (b) S. pneumoniae was incubated with Fab or F(ab′)2 fragments and in vitro agglutination was visualized using Nomarski microscopy with × 200 magnification, with 20 μm scale bars. (c) Binding of Fab and F(ab′)2 fragments to immobilized capsular polysaccharide was quantified using enzyme-linked immunosorbent assay (ELISA), and shown as the % binding compared with whole IgG±s.e.m. (d) Mice were passively immunized with equivalent concentrations of IgG fragments and challenged with 104 pneumococci (type 14) 4 h later. CFU per animal was determined 24 h post immunization±s.e.m. n⩾ 12 mice per group. *P<0.05. (e) Western blot analysis of nasal lavages from mice immunized 4 h prior with either Fab or F(ab′)2. Antibody fragments were detected with anti-rabbit IgG (raised against F(ab′)2) using horseradish peroxidase under reducing conditions. Size marker in kilodaltons.
To further examine the role of agglutination in protection, we took advantage of the susceptibility of IgG to partial reduction. Treatment with a mild reducing agent, such as dithiothreitol (DTT), preferentially reduces the hinge region disulfide bonds to generate reduced IgG (rIgG, or half IgG), which is monovalent and no longer agglutinates its target, but does not affect antigen binding14 (Figure 8a,b). In contrast to results with the whole IgG molecule, rIgG was not protective (Figure 8c), although both accessed the mucosal surface in similar quantities (Figure 8d). These data demonstrate that agglutination resulting from the multivalent character of immunoglobulin promotes mucosal protection.

Lack of protection by reduced IgG antibody. IgG was partially reduced to reduced IgG (rIgG) by incubation with dithiothreitol (DTT). (a) Whole IgG and rIgG fragments were separated by SDS-PAGE gel and detected with coomassie staining to determine the purity of rIgG under non-reducing conditions. Size markers in kilodaltons. Schematic of IgG reduction is shown, with dashed line indicating separation site and disulfide bonds represented by thin lines. (b) S. pneumoniae was incubated with whole IgG or rIgG and in vitro agglutination was visualized using Nomarski microscopy with × 200 magnification, with 20 μm scale bars. (c) Mice were immunized with equal amounts of whole IgG or rIgG, and challenged with 104 pneumococci (type 6A) 4 h later. CFU per animal was determined 24 h post immunization±s.e.m. n⩾ 4 mice per group. *P<0.05. Dotted line indicates limit of detection (4 CFU). (d) Western blot analysis of nasal lavages from mice immunized 4 h prior, detecting rabbit antibody with anti-rabbit IgG (raised against F(ab′)2) using alkaline phosphatase. Size marker in kilodaltons.
Many successful respiratory pathogens express a protease with specificity for bonds within the hinge region of human IgA1, the most abundant immunoglobulin subclass on airway mucosal surfaces.15, 16 Cleavage of di- or polyvalent IgA1 generates monovalent Fabα fragments. In the case of the pneumococcus, its IgA1-protease (Iga) is cell-surface attached and preferentially cleaves bound antibody.17 To determine whether Iga functions by abrogating the agglutinating effect of human IgA1, previously described human IgA (both IgA1 and IgA2) monoclonal antibodies (mAbs) generated to pneumococcal capsular polysaccharide18 were tested for protection against a protease-expressing wild-type strain and an isogenic mutant lacking the protease (Δiga). Cleavage of the human IgA 1 (hIgA1) mAb correlated with a loss of its ability to agglutinate pneumococci of the serotype recognized by the antibody (Figure 9a,b). Mice were then passively immunized with the TS hIgA1 mAb or an equivalent concentration of an agglutinating TS hIgA2 mAb, which is not cleaved by the protease and served as a control (Figure 9c). Expression of the protease eliminated the protective effect of the IgA1 on the wild-type strain – an effect that correlated with the loss of agglutination after exposure to the protease. In contrast, protection was observed against the Δiga strain immunized with the TS hIgA1 mAb, and the protease had no effect on protection by the TS hIgA2 mAb. As the protease preferentially cleaves antibody bound to the bacterial surface, its ability to mediate evasion of protective protease-sensitive antibody was unlikely to be caused by differences in the binding of intact versus digested antibody or their ability to access the mucosal surface. These data provided further evidence for the role of agglutination in antibody-mediated protection against colonization and also highlighted a common bacterial mechanism for overcoming this host defense.

