
Abstract
Mast cells (MCs) are cells of hematopoietic origin that normally reside in mucosal tissues, often near epithelial cells, glands, smooth muscle cells, and nerves. Best known for their contributions to pathology during IgE-associated disorders such as food allergy, asthma, and anaphylaxis, MCs are also thought to mediate IgE-associated effector functions during certain parasite infections. However, various MC populations also can be activated to express functional programs—such as secreting preformed and/or newly synthesized biologically active products—in response to encounters with products derived from diverse pathogens, other host cells (including leukocytes and structural cells), damaged tissue, or the activation of the complement or coagulation systems, as well as by signals derived from the external environment (including animal toxins, plant products, and physical agents). In this review, we will discuss evidence suggesting that MCs can perform diverse effector and immunoregulatory roles that contribute to homeostasis or pathology in mucosal tissues.
Similar content being viewed by others


Introduction
Mast cells (MCs) are normal residents of mucosal tissues, but their numbers and anatomical location can change markedly during immune responses, infections, and other disorders affecting such sites, in humans, mice, and other species.1, 2, 3, 4, 5 MCs stimulated via the high-affinity receptor for IgE (FcɛRI) or by any of multiple other mechanisms can release a diverse spectrum of biologically active mediators, and such products, individually or in aggregate, can have many different effects on immune or structural cells present in mucosal tissues.4, 6, 7, 8 As a result, there is no lack of ideas about the potential effector or immunoregulatory functions MCs might have during mucosal immune responses.3, 4, 8
However, it can be quite challenging to prove that MCs can perform such proposed functions in vivo, and even more difficult, in light of the potential redundancy of effector and immunoregulatory mechanisms, to assess the biological importance of such MC functions in particular settings. In this review, we will outline some basic principles of MC biology and then consider evidence that implicates MCs in physiological, immunological, and pathological processes affecting mucosal sites. We will particularly focus on findings derived from studies in mice, a species in which biological responses can be analyzed in animals that lack MCs or specific MC-associated products.
MC Development, Phenotype, Tissue Distribution, and Plasticity
MCs are tissue-resident cells that arise from hematopoietic progenitors.9 Unlike other immune cells, MCs normally do not mature before leaving the bone marrow but circulate through the vascular system as immature progenitors that complete their development peripherally within the connective or mucosal tissues, or in serosal cavities, in a process potentially regulated by multiple local factors.3, 4, 5, 10, 11
The KIT ligand stem cell factor (SCF) has a critical role in MC biology by regulating the development, migration, growth, survival, and local activation of MCs.12, 13, 14 Various other factors also can modulate MC growth and survival, including interleukin (IL)-3,15 IL-4,16, 17, 18, 19 IL-9,20, 21 IL-10,22, 23, 24 IL-33,25, 26, 27 CXCL12,28, 29 transforming growth factor-β,30 and nerve growth factor.31
MCs reside in almost all vascularized tissues and can be especially numerous in those exposed to the external environment, such as the skin and mucosal sites.3 MCs are, therefore, well positioned to respond to various allergens, pathogens, and other agents that can be ingested, inhaled, or encountered after disruption of the epithelial barrier.8 Moreover, many phenotypic and functional characteristics of MCs, such as their proliferation, survival, and ability to store and/or secrete various products, can be modulated or “tuned” by many genetic and environmental factors, including changes in the cytokine milieu associated with inflammatory or immune responses.8
Despite their potential phenotypic “plasticity”, MCs are often subclassified based on certain of their “baseline” phenotypic characteristics and their anatomic locations (Table 1). In mice, two types of MCs have been described: “connective tissue-type” MCs (CTMCs) and mucosal MCs (MMCs).4, 5, 8 CTMCs are often located around venules and near nerves and reside in serosal cavities, whereas MMCs occupy the mucosae of the gut and respiratory mucosa.5 MMCs are found at relatively low numbers in most mucosal tissues (in mice, they are normally present in higher numbers in the glandular stomach mucosa than in the intestines), but the expansion of MMC populations can be induced in a T-cell-dependent manner.5, 32 CTMCs and MMCs often are distinguished based on their protease content (Table 1). Mouse intestinal MMCs elicited during parasite infection express the chymase mouse MC protease-1 (MCPT1) but not the elastase MCPT5, whereas CTMCs do not express MCPT1 but express MCPT5, the chymase MCPT4, and the tryptases MCPT6 and MCPT75, 33, 34 (notably, C57BL/6 mice do not express MCPT7 because of a point mutation in the exon/intron 2 splice of the Mcpt7 gene35). However, the plasticity of MC phenotype can make such a classification challenging, as features of the cells, including their protease content, may vary during the course of immune responses.5, 8, 33, 36, 37
In humans, MCs can be subcategorized into MCT, which express high levels of the MC-specific protease tryptase but little or no chymase (these, therefore, are thought to resemble rodent MMCs), and MCTC, which express both tryptase and chymase (and in that respect resemble rodent CTMCs)38, 39 (Table 1). MCC (which express chymase but little or no tryptase) also have been described, but they appear to be infrequent.40 Clinical evidence suggests that human MCT (like mouse MMCs) may be dependent on T cells, at least in part, to maintain normal numbers in mucosal sites.41 The majority of human lung MCs ordinarily are MCT (∼90%) and these cells are found in the bronchial/bronchiolar lamina propria and alveoli.42 MCTC typically are located beneath the epithelium in the lamina propria and submucosa, in close proximity to submucosal glands, and some MCTC are found within and around the airway smooth muscle layers of major bronchi.43 The lamina propria of the human intestinal mucosa normally contains ∼1.5–3% MCs.44, 45 In the human small intestine, MCT represent ∼98% of all MCs in the mucosa and ∼13% of MCs in the submucosa are MCT.42 In naïve mice, relatively low numbers of MCs are found in the lung and these cells are located around the larger airways and blood vessels. As noted above, in naïve mice, few MCs are found in the mucosa of the gastrointestinal tract except for the glandular stomach and small numbers can be found in the submucosa and muscularis propria.
However, MC numbers at mucosal sites can increase in both humans and mice in pathological settings such as inflammatory bowel disease,46, 47 food allergy,48, 49 parasite infections,50, 51 asthma,52, 53, 54, 55, 56 or various types of lung fibrosis.57, 58, 59, 60 Such increases in MC numbers could reflect, at least in part, the division of mature MCs at mucosal sites. Although MCs are often considered as terminally differentiated cells that cannot divide, we and others have provided evidence that at least certain “mature” MCs, i.e., those that can be identified morphologically based on their abundant cytoplasmic granules retain some proliferative ability.61, 62, 63, 64 Increased MC numbers in such settings also may reflect the maturation of increased numbers of MC progenitors, whose numbers in tissues may increase due to their increased recruitment and/or survival in such tissues, and/or via the local proliferation of such progenitors.5, 65 Although it is not yet clear to what extent MC progenitors can proliferate in tissues, increased numbers of such progenitors have been observed at mucosal sites under various pathological conditions. For example, Arinobu et al.66 observed a fourfold expansion of MC progenitors in the intestine following sensitization and challenge of mice with the antigen ovalbumin (OVA). Antigen-dependent expansion of MC progenitors also was observed in mouse lung following sensitization and challenge with aerosolized OVA, and IL-9 and CD4+ T cells were found to contribute to such expansion.67 Finally, it has been reported that certain MC progenitors can proliferate in vitro;66 however, whether they can also proliferate within mucosal tissues remains to be proven.
MC Activation
MCs ordinarily express on their surface large numbers of the high-affinity IgE receptor, FcɛRI. During IgE-dependent immune responses, the antigen-dependent cross-linking of antigen-specific IgE bound to FcɛRI induces the aggregation of FcɛRI, promoting the activation of downstream signaling events that lead to the secretion of biologically active products implicated in allergic reactions.6, 68, 69 The IgE-dependent stimulation of MCs has been extensively reviewed.6, 69, 70, 71, 72 It was recently reported that perivascular MCs can “sample” circulating IgE directly in the blood by extending cell processes across the vessel wall.73 Moreover, MC FcɛRI were shown to be able to distinguish between high- or low-affinity stimuli, permitting the MCs to respond differentially to such signals by releasing distinct spectra of secretory products in vitro and by orchestrating distinct in vivo outcomes.74
Our group recently reported a beneficial role for IgE, FcɛRIα, and FcɛRIγ in defense against honeybee venom-induced mortality in mice.75 Together with the evidence that expression of the FcɛRIα chain is important for the full expression of acquired resistance to the hypothermia-inducing effect of honeybee venom-derived phospholipase A2,76 these findings support the hypothesis that IgE, which contributes to allergic disorders, also has an important function in protection of the host against noxious substances.77, 78
MCs can respond to many stimuli beside IgE. MCs can respond to various pathogens through the activation of TLRs, including TLR-2 and TLR-479, 80 and, via G-protein-coupled receptors, to certain peptides found in venoms,81, 82, 83 or can be activated by various complement peptides84, 85 and platelet-activation factor.86 There is evidence that MCs also can be directly or indirectly activated by some plant products, including aqueous pollen extracts from birch,87 and by products of the coagulation system, including Factor Xa88 and thrombin receptor-activating peptide.89 MCs also can respond to certain chemokines and cytokines (including IL-3325, 26, 27, 90, 91 and TSLP92), or be activated through the aryl hydrocarbon receptor,93, 94 the CD40 ligand,95 or the OX40 ligand96, 97, 98 or by immune complexes of IgG.99, 100 MC activation (e.g., via the FcɛRI) can also be modulated by various mechanisms, including interactions with other cells such as granulocytes,101 regulatory T cells,102 and other lymphocytes,103 via a variety of negative regulatory receptors expressed on their surface,8, 104, 105, 106 or by exposure to certain cytokines, including the KIT ligand SCF,8, 12, 13, 14, 107, 108 IL-33,25, 26, 27, 109 and interferon (IFN)-γ.56, 110
MC-Derived Mediators
MCs store preformed mediators in their granules and can release some of them almost instantly on degranulation. These stored mediators include vasoactive amines such as histamine111, 112 (although MCs are considered the main source of histamine outside of the central nervous system, other cells also can produce histamine, including basophils113 and neutrophils114, 115), and, in rodents, serotonin.112 MC granules also contain many neutral proteases (tryptases, chymases, and carboxypeptidase A3 (CPA3))42, 116, 117, 118, 119, 120, 121, 122 (Table 1). As noted above, MC protease content can vary depending on the cells’ tissue location and microenvironment. Only one chymase is expressed in human MCs but there are 13 known mouse chymase genes.123 Among those, the β-chymase MC protease 4 (MCPT4) appears to be most functionally similar to human chymase.124, 125 MC granules also contain some preformed cytokines and growth factors, including tumor necrosis factor (TNF) in both humans126, 127 and mice.128, 129 MCs can also synthesize and secrete certain lipid mediators, such as prostaglandins and leukotrienes.130, 131 Finally, MCs are also able to synthesize and secrete a large number of cytokines, chemokines, and growth factors, including TNF,128, 132, 133, 134 IL-1,135, 136, 137 IL-6,135, 138, 139 IL-10,140, 141, 142 IL-17,143, 144, 145 vascular endothelial growth factor and other vascular growth factors,146, 147, 148 SCF,149, 150 and many others. Release of lipid mediators typically occurs within 1–2 h after MC activation and is associated with immediate responses, whereas the synthesis and secretion of cytokines and chemokines characteristically occurs over a longer time frame, associated with the development of late phase or more chronic responses.8, 151
Mouse Models to Study MC Functions in vivo
Pharmacological agents thought to target MC activation or MC proteases have been used in vivo to assess the functions of MCs. However, none of the drugs or antibodies described to date is fully specific for MCs or for particular MC proteases.70, 152, 153 Therefore, we favor using genetic approaches to gain insights into MCs functions in vivo.