Bacterial IgA1 protease eliminates mucosal protection by human IgA1. (a) Human monoclonal IgA1 was incubated overnight with wild-type (WT) or IgA1 protease-deficient (Δiga) pneumococci (type 2). The supernatant was analyzed by western blot analysis detecting human IgA 1 (hIgA1) by anti-human IgA antibody (α-chain-specific) using alkaline phosphatase under non-reducing conditions. Size markers in kilodaltons. Schematic of IgA1 digestion to generate fabα is shown, with open boxes indicating the hinge region, and dashed line indicating protease cleavage site relative to disulfide bonds (thin lines). (b) S. pneumoniae was incubated with TS hIgA1 monoclonal antibody (mAb) following cleavage from WT or Δiga pneumococci, and in vitro agglutination was then visualized using Nomarski microscopy with × 200 magnification, with 20 μm scale bars. (c) Infant mice were passively immunized with 25 μg per animal anti-type 2 hIgA1 or hIgA2 mAbs (or phosphate-buffered saline (PBS) vehicle control) and challenged 4 h later with 103 WT or Δiga pneumococci (type 2). CFU per animal was determined 24 h post immunization. The dashed line represents the limit of detection (4 CFU). n⩾3 mice per group. ns, not significant. *P<0.05.
DISCUSSION
The agglutinating effect of immune serum was first cited in 1891 by Élie Metchnikoff19, who described large ‘packets’ of streptococci in the blood of vaccinated rabbits. These observations on ‘clumping’ as a marker of immunity soon lead Neufeld to develop the Quelling reaction, an assay that was the basis for the selection of therapeutic sera in the pre-antibiotic era and is still used today for typing pneumococci.20 Findings in our report suggest that the agglutinating effect of antibody, in fact, has a major role in host defense during the initial phase of infection, colonization of mucosal sites. Many studies have correlated agglutination and protection; however, the contribution of the agglutinating effect of antibody has largely been assumed, and to our knowledge never directly demonstrated.21, 22, 23 In the case of H. influenzae, for instance, protection against colonization by antibody was also shown to be Fc-independent, although the mechanism of this effect with F(ab′)2 was not analyzed.24
Several independent lines of experimental evidence contributed to our conclusions. First, only specific antibody that agglutinated the challenge strain was protective against that strain. Second, protection of previously colonized immune animals correlated with the rapid agglutination of bacteria in vivo during rechallenge. Third, prior treatment of IgG to remove its divalent binding, either by generating Fab or rIgG fragments, prevented both agglutination and protection but still allowed binding to the organism. Fourth, the bacterial protease that cleaves human IgA1 to yield monovalent Fabα fragments also eliminated both agglutination and protection.
Our findings demonstrate that agglutinating antibody contributes significantly to mucosal defense by blocking the acquisition of colonization, including through host-to-host transmission. We cannot rule out a lesser role of Fab fragments in mucosal protection; it is, however, difficult to assess any potential effects on bacterial adherence to the epithelium or neutralization with in vivo studies. Passive immunization with agglutinating antibody that protected against primary colonization had little impact once colonization was established. This temporal difference is distinct from the requirement for Th17-dependent cell-mediated immune clearance of established colonization.25, 26 Our observations suggest that agglutination may prevent bacteria from accessing the epithelial surfaces where stable colonization occurs. Large aggregates of bacteria could be more easily swept away by mucociliary flow or other mechanical factors. Bacterial characteristics that promote association with mucus have previously been shown to enhance their removal.27 We cannot exclude the possibility that bacteria are also neutralized more effectively within aggregates, as it is difficult to compare growth characteristics under agglutinating conditions. Although we have previously reported that pneumococci that grow as chains of increased length, with more surface area per particle, adhere better to epithelial cells in vitro and colonize more efficiently, these forms are far smaller than the large aggregates described in the current study.28 We have also reported that aggregation of bacteria promotes complement activation and complement-dependent killing and that this effect is enhanced by agglutinating antibody.29 However, in the current study mucosal protection by passively administered antibodies did not require complement or Fc components required for phagocytic killing.