c-kit mutant MC-deficient mice and the “MC knock-in model”
For many years, c-kit mutant MC-deficient mice, such as WBB6F1-KitW/W-v and C57BL/6-KitW-sh/W-sh mice, have been used to analyze the functions of MCs in vivo.7, 8, 141, 154, 155, 156, 157, 158 These two types of mice are profoundly MC deficient but also have several other phenotypic abnormalities,155, 157, 158, 159, 160, 161, 162, 163 including a marked reduction in intestinal cells of Cajal, which results in abnormal electrical pacemaker activity in the small intestine.155, 164 Abnormalities in biological responses in c-kit mutant mice may reflect their MC deficiency and/or one or more of their other phenotypic abnormalities. However, at many anatomical sites, the deficiency in MCs can be selectively “repaired” by the adoptive transfer of genetically compatible, in vitro-derived MCs such as bone marrow-derived cultured MCs (BMCMCs), to create the so-called “MC knock-in mice”.8, 10, 60, 155, 156, 165, 166
c-kit-independent MC-deficient mice and mice deficient for MC-associated products
More recently, several groups have generated new strains of mice permitting the constitutive or inducible deletion of MCs independently of mutations affecting c-kit structure or expression.60, 167, 168, 169, 170, 171, 172 Most of these groups used a strategy consisting of generating transgenic mice expressing the Cre recombinase under the control of promoters for MC proteases, such as those for Cpa3 or MC protease 5 (Mcpt5).167, 168, 172 Such mice then were crossed with mice in which the genes of interest have been “floxed” to delete the expression of these gene products in the MCs.168, 173 Our group mated Cpa3-Cre mice with mice expressing the floxed survival factor Mcl-1: the resulting Cpa3-Cre; Mcl-1fl/fl mice were severely deficient in MCs but had also markedly reduced basophil levels.167 Feyerabend et al.172 reported a severe MC deficiency (and a more modest deficiency in basophils) in another line of Cpa3-Cre mice due to Cre-mediated cytotoxicity. Mcpt5-Cre mice, which express Cre in connective tissue-type MCs but not MMCs,168, 169 were mated with transgenic mice expressing Cre inducible diphtheria toxin A (DTA) or diphtheria toxin receptor (iDTR) genes to achieve constitutive (in Mcpt5-Cre; DTA+ mice) or inducible (after DT injection in Mcpt5-Cre; iDTR+ mice) ablation of CTMCs.168 All of these mice and some additional new types of MC-deficient mice have been recently reviewed in detail.152, 174, 175
Several strains of mice that are deficient for one or multiple MC-associated proteases, or are unable to synthesize histamine (due to a deficiency in histidine decarboxylase) or heparin (due to a deficiency in N-deacetylase/N-sulfotransferase-2), also have been developed. Although each of these strains of mice can provide important information concerning the roles of particular products released by MCs, some of them have a complex phenotype and there are a number of considerations that should be kept in mind when interpreting findings obtained with these animals, as reviewed in Reber et al.,152 Galli et al.175, and Wernersson and Pejler.176
Role of Mast Cells in the Regulation of Intestinal Epithelial Permeability
The intestinal epithelium forms a selectively permeable barrier against the external environment.177 Disruption or dysregulation of this barrier is associated with many intestinal disorders, including bacterial, viral and parasitic infections, inflammatory bowel disease, and food allergies.177, 178 Groschwitz et al.177 demonstrated that naïve c-kit mutant MC-deficient KitW-sh/W-sh mice and mice deficient for the chymase MCPT4 have altered intestinal barrier structure and function, with decreased intestinal epithelial cell migration along the villus/crypt axis of jejunum, increased crypt depth in the jejunum (without differences in villus length), and increased intestinal permeability as compared with wild-type (WT) mice. Engraftment of KitW-sh/W-sh mice with WT BMCMCs but not Mcpt4−/− BMCMCs restored these features to levels observed in WT mice, evidence that MCs can contribute to the homeostatic regulation of the intestinal barrier through MCPT4-dependent mechanisms.177
Other studies have provided evidence that MCs can control intestinal epithelial ion transport or permeability during effector phases of inflammatory responses,179, 180, 181 including during anaphylaxis.179 Isolated intestinal preparations from OVA-sensitized WT mice displayed increases in short-circuit current (Isc) following ex vivo stimulation with OVA or following electrical transmural stimulation of intestinal neurons. Such responses were significantly diminished in MC-deficient KitW/W-v or WCB6F1-MgfSl/Sl-d mice (Sl-d is a deletion in the transmembrane domain of the Scf gene182 and MgfSl/Sl-d mice do not express the membrane form of SCF183). Moreover, transfer of BM cells from WT mice to KitW/W-v mice “normalized” the Isc responses to both antigen and transmural stimulation, indicating a role for MCs and/or other BM-derived cell type in this process.179 A role for MCs in this setting also was suggested by tests of pharmacological agents, which antagonize the factions of certain MC-derived mediators.179
Infection with the parasite Trichinella spiralis (T. spiralis) increases paracellular permeability of the jejunum and decreases the expression of occludin in the tight junctions of enterocytes.181 Treatment of WT mice with a c-kit blocking antibody abrogated MC hyperplasia during T. spiralis infection and blocked parasite-induced increases in intestinal permeability.181 Mice deficient in the chymase MCPT1 also exhibited diminished intestinal permeability during T. spiralis infection, even though numbers of intestinal MMCs were higher during infection in Mcpt1−/− mice than in WT mice.181
Injection of the neuropeptide substance P induces intestinal ion secretion with increase in Isc responses. In intestinal preparations from MC-deficient KitW/W-v mice, substance P-induced Isc responses were diminished to ∼50% of those observed in WT mice and were normalized by the adoptive transfer of WT BM cells, suggesting that MCs can contribute to substance P-induced changes in intestinal ion secretion.184 By contrast, our group demonstrated that MCs can limit the toxicity associated with high concentrations of another neuropeptide, VIP (vasoactive intestinal polypeptide).82 In that setting, our evidence indicated that VIP induced MC degranulation, releasing the chymase MCPT4, which then degraded VIP.82
Roles of Mast Cells in Allergic Responses at Mucosal Sites
Asthma
Asthma is a multifaceted disorder characterized by reversible airway narrowing (in many patients in response to particular allergens), immunologically nonspecific airway hyperresponsiveness (AHR), chronic inflammation of the airways, and airway remodeling, including fibrosis, goblet cell hyperplasia/metaplasia, increased mucus production, smooth muscle thickening, and increased vascularity.185, 186, 187 Early manifestations of the disorder can appear in childhood, and both genetic188 and environmental factors189 contribute to the development and progression of asthma. Rather than being a single “disease”, the disorder called asthma is likely comprised of distinct subphenotypes with different clinical characteristics and underlying mechanisms.190, 191, 192 Analysis of lung epithelial brush biopsies, bronchoalveolar lavage fluids, lung biopsies, and autopsies have shown increased numbers of MCs in the airways of some asthmatic subjects54, 193, 194, 195 but not others.194, 196, 197 One feature more often seen in asthmatic subjects than in those without the disease is the presence of MCs within the bronchial epithelium.198, 199, 200
In subjects with asthma, B-cell class switching to IgE occurs in the lymph nodes,201 as well as locally in the respiratory mucosa.202 IgE binds to FcɛRI, highly expressed on MCs and basophils, but also, in certain settings by eosinophils and neutrophils; evidence has been reported that FcɛRI also can be expressed by airway epithelial and smooth muscle cells and by certain nerves (reviewed in Galli and Tsai69). IgE not only permits allergen-dependent MC activation, but also enhances the stability of FcɛRI on the MC surface, thus increasing the levels of receptor expression of FcɛRI, contributing to the maintenance of a positive amplification loop (reviewed in Kalesnikoff and Galli6).