Our observations were based largely on the use of passive immunization with IgG, which can bridge its targets because of its divalent binding characteristic. Most mucosal antibody, however, is secretory polymeric IgA (tetravalent) or IgM (deca- or dodecavalent), which should agglutinate bacteria even more efficiently. Polymeric anticapsular IgA has been shown to mediate agglutination of pneumococci more effectively than its monomeric form.30 Our data confirm that bacterial IgA1 protease hinders the agglutinating and protective effect of protease-sensitive human IgA1 and extend these results by confirming this effect in vivo on mucosal surfaces of the upper respiratory tract. The importance of circumventing IgA1, which accounts for ∼90% of airway immunoglobulins in humans, may contribute to the successful colonization by members of the genera Streptococcus, Haemophilus, and Neisseriae, each of which expresses an IgA1 protease.15, 16 These proteases cleave only within the hinge region to generate non-agglutinating Fabα. Despite their shared function, these proteins encompass different enzymatic classes and cleave different peptide bonds, suggesting that the bacterial proteases that relieve the agglutinating effect of human IgA1 are an example of convergent evolution. As protease-expressing organisms are able to evade the protective effect of IgA1, other antibodies on mucosal surfaces, such as IgG derived from the plasma pool, are critical for mucosal defense. Note that our experiments using immune mice, which generated antipneumococcal IgA, would not have allowed for evasion of protection by the protease as mouse IgA lacks the enzymatic target.
Encapsulation by a thick layer of polysaccharide protects organisms from recognition of its underlying structures by antibody. However, the capsule is also the Achilles heel of encapsulated bacteria, as it is an abundant antigen that induces agglutinating antibody. When given systemically, sufficient amounts of protease-resistant immunoglobulin are generated to access and protect mucosal surfaces either by passive transudation or possibly via active transport of IgG by FcRn. In response to the selective pressure of immunoglobulin, encapsulated bacteria vary the composition of their surface polysaccharide (antigenic variation). Our results suggest that alternative vaccine strategies to target more conserved bacterial antigens would similarly need to generate agglutinating antibody in order to provide comparable levels of mucosal protection. Moreover, protective immune responses to other pathogens might also benefit from strategies that induce agglutinating antibodies.
Methods
Bacterial strains and culture conditions. S. pneumoniae strains were grown in tryptic soy broth (BD, Franklin Lakes, NJ, USA) at 37 °C in a non-shaking water bath to an optical density (OD)620=0.5. Strains used were clinical isolates selected for their ability to colonize the murine nasopharynx and included pneumococcal serotypes 2 (D39),31 4 (TIGR4),32 6A,13 9V (Collection of R. Austrian), 14 (Collection of E. Janoff), and 23F.9 D39Δiga, an insertion–deletion mutant lacking the pneumococcal IgA1-protease, was previously described.33 All strains were passaged IN in mice before preparation of frozen stocks.
Immunization reagents. Hyperimmune rabbit antipneumococcal sera were obtained from the collection of the late Dr Robert Austrian (University of Pennsylvania). Antisera were generated using serial immunization of rabbits with formalin-treated heat-killed whole bacteria of the specified type as previously described.34 Each strain used for generating antipneumococcal serum was distinct from the type-matched challenge isolates described above and would be recognized by anticapsular antibodies but not necessarily antibodies against other underlying antigens.
IgG was purified from rabbit antipneumococcal sera using a HiTrap Protein G column and AKTA FPLC (GE Healthcare Life Sciences, Pittsburgh, PA). IgG was buffer-exchanged into phosphate-buffered saline (PBS) using Amicon 10 k MWCO centrifugal filters (Millipore, Billerica, MA).
F(ab′)2 fragments were prepared from IgG purified from rabbit antipneumococcal serum using immobilized pepsin (Thermo Fisher Scientific, Waltham, MA) which digests the Fc fragment into small peptides. Briefly, IgG was incubated with immobilized pepsin at 37 °C for 5.5 h with continuous end-over-end rotation. Beads were removed by centrifugation and undigested IgG was removed using the Protein A Nab column (Thermo Fisher Scientific). F(ab′)2 fragments were buffer-exchanged into PBS and concentrated using Amicon 10 k MWCO spin filters.
Fab fragments were prepared from IgG purified from rabbit antipneumococcal serum using immobilized papain (Thermo Fisher Scientific). Briefly, IgG was incubated with immobilized papain at 37 °C for 17 h with continuous end-over-end rotation. Beads were removed using centrifugation and undigested IgG was removed by size exclusion chromatography using a HiPrep 26/60 Sephacryl S200 HR column (GE Healthcare Life Sciences) and an AKTA FPLC. Owing to the similar size of cleaved Fc and Fab, these fragments co-eluted. In pilot experiments, in which the papain-digested IgG was serially passed through a Protein A column until all Fc fragments were removed, there was no difference in protection in the presence or absence of this fragment.
Monovalent, rIgG was generated from rabbit antipneumococcal serum by partial reduction under mild reducing conditions that preferentially reduces the hinge region disulfide bonds.14, 35 Briefly, whole IgG was incubated in 25 mM DTT for 1.5 h at 37 °C. The rIgG was then alkylated with 25 mM iodoacetamide for 30 min at 37 °C to prevent re-oxidation to whole IgG. Undigested IgG and overly rIgG species were then removed using size exclusion chromatography as described above. The ∼75-kD rIgG fraction consisting of one heavy and one light chain was buffer-exchanged into PBS.
Human mAbs specific for S. pneumoniae serotypes 2 (IgA1 and IgA2) and serotype 8 (IgA1) were purified to >98.5% purity from B cells of immunized healthy adults that had been fused with the K6H6/B5 mouse–human heteromyeloma, as previously described.30, 33
Passive immunization experiments. C57Bl/6J (Jackson Laboratories, Bar Harbor, ME) and B6;129S4-C3tm1Crr/J (C3−/−, kind gift of Elizabeth Grice, University of Pennsylvania) mice were housed in accordance with Institutional Animal Care and Use Committee protocols. Five- to six-week-old adult mice were immunized IP with rabbit antipneumococcal sera, IgG, IgG fragments, mAbs, or vehicle controls. Unless specified otherwise, 4 h post immunization mice were inoculated IN, with 10 μl containing ∼2 × 104 CFU of the pneumococcal strain indicated in PBS. At 24 h post immunization, mice were killed, the trachea cannulated, and 200 μl of PBS instilled and lavage fluid was collected from the nares for quantitative culture. Lavage fluid was vortexed vigorously before plating to ensure any bacterial aggregates were dissociated. In pilot experiments we confirmed that agglutination by TS antiserum did not reduce colony counts using this plating procedure. Selective media were used to inhibit the growth of contaminants: neomycin (5 μg ml−1) for wild-type strains or spectinomycin (200 μg ml−1) for the Δiga mutant. The limit of detection was 2 CFU per animal, unless otherwise indicated. The success of passive immunization was confirmed using western analysis of serum obtained at the time of killing for each animal tested. The infant mouse protection experiment was performed on 14-day-old mice passively immunized IP with a volume of 25 μl or less and challenged IN with 3 μl containing 1–2 × 103 CFU. Protection was assessed as described above for adult mice.