Several mouse models of allergic airway inflammation have been developed to recapitulate many aspects of asthma. Studies using the MC knock-in model in c-kit mutant mice sensitized with an antigen in the absence of artificial adjuvant,55, 56, 203, 204, 205, 206 or employing relatively low doses of antigen for sensitization or challenge,207, 208 have provided evidence that MCs and MC-derived TNF can amplify multiple features of allergic airway inflammation, including airway responsiveness, inflammation, and tissue remodeling.55, 56, 203, 204, 205, 207, 208 However, contributions of MCs to various features of allergic asthma are not observed (perhaps because they are redundant) in some models of allergic airway inflammation employing strong artificial adjuvants (such as alum) and relatively high doses of antigen for sensitization and challenge.203, 209, 210, 211, 212
Genetic background also can influence the contribution of MCs to allergic airway inflammation. Becker et al.213 confirmed that KitW-sh/W-sh mice on the C57BL/6 background have reduced airway inflammation and AHR in an adjuvant-free model of asthma, but found no significant differences between BALB/c-KitW-sh/W-sh and BALB/c WT mice in their model. These findings clearly indicate that roles of MCs in this asthma model that are important in one strain background (the “Th1-biased” C57BL/6 background) may not be important (or may be redundant) in the more “Th2-biased” BALB/c background. These findings are of substantial interest, given the strong evidence that genetic factors have an important role in human asthma.
In a mouse model of chronic allergic airway inflammation, studies in MC knock-in mice indicated that MC expression of the IFN-γR contributes to the development of many features of the model that also require MCs and FcɛRIγ for optimal expression, including AHR, neutrophil, and eosinophil infiltration in the lung, lung collagen deposition, and increased expression of lung IL-6, IL-13, IL-33, multiple chemokines, arginase-1, and the acute-phase protein serum amyloid A3. However, the expression of IFN-γR also contributes to some features of the model, which require MCs for optimal expression but that occur relatively independently of FcɛRIγ, such as elevations of levels of integrin α7 and the macrophage receptor with collagenous structure in the affected lungs.56 In a passive model of OVA-induced allergic airway inflammation, transfer of OVA323–339-peptide-specific, IFN-γ-producing Th1 cells to naïve mice primed them to develop airway neutrophilia and AHR that was most prominent in mice challenged with LPS as well as antigen.214 It also has been reported that co-stimulation of mouse pulmonary macrophages with LPS and IFN-γ induces the production of IL-27,215 that in turn can enhance production of IL-1 and TNF by MCs.216 Such studies provide support for the hypothesis that bacterial infections can sustain or enhance inflammation driven by Th1 responses in asthma.
Some patients with severe asthma exhibit enhanced sputum neutrophilia (but not eosinophilia) and enhanced serum and sputum levels of IL-17.217 In diseases with a prominent Th17 signature such as atopic dermatitis,218 chronic exposure to antigens, such as via epicutaneous sensitization,219 can enhance airway inflammation and “local” Th17 inflammation in the lung.220 Evidence from our mouse models55, 56 and those of others221 show that chronic airway exposure to OVA can increase bronchoalveolar lavage neutrophils and lung levels of IL-17 (in addition to Th2 cytokines), and that the presence of MCs is essential for the development of such features. Some mouse or human MCs can produce IL-17 on non-IgE-dependent stimulation (e.g., with 6-formylindolo(3,2-b)carbazole (FICZ)) or when exposed FICZ in combination with IgE/antigen and, based on immunohistochemical findings, MCs appear to represent a major in vivo source of IL-17 in the chronically inflamed bronchial lamina propria of patients with chronic obstructive pulmonary disease,94 and in other settings.145, 222, 223
IL-33 is also thought to contribute to the pathology of asthma.100, 224, 225, 226, 227, 228 The IL-33 receptor, ST2, is expressed by MCs and basophils,229 but not by airway smooth muscle cells or lung fibroblasts.230 In mice, IL-33-induced enhanced airway inflammation is partly dependent on IL-33-dependent MC production of IL-13.231 IL-33 is considered an alarmin or a proinflammatory cytokine,232 but its biology might be more complex as it has been reported that chronic exposure of human and mouse MCs to IL-33 in vitro can induce a hyporesponsive MC phenotype, raising the intriguing possibility that IL-33 might actually have certain protective roles in chronic airway inflammation.233
In summary, evidence from studies of human asthma and mouse models of the disorder support the general conclusion that MCs can not only have critical roles in amplifying acute immunological responses to antigen and in helping to orchestrate the later development of multiple features of the disorder, but also suggest that the roles of MCs in particular subphenotypes of asthma may vary, in part, due to differences in the cytokines present in those settings (Figure 1). Moreover, recent data raise the interesting possibility that some individual MC mediators may have effects that can restrain the development of certain features of the pathology. For example, Waernet al.234, 235 reported that mice deficient for the chymase MCPT4 exhibit increased pathology (i.e., airway inflammation, AHR, and smooth muscle thickening) in two different models of allergic lung inflammation, and that such protective effects might reflect, at least in part, degradation of IL-33 by the chymase.