Quantitative real-time PCR. In order to confirm bacterial load in nasal lavages, pneumococcal gyrA was amplified with qPCR using the following primers: forward, CCCTTTGGCAGTCCGACCA; reverse, ACGTGGGGGTCGTGGTGTCC. Nasal lavages (2 μl) from passive immunization experiments were added directly to a qPCR reaction. The qPCR reaction was performed in a total volume of 20 μl using the Power SYBR Green PCR Master Mix (Life Technologies, Grand Island, NY), containing 60 nM of each primer. The reaction conditions for amplification of DNA were 95 °C for 10 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min, run on a StepOne Plus Real Time PCR System (Life Technologies). All reactions were performed in duplicate and the mean values were calculated based on a standard curve of DNA from known bacterial quantities. Data were analyzed using the StepOne Software v2.0 (Life Technologies).
Pneumococcal transmission. One in four mice per litter of 4-day-old pups were inoculated IN with 1–2 × 103 CFU of the serotype 23F isolate. These mice were considered ‘index’ mice, and the remaining un-inoculated mice in the litter ‘contact’ mice. In the immune group, on days 8, 10, and 12 (age), contact mice were passively immunized IP with rabbit antipneumococcal antisera (25 μl). Both index and contact mice received 2 × 104 TCID50 of influenza A strain X31, a H3N2 recombinant of mouse adapted PR8 in 3 μl IN on day 8 (age) to promote nasal shedding. Influenza A is required for pneumococcal transmission between littermates11 and was only used in transmission experiments. All mice were killed on day 14 (age) and lavages were plated to determine pneumococcal transmission from index to contacts.
Western blot analysis. Mouse samples (10 μl serum diluted 1:1,000, or 20 μl undiluted nasal lavage) were resolved on a 10% Mini-PROTEAN TGX gel using a Tetra cell (Bio-Rad, Hercules, CA). Reduced samples were prepared with Laemmli sample buffer containing β-mercaptoethanol and boiled for 5 min. Non-reduced samples were prepared without β-mercaptoethanol and boiling. Gels were transferred onto polyvinylidene fluoride membranes using semi-dry transfer apparatus (Bio-Rad). Membranes were blocked in Pierce Protein-Free (TBS) Blocking Buffer (Thermo Fisher Scientific). Whole rabbit IgG was detected in lavages using anti-rabbit IgG (whole molecule; Sigma, St Louis, MO). Rabbit IgG fragments were detected using anti-rabbit IgG (rasied against F(ab′)2; Jackson ImmunoResearch Laboratories, West Grove, PA). hmAbs were detected using anti-human IgA (α-chain-specific; Sigma).
Enzyme-linked immunosorbent assays. To determine target-specific (anticapsular) antibody binding, capsular polysaccharide of the type specified (ATCC) was coated (2.5 μg ml−1) onto Immulon 1B 96 well plates (Thermo Fisher Scientific) for 5 h at 37 °C. TS antisera, intact IgG, or IgG fragments were added in doubling dilutions and incubated at room temperature overnight. Binding of antibody or fragments was detected with biotin-conjugated anti-rabbit IgG (light chain-specific; Cell Signaling Technology, Danvers, MA) followed by streptavidin conjugated to alkaline phosphatase. Signal was developed with p-nitrophenyl phosphate (Sigma), and the absorbance at 415 nm was recorded after a standardized period of 30 min. A capture enzyme-linked immunosorbent assay was used to calculate the concentration of total rabbit IgG in TS antisera. Briefly, Immulon 2HB plates (Thermo Fisher Scientific) were coated with anti-rabbit IgG (light chain-specific; 2.5 μg ml−1) overnight at 4 °C. TS rabbit antisera were added in doubling dilutions overnight at room temperature. Bound total rabbit IgG was detected using anti-rabbit IgG (whole molecule-specific) conjugated to alkaline phosphatase (Sigma). Polysaccharide capsule-specific antibody titer was then calculated by comparing the titers of total rabbit IgG to target-specific IgG. Murine titers of IgA in lavage fluid and IgG in serum were determined as previously described.13 Human type 2 capsule-specific IgA was measured as described.30
Agglutination assays. To compare Fab and F(ab′)2, the serotype 14 isolate was diluted in tryptic soy broth to OD620=0.1 and incubated with 100 μg ml−1 of fragments for 1 h at 37 °C. Samples were resuspended and placed onto glass slide to visualize agglutination. For rIgG the serotype 6A isolate was grown to mid-log phase and mixed with antibody (at 3.5 mg ml−1) on glass slide at 3:1 (v:v) ratio. Agglutination was visualized at × 200 magnification on a Nikon E600 Eclipse microscope equipped with a liquid crystal (Micro-color RGB-MS-C, CRI, Boston, MA) and a high-resolution charge-coupled device digital camera (CoolSnap CF, Roper Scientific, Tucson, AZ) with Nomarski optics. All image analyses were performed with iVision-Mac (BioVision Technologies, Exton, PA).
Cleavage of human IgA in vitro. hMAb (IgA1 to serotype 8, 960 μg ml−1) was incubated with mid-log phase D39 or D39Δiga at a ratio of 1:30 (v:v) and incubated at 37 °C overnight. Cultures were centrifuged to pellet bacteria and supernatant was collected and separated on non-reducing gel to identify pneumococcal IgA1–protease-dependent cleavage of unbound IgA1 (see Western protocol).17 Supernatant was also used for agglutination assay as described above for rIgG samples.
Immunofluorescence staining of mouse nasal tissue. C57Bl/6J and B6.129S2-Ighmtm1Cgn/J (μMT) mice (Jackson Laboratories) were immunized IN with three doses of strain TIGR4 (106–107 CFU per dose; immune) or PBS (naive) on days 0, 7, and 14. Two weeks after the final dose, when the initial colonizing strain has been cleared, mice were challenged IN with 107 CFU strain TIGR4. At 30 min post challenge, the mice were killed, heads collected, fixed, decalcified, frozen embedded, and 5-μm nasal tissue sections were cut. Frozen tissue sections were stained as previously described.27 Briefly, sections were fixed in 1:1 acetone:methanol and blocked with StartingBlock protein blocker (Thermo Fisher Scientific). Bacteria were stained with typing sera (Staten Serum Institut, Copenhagan, Denmark) and detected with donkey anti-rabbit cy3. Host cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies). Fluorescent image acquisition was performed as described above for agglutination assays.
Complement fixation assay. Serotype 14 and 23F isolates were incubated with 2% fresh mouse serum as a source of complement, with or without TS antisera (1:1,000 dilution) and incubated at 37 °C for 30 min. Bacteria were washed and C3 deposited on the bacteria was detected by fluorescein isothiocyanate-conjugated anti-mouse C3 (Cedarlane, Burlington, Canada) using a FacsCalibur (Becton Dickinson, Franklin Lakes, NJ, USA). Inactivated complement, heated to 56 °C for 30 min, and serum from C3−/− mice were used as negative controls. Flow cytometry data were analyzed using FlowJo 10 (Tree Star, Ashland, OR).
Animal study approval. All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Statistical analysis. Colonization density was expressed as the log CFU per animal for calculation of medians. Mann–Whitney test was performed when two groups were compared; Kruskal–Wallis test with Dunn’s post test was performed when groups of three or more were compared (GraphPad Prism 5, GraphPad Software, La Jolla, CA).
References
Durandy, A., Kracker, S. & Fischer, A. Primary antibody deficiencies. Nat. Rev. Immunol. 13, 519–533 (2013).
Kauppi, M., Eskola, J. & Kayhty, H. Anti-capsular polysaccharide antibody concentrations in saliva after immunization with Haemophilus influenzae type b conjugate vaccines. Pediatr. Infect. Dis. J. 14, 286–294 (1995).
Whitney, C. et al. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N. Engl. J. Med. 348, 1737–1746 (2003).
Dagan, R., Givon-Lavi, N., Fraser, D., Lipsitch, M., Siber, G.R. & Kohberger, R. Serum serotype-specific pneumococcal anticapsular immunoglobulin G concentrations after immunization with a 9-valent conjugate pneumococcal vaccine correlate with nasopharyngeal acquisition of pneumococcus. J. Infect. Dis. 192, 367–376 (2005).