Schematic, highly simplified representation of the potential roles of MCs in airway chronic allergic inflammation and remodeling. Individuals not yet sensitized to environmental allergens do not have specific IgE to such allergens, and few MCs are present within the epithelium (left panel). During allergic sensitization, environmental antigens (Ag) are captured by APCs in the airway lumen or in the epithelium of the airway mucosa, and Ag-activated APCs mature and migrate to regional lymph nodes, where priming of T cells occurs (not shown). The presence of IL-4 or IL-13, which may be derived from a variety of potential cellular sources, induces T cells to become TH2 cells (right panel). In some cases, allergens also can reach APCs in the submucosa through damaged epithelium. Cytokines induced by epithelial damage (such as IL-33 and TSLP) can activate ILC2 cells, which secrete type 2 cytokines, such as IL-4 and IL-13. The Th2 environment promotes heavy-chain class switching from IgM or IgG to IgE for Ag-specific IgE production in B cells. IgE binds to FcɛRI on MCs (and basophils) and sensitizes these cells to respond to subsequent Ag exposures. Ag-induced aggregation of IgE-bound FcɛRI causes the prompt release of pre-stored MC mediators, including histamine and TNF, which can promote bronchoconstriction and, more slowly, fibroblast proliferation. FcɛRI activation also induces the production and the release of de novo-synthesized compounds, such as leukotrienes, prostaglandins, and proinflammatory cytokines (e.g., IL-5, IL-6, IL-8, IL-13, and TNF) and chemokines (not shown), which contribute to the development of local inflammation. Both soluble factors, such as IFN-γ, TSLP, IL-33, S1P, LPS (through PRRs) and cells present at the site, such as TH cells and various Treg cells (not shown), which can interact with OX40L on MCs, modulating IgE-dependent MC activation, or B cells, which can interact with CD40L on MCs, which may enhance B-cell IgE production. At least one MC-secreted product, MCPT4 (not shown), can negatively regulate the inflammatory environment, in part through the degradation of IL-33. Repetitive exposure to specific Ag favors persistent inflammation (with large numbers of eosinophils, and with MCs appearing within the epithelium), goblet cell hyperplasia and increased mucus secretion, smooth muscle cell proliferation, increased vascular permeability (and increased numbers of blood vessels) and airway edema, thickening, and remodeling. In some asthma subtypes, genetic or environmental factors, including pathogen-derived products, tissue damage, airway pollutants, and oxidative stress, may confer strong TH1 and/or TH17 signatures associated with large numbers of neutrophils at the site of inflammation. Studies in MC knock-in mice have indicated that some actions of MCs, such as increasing the number of epithelial goblet cells, can occur in a model of chronic allergic inflammation by MC-dependent mechanisms that do not require MC signaling through the FcɛRIγ chain, whereas MCs must express both the FcɛRIγ chain and the IFN-γ receptor 1 (IFN-γR1) to mediate substantial increases in lung eosinophils and neutrophils. Note: downregulatory mechanisms that can be engaged in this setting, such as co-engagement by multivalent Ag of both FcɛRI and FcγRIIb, or effects of regulatory T-cell populations, are not shown. AhR, Aryl hydrocarbon receptor; APC, antigen-presenting cell; Baso, basophils; Eos, eosinophils; FcɛRI, high-affinity receptor for IgE; IL, interleukin; ILC2, innate lymphoid cells type 2; LPS, lipopolysaccharide; MC, mast cell; Neu, neutrophils; PRR, pattern-recognition receptor; S1P, sphingosine-1-phosphate; TH, T helper; TNF, tumor necrosis factor; TSLP, thymic stromal lymphopoietin.
Food allergy and anaphylaxis
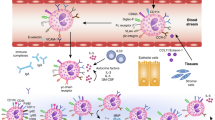
Food allergies are caused by adverse acquired immune responses to food components, primarily proteins.236 Their prevalence has recently increased and food allergies now affect ∼6% of children and 3–4% of adults in developed countries.236 The manifestations of food allergy can range from mild to severe, with the most severe form being anaphylaxis, an acute and potentially life-threatening multisystem reaction to allergen exposure. In the United States, the majority of cases of food-induced fatal or near-fatal anaphylaxis are caused by peanuts or tree nuts.237, 238 Studies in mice indicate that MCs are critical effector cells of both food-induced intestinal inflammation and anaphylaxis (Figure 2).

Schematic, highly simplified representation of the potential roles of MCs in food allergy and parasite infections. In the normal intestine, MCs can contribute to the homeostatic regulation of the epithelial barrier through chymase- (MCPT4-) dependent mechanisms and few MCs are present within the epithelium (left panel). During sensitization with food allergens (middle panel) or primary infections with parasites (right panel), antigens (Ag) are captured by APCs and Ag-activated APCs mature and migrate to regional lymph nodes, where priming of T cells occurs (not shown). The presence of IL-4 or IL-13, which may be derived from a variety of potential cellular sources, induces T cells to become TH2 cells. TH2 cells and ILC2 cells release IL-3 and IL-9, which promote the expansion of mucosal MCs (MMCs), and some of these MMCs are found in the intestinal epithelium. IgE binds to FcɛRI on MCs (and basophils) and sensitizes these cells to respond to subsequent Ag exposures. Ag-induced aggregation of IgE-bound FcɛRI causes the prompt release of prestored MC mediators, including histamine that can promote vasodilatation and increased vascular permeability. FcɛRI activation also induces the production and release of de novo-synthesized compounds, such as leukotrienes, prostaglandins, and proinflammatory cytokines (such as IL-13) and chemokines (not shown). Such MC-derived products contribute to intestinal inflammation (including the recruitment and activation of neutrophils, basophils, and eosinophils and other leukocytes), increased intestinal permeability and motility, and, in the case of parasite infections, worm expulsion. During food allergy, the activation of MCs also can promote diarrhea and, in some unfortunate individuals, anaphylaxis (not shown). IgG–Ag immune complexes can potentially modulate MC activation through Fcγ receptors (MCs express the activating receptor FcγRIII and the inhibitory receptor FcγRIIb). Macrophages, basophils, and neutrophils are also activated by IgG–Ag immune complexes and release PAF, which is thought to contribute to diarrhea and anaphylaxis in food allergy. Note: downregulatory mechanisms that can be engaged in these settings, such as co-engagement by multivalent Ag of both FcɛRI and FcγRIIb, or effects of regulatory T-cell populations, are not shown. APC, antigen-presenting cell; Baso, basophils; Eos, eosinophils; FcɛRI, high-affinity receptor for IgE; FcγRs: receptors for IgGs; IL, interleukin; ILC2, innate lymphoid cells type 2; Neu, neutrophils; PAF, platelet-activating factor; PRR, pattern-recognition receptor; TH2, T helper 2.
Multiple mouse models of anaphylaxis have been developed to investigate the contribution of MCs and other effector cells. Two main pathways of active anaphylaxis have been described in mice: a “classical” pathway consisting of antigens, IgE, FcɛRI, MCs, and histamine, and an “alternative” pathway involving IgG-antigen immune complexes, FcγRIII, platelet-activating factor (PAF), and, depending on the exact model used, macrophages, basophils, and/or neutrophils.239, 240, 241, 242, 243, 244, 245 Several studies using KitW/W-v and/or KitW-sh/W-sh MC-deficient mice have provided evidence that MCs can contribute significantly to peanut-induced active anaphylaxis.242, 244, 245 We recently confirmed these findings using c-kit-independent MC-deficient mice, by showing that selective ablation of CTMCs (induced by repeated injections of diphtheria toxin in Mcpt5-Cre; iDTR mice168) significantly diminished the hypothermia induced by peanut challenge in mice sensitized orally with peanut together with the mucosal adjuvant cholera toxin.243 However, antigen challenge induced significant hypothermia (albeit less than that in the corresponding WT mice) in Cpa3-Cre+; Mcl-1fl/fl mice, which have a marked MC deficiency and a substantial reduction in basophils.243 Antigen-induced elevations in serum histamine were abolished in MC- and basophil-deficient Cpa3-Cre+; Mcl-1fl/fl mice, whereas small but significant increases in PAF levels were still detected in spleen specimens from these mice.243 Together these findings implicate the involvement of both the classical and alternative pathways of anaphylaxis in this mouse model of peanut-induced active anaphylaxis.
The reaction to peanut in some mouse models might be even more complex, as Khodoun et al.246 found that peanut, but not milk or egg proteins, can induce shock reactions through an innate immune mechanism in mice. The authors found that this response was almost absent in mice lacking the complement factor C3 or the receptor C3aR, but developed fully in antibody-deficient Rag1 mice (which lack mature T and B cells) and μMT mice (in which the development of conventional B cells is arrested at the pro-B-cell stage).246 However, some reports indicate that μMT mice have B1 B cells and can produce IgE and IgG.247, 248, 249 Macrophages, basophils, and PAF contributed to this shock reaction to a greater extent than did MCs and histamine.246 Therefore, depending on the model used, innate components might also participate importantly in peanut-induced anaphylaxis, which perhaps accounts for the fact that peanut allergy is more likely than most other forms of food allergy to cause lethal anaphylaxis. However, it is important to recognize that Khodoun et al.246 increased the sensitivity of the mice to develop shock reactions in these experiments by pretreating the animals intravenously with a long-acting form of IL-4 (consisting on IL-4/anti–IL-4 mAb complexes, which slowly dissociate in vivo to release free IL-4) and with the β-adrenergic antagonist propranolol.
Although IgE-dependent activation of MCs is widely thought to contribute importantly to anaphylaxis in humans, subjects with food allergy-associated anaphylaxis, unlike those with insect venom-induced anaphylaxis, typically exhibit little or no elevations in blood levels of the MC-associated protease, tryptase.250 By contrast, levels of PAF in the serum have been directly correlated with the severity of organ system involvement in patients with acute allergic reactions triggered by foods, medications, or insect stings.251, 252 Moreover, the serum activity of PAF acetylhydrolase (an enzyme that converts PAF to the biologically inactive lyso-PAF) was significantly lower in peanut-allergic patients with fatal peanut anaphylaxis than in those with mild allergic reactions to peanuts or in the control groups.251 Although they do not constitute proof, these results are consistent with the possibility that activation of both the “classical” pathway and the “alternative” pathway might be involved in at least some examples of anaphylaxis in humans. The existence of IgG-mediated anaphylaxis in humans is perhaps best supported by the occurrence of anaphylaxis in patients infused with monoclonal antibodies, such as the chimeric mouse/human anti-TNF mAb infliximab.239, 253 One study showed that 11 out of 165 patients with Crohn’s disease treated with infliximab developed signs of anaphylaxis. All these patients had IgG antibodies to the mouse immunoglobulin determinants on infliximab. Although none of the patients had detectably increased serum levels of total IgE, the authors did not report whether they attempt to measure levels of infliximab-specific IgE. However, none of these patients had increased tryptase levels in blood 20 min after the onset of the reaction.239, 253
Anaphylaxis represents the extreme end of a spectrum of responses to food allergens in allergic patients. In most patients, reactions are manifested mainly by local signs and symptoms, and the skin is affected in ∼80% of subjects.254 Up to 50% of patients also develop gastrointestinal symptoms (abdominal pain, vomiting, and diarrhea) and a significant portion of patients also experience respiratory symptoms (cough, chest tightness, and wheezing).255, 256 Multiple lines of evidence suggest that IgE-dependent MC activation can have an important role in these local manifestations of food allergy. Cafarelli et al.256, 257 found elevated numbers of IgE-positive cells (plasma cells and 2.7% MCs) in duodenal biopsies from children with food allergies, whereas MCs were virtually absent in the control biopsies. Moreover, when stimulated ex vivo with anti-IgE, intestinal MCs obtained from enzymatically dispersed duodenal biopsies from food allergic patients released more histamine in comparison with cells from nonallergic individuals.256, 258
Brandt et al.48 developed a mouse model of allergen-induced gastrointestinal inflammation consisting of sensitization with OVA together with alum and repeated oral challenges with OVA. In this model, sensitized and challenged BALB/c mice (but not C57BL/6 mice) developed large increases in numbers of MMCs in the jejunum, ileum, and colon and increased levels of MCPT1 in the plasma. These mice also exhibited a strong Th2 response in the intestine, with signs of allergy such as diarrhea and increased intestinal permeability, but without hypothermia.48 However, systemic (intravenous) OVA challenge of OVA/alum-sensitized mice induced hypothermia that was significantly more severe in animals which had been previously challenged with OVA intragastrically compared with those mock challenged with saline. Notably, lethal anaphylactic shock occurred only in mice that previously had developed gastrointestinal allergy, suggesting that gastrointestinal allergic inflammation can prime mice for more severe anaphylaxis following systemic antigen challenge.48 The authors showed that treatment with an anti-KIT antibody (ACK2) abrogated the diarrhea, diminished intestinal permeability, and eliminated MMCs in the jejunum.48 These features were also diminished in mice treated with an anti-IgE antibody and in mice deficient for the high-affinity IgE receptor FcɛRI (but not in mice treated with a blocking antibody against the IgG receptors FcγRII/III). Finally, they demonstrated that treatment with a combination of pharmacological inhibitors of PAF and serotonin blocked diarrhea, whereas the blockade of histamine had no effect on diarrhea.48 Wang et al.259 reported that, in a model of peanut allergy in BALB/c mice, allergen-induced diarrhea and other features of the response were also partially diminished in mice deficient for the FcɛRIα chain. Adoptive transfer of WT BMCMCs, but not FcɛRIα−/− or Il-13−/− BMCMCs, restored diarrhea in FcɛRIα-deficient mice, suggesting that this feature is dependent on IgE-mediated activation of MCs and on the release of IL-13 by MCs.