Trotter, C.L. et al. Optimising the use of conjugate vaccines to prevent disease caused by Haemophilus influenzae type b, Neisseria meningitidis and Streptococcus pneumoniae. Vaccine 26, 4434–4445 (2008).
Lexau, C. et al. Changing epidemiology of invasive pneumococcal disease among older adults in the era of pediatric pneumococcal conjugate vaccine. JAMA 294, 2043–2051 (2005).
Griffin, M.R., Zhu, Y., Moore, M.R., Whitney, C.G. & Grijalva, C.G. U.S. hospitalizations for pneumonia after a decade of pneumococcal vaccination. N. Engl. J. Med. 369, 155–163 (2013).
Weiser, J.N. & Nahm, M.N. Immunity to extracellular bacteria In Fundamental Immunology (Paul W.E., ed) 1182–1203 Lippincott Williams & Wilkins: Philadelphia, PA, USA, (2008).
McCool, T., Cate, T., Moy, G. & Weiser, J. The immune response to pneumococcal proteins during experimental human carriage. J. Exp. Med. 195, 359–365 (2002).
Kadioglu, A., Weiser, J.N., Paton, J.C. & Andrew, P.W. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6, 288–301 (2008).
Diavatopoulos, D.A. et al. Influenza A virus facilitates Streptococcus pneumoniae transmission and disease. FASEB J. 24, 1789–1798 (2010).
Johnson, S. et al. Correlation of opsonophagocytosis and passive protection assays using human anticapsular antibodies in an infant mouse model of bacteremia for Streptococcus pneumoniae. J. Infect. Dis. 180, 133–140 (1999).
Roche, A.M., King, S.J. & Weiser, J.N. Live attenuated Streptococcus pneumoniae strains induce serotype-independent mucosal and systemic protection in mice. Infect. Immun. 75, 2469–2475 (2007).
Schreiber, J.R., Barrus, V.A. & Siber, G.R. Decreased protective efficacy of reduced and alkylated human immune serum globulin in experimental infection with Haemophilus influenzae type b. Infect. Immun. 47, 142–148 (1985).
Kilian, M. & Reinholdt, J. Immunoglobulin A1 proteases of pathogenic and commensal bacteria of the respiratory tract In Colonization of Mucosal Surfaces (Nataro J.P., Cohen P.S., Mobley H.L.T., Weiser J.N., eds) 119–130 ASM Press: Washington, DC, USA, (2005).
Woof, J.M. & Russell, M.W. Structure and function relationships in IgA. Mucosal immunol. 4, 590–597 (2011).
Bender, M.H. & Weiser, J.N. The atypical amino-terminal LPNTG-containing domain of the pneumococcal human IgA1-specific protease is required for proper enzyme localization and function. Mol. Microbiol. 61, 526–543 (2006).
Janoff, E.N. et al. Pneumococcal IgA1 protease subverts specific protection by human IgA1. Mucosal Immunol. 7, 249–256 (2013).
Metchnikoff, E Etudes sur l'immunité. IV. L'immunité des cobayes vaccinées contre le Vibrio Metchnikowii. Ann. Inst. Pasteur (Paris) 5, 465–478 (1891).
Neufeld, F. Verber die Agglutination der Pneumokokken und uber die Theorie der Agglutination. Z. Hyg. Infektionskr. 40, 54 (1902).
Sawada, S., Suzuki, M., Kawamura, T., Fujinaga, S., Masuho, Y. & Tomibe, K. Protection against infection with Pseudomonas aeruginosa by passive transfer of monoclonal antibodies to lipopolysaccharides and outer membrane proteins. J. Infect. Dis. 150, 570–576 (1984).
Cherry, J.D., Gornbein, J., Heininger, U. & Stehr, K. A search for serologic correlates of immunity to Bordetella pertussis cough illnesses. Vaccine 16, 1901–1906 (1998).