Little is known about the mechanism(s) leading to sensitization with food allergens. Forbes et al.260 showed that transgenic mice that overexpress IL-9 have increased numbers of intestinal MMCs, associated with an increased intestinal permeability, which can enhance oral sensitization to OVA administered without an adjuvant. Epidemiologic studies have demonstrated that cutaneous inflammation associated with atopic dermatitis is a significant risk factor for the development of food allergies.261, 262, 263 Recently, Noti et al.263 reported that epicutaneous sensitization of mice to food antigens (OVA or peanut extract) applied to an atopic dermatitis-like skin lesion (which leads to increased levels of TSLP in the skin) followed by an oral challenge with the antigen promoted intestinal Th2-driven inflammation and increased numbers of intestinal MMCs. Such features are much diminished in mice deficient for the TLSP receptor or IgE, or in mice in which basophils have been depleted (but the authors did not assess responses of MC-deficient mice in this model). These results indicate that a “TSLP-basophil axis” can contribute to the development of IgE-mediated intestinal MMCs expansion and food allergy in mice sensitized epicutaneously with food allergens.263
Burton et al.264 recently developed an adjuvant-free model of peanut allergy using mice with a disinhibiting mutation in the IL-4 receptor α chain (il4raF709 mice), which results in amplified signaling on interaction of the receptor with the Th2 cytokines IL-4 or IL-13 but not constitutive activation. Oral sensitization of il4raF709 mice with peanut, followed by oral challenge with peanut, led to the expansion and activation of intestinal MMCs, and the development of diarrhea, intestinal inflammation, and hypothermia. The authors used MC-deficient Mcpt5-Cre; DTA mice and IgE-deficient mice to demonstrate that, in this model, both MCs and IgE were required for induction of antibody and Th2-cell-mediated responses to peanut ingestion, as well as for the suppression of expansion of regulatory T (Treg) cells. MC-targeted genetic deletion of the FcɛRI signaling kinase Syk in Mcpt5-Cre;Sykfl/fl mice also prevented peanut sensitization. Therefore, in addition to their key effector role during many allergic reactions, under certain circumstances, MCs and IgE also appear to be able to amplify sensitization to certain food allergens such as peanut, as well as participate in the suppression of tolerance.
Roles of Mast Cells in Defense Against Mucosal Pathogens
MCs are located at sites exposed to invading pathogens, such as the skin, the gut, the lung, and the genitourinary mucosa. MCs are, therefore, likely to be among the first innate cells (together with macrophages and dendritic cells) to respond to such pathogens. Studies in mice indicate that MCs can contribute to multiple defense strategies against various pathogens, including parasites (Figure 2), bacteria, and viruses,79, 265, 266, 267, 268 but that, in certain settings, MCs can contribute to the pathology associated with such infections.
Parasite infections
Parasite infections that involve the intestines and provoke the development of Th2 responses are often associated with a large expansion in MMCs in rodents,269, 270, 271 and with an expansion of MMC populations in monkeys272 and humans.273 Space does not permit a comprehensive discussion of the complex innate and adaptive immune mechanisms that are thought to contribute to helminth clearance.274, 275, 276, 277 Instead, we will review briefly some of the evidence indicating that MCs can influence aspects of these responses. Woodbury et al.269 demonstrated that, in rats infected with T. spiralis or Nippostrongylus brasiliensis, systemic secretion of the rat MC-associated chymase rMCP-2 coincides with the immune expulsion of these nematodes. Many groups have assessed the responses of KitW/W-v and/or KitW-sh/W-sh mice to primary infection with various parasites, including N. brasiliensis,278, 279 Strongyloides ratti,280 Strongyloides venezuelensis,51, 281, 282 T. spiralis,283, 284 and Trichinella muris.285, 286 Most of these studies show that such c-kit mutant MC-deficient mice have a delay in intestinal worm clearance during the primary infection. However, due to the inability to engraft intestinal MMCs in such c-kit mutant mice by the systemic adoptive transfer of MCs,155, 177, 287, 288 it is not possible to know to what extent the delays in parasite clearance detected in these MC-deficient mice reflect their lack of MMCs vs. one or more of their other phenotypic abnormalities (including their deficiency on intestinal cells of Cajal, which results in abnormal gut motility164).
However, other lines of evidence support an important contribution for MCs in intestinal worm clearance. Ha et al.283 showed that engraftment with total BM cells accelerated expulsion of T. spiralis in KitW/W-v mice. Expulsion of T. spiralis was significantly delayed in mice lacking the chymase MCPT1, which suggests an important contribution of intestinal MMCs and MCPT1 in the clearance of this infection.271 Although the kinetics of T. spiralis expulsion from the small intestine were similar between MCPT6-deficient and WT mice, the MCPT6-deficient mice had diminished levels of eosinophils in infected skeletal muscle.289 Recently, Blankenhaus et al.290 showed that c-kit-independent MC-deficient BALB/c-Cpa3Cre/+ mice (which, beside their MC deficiency, also have reduced basophil numbers172) exhibited increased parasite burden in the small intestine following infection with S. ratti.