Mathias, A., Longet, S. & Corthesy, B. Agglutinating secretory IgA preserves intestinal epithelial cell integrity during apical infection by Shigella flexneri. Infect. Immun. 81, 3027–3034 (2013).
Kauppi, M., Saarinen, L. & Kayhty, H. Anti-capsular polysaccharide antibodies reduce nasopharyngeal colonization by Haemophilus influenzae type b in infant rats. J. Infect. Dis. 167, 365–371 (1993).
Zhang, Z., Clarke, T.B. & Weiser, J.N. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Invest. 119, 1899–1909 (2009).
Malley, R., Trzcinski, K., Srivastava, A., Thompson, C., Anderson, P. & Lipsitch, M. CD4+ T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc. Natl Acad. Sci. USA 102, 4848–4853 (2005).
Nelson, A.L., Roche, A.M., Gould, J.M., Chim, K., Ratner, A.J. & Weiser, J.N. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infect. Immun. 75, 83–90 (2007).
Rodriguez, J.L., Dalia, A.B. & Weiser, J.N. Increased chain length promotes pneumococcal adherence and colonization. Infect. Immun. 80, 3454–3459 (2012).
Dalia, A.B. & Weiser, J.N. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe 10, 486–496 (2011).
Fasching, C.E., Grossman, T., Corthesy, B., Plaut, A.G., Weiser, J.N. & Janoff, E.N. Impact of the molecular form of immunoglobulin A on functional activity in defense against Streptococcus pneumoniae. Infect. Immun. 75, 1801–1810 (2007).
Avery, O.T., MacLeod, C.M. & McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. J. Exp. Med. 79, 137–157 (1944).
Tettelin, H. et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293, 498–506 (2001).
Weiser, J., Bae, D., Fasching, C., Scamurra, R., Ratner, A. & Janoff, E. Antibody-enhanced pneumococcal adherence requires IgA1 protease. Proc. Natl Acad. Sci. USA 100, 415–420 (2003).
Schiffman, G., Bornstein, D.L. & Austrian, R. Capsulation of Pneumococcus with soluble cell wall-like polysaccharide. II. Nonidentity of cell wall and soluble cell wall-like polysaccharides derived from the same and different pneumococcal strains. J. Exp. Med. 134, 600–617 (1971).
Machino, Y. et al. Effect of immunoglobulin G (IgG) interchain disulfide bond cleavage on efficacy of intravenous immunoglobulin for immune thrombocytopenic purpura (ITP). Clin. Exp. Immunol. 162, 415–424 (2010).
Acknowledgements
We thank Jan Erikson from The Wistar Institute for providing X31 Influenza virus. This work was supported by the US Public Health Service, grant number AI38446 and AI05168 (J.N.W.), AI092468 and AI108479 (E.N.J), and the Robert Austrian Research Award in Pneumococcal Vaccinology (A.M.R). Additional support was provided by the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases (P30-DK050306) and its Molecular Pathology and Imaging core facilities, and the Mucosal and Vaccine Research Program Colorado (MAVRC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Roche, A., Richard, A., Rahkola, J. et al. Antibody blocks acquisition of bacterial colonization through agglutination. Mucosal Immunol 8, 176–185 (2015). https://doi.org/10.1038/mi.2014.55
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.55
This article is cited by
-
Immunofluorescent-aggregation assay based on anti-Salmonella typhimurium IgG-AuNCs, for rapid detection of Salmonella typhimurium
Microchimica Acta (2022)
-
Pneumococcal capsule blocks protection by immunization with conserved surface proteins
npj Vaccines (2021)
-
Applications of nanobodies in plant science and biotechnology
Plant Molecular Biology (2021)
-
Topical application of nebulized human IgG, IgA and IgAM in the lungs of rats and non-human primates
Respiratory Research (2019)
-
Pneumococcal quorum sensing drives an asymmetric owner–intruder competitive strategy during carriage via the competence regulon
Nature Microbiology (2018)