Although the results described above suggest potentially important roles for MMCs and some of their associated chymases in worm expulsion, it is possible that in some parasite infections the effects of MCs might actually favor the parasite. For example, anti-SCF treatment diminished intestinal MMC hyperplasia in rats infected with N. brasiliensis or T. spiralis, but such anti-SCF treatment decreased parasite egg production during N. brasiliensis infection.291 Similarly, during a primary infection with N. brasiliensis, c-kit mutant MC-deficient Ws/Ws rats exhibited reduced numbers of eggs in the feces at day 8 of infection versus the correponding WT rats.292 Neither study proved that MCs were responsible for the observed effects, but the results are intriguing in suggesting that some parasites may have learned how to exploit MC-associated effector mechanisms to their own advantage.
IL-3 can promote the expansion of intestinal MMCs in mice51, 283 and treatment with IL-3 accelerates expulsion of S. ratti.283 Both KitW/W-v mice and mice lacking IL-3 exhibited a delay in S. venezuelensis expulsion, and this delay was greatly enhanced when these deficiencies were combined (i.e., in Il-3−/−; KitW/W-v mice, in which infection provoked little or no expansion of basophil or MMC populations).51 These findings indicate that one of the functions of IL-3 in this setting is to expand populations of hematopoietic effector cells, and are consistent with the possibility that both MCs and basophils contribute to expulsion of S. venezuelensis during the primary infection.
IL-9 also has an important role in the expansion of intestinal MMCs during parasite infection and transgenic mice that overexpress IL-9 have increased intestinal MMCs and increased efficiency of worm expulsion during infection with T. spilaris.181 There is evidence that IL-9-mediated MC activation is also a key mechanism mediating S. ratti expulsion in mice.290 This mechanism cannot be generalized to all parasites, as in the case of infection with N. brasiliensis, it appears that neither IL-9 nor MCPT1 influences worm expulsion.271, 293
Most studies of the roles of MCs in parasite infection have focused on the primary responses to the infection (Figure 2). Many parasites induce strong antibody responses, including high levels of antigen nonspecific IgE as well as antigen-specific IgE and IgG antibodies, and secondary infections are often associated with a more rapid expulsion of the parasites than occurs in the primary infection.294, 295, 296 However, it is not yet clear to what extent the interactions of such antibodies with MCs importantly contribute to such secondary responses. Although there have been few studies of secondary parasite infections in genetically MC-deficient mice, numbers of MMCs and serum MCPT1 levels were significantly higher in BALB/c WT mice at day 3 after secondary vs. primary infections with T. spiralis, and worm burden at that time was significantly less in the secondary than in the primary infection.181 Given that the co-engagement of FcγRIIB with FcɛRI can diminish antigen-dependent MC activation,297, 298 it will be important to investigate whether this or other mechanisms can downregulate or otherwise alter MC responses during secondary parasite infections, as well as to determine whether MCs can confer benefits to the host or the parasite in such settings.
Bacterial infections
Several studies have indicated that MCs can have an important role in enhancing survival during models of experimental bacterial sepsis in mice. Many of these data were obtained using the cecal ligation and puncture (CLP) model in which commensal bacteria are allowed to escape from the cecum into the peritoneum, and most of the studies employing MC-deficient mice have used KitW/W-v and/or KitW-sh/W-sh mice, which have multiple defects in immune responses other than their MC deficiency.80, 158, 299, 300, 301, 302, 303, 304, 305 Experiments assessing responses of MC-deficient KitW/W-v mice engrafted with WT or various mutant BMCMCs have demonstrated that such MCs are mainly activated through TLR4 (but not TLR-2) during polymicrobial sepsis,80 and that MC-derived IL-12,306 as well as MC expression of the cysteinyl protease dipeptidyl peptidase I302 and the transcription factor Smad3,307 are also required for optimal survival during the CLP model.
MCs also can be activated by the endogenous peptide endothelin-1 (ET-1), primarily through the ET(A) receptor. Activation by ET-1 promotes MC degranulation and the release of proteases, which in turn can degrade ET-1. MC protease-dependent degradation of ET-1 can contribute to optimal survival during CLP, which is associated with markedly elevated levels of ET-1,301 and CPA3 is the critical protease that mediates degradation of ET-1.81, 83 Other MC-associated proteases also have been implicated in defense against bacteria. Studies in Mcpt4−/− mice indicate that the chymase MCPT4 has effects that can enhance survival in a moderately severe model of CLP, perhaps, in part, through the degradation of TNF.305 Orinska et al.308 reported evidence that intracellular IL-15 expression in MCs can transcriptionally limit the amount of the chymase MCPT2 in the cells, resulting in decreased MC antibacterial properties and reduced survival of the mice after CLP.
MCs can mediate neutrophil recruitment after intraperitoneal (i.p.) injection of Klebsiella pneumoniae, probably via multiple mechanisms including the release of TNF,133 IL-6,304 and the tryptase MCPT6.309, 310 There is evidence that MCs can enhance resistance to pulmonary infection with Mycoplasma pneumonia.311 Histamine has an important role in this model, but neutrophils, rather than MCs, were the major source of histamine in the lungs of the infected mice.115 MCs also can contribute to Clostridium difficile toxin A-induced intestinal fluid secretion and neutrophil infiltration.312 Malaviya et al.313 reported that, during infection with E. coli, neutrophil recruitment and bacterial clearance is controlled by JAK3 activation in MCs; this effect was attributed to the diminished ability of Jak3−/− MCs to produce TNF. By contrast, there is evidence from work in MC knock-in mice that MC-derived TNF can enhance bacterial growth and hasten death after i.p. inoculation of Salmonella typhimurium.158
Urinary tract infections, mainly caused by uropathogenic E. coli, represent one of the most common bacterial infections in humans.314 Using the MC knock-in approach in c-kit mutant mice, Shelburne et al.315 demonstrated that MCs and MC-derived TNF can amplify the protective adaptive immune response to infection with uropathogenic E. coli by promoting: (1) recruitment of dendritic cells at the site of infection (in this case the footpad), (2) migration of dendritic cells into the draining lymph nodes, and (3) production of E. coli-specific IgG and IgM antibodies. Increased numbers of surviving bacteria were found in the urinary bladder of c-kit mutant MC-deficient KitW/W-v mice as compared with Kit+/+ mice following experimental infection with E. coli.316 Chan et al.317 compared the kinetics of E. coli clearance in the bladder and kidneys of infected mice and found that, although all bacteria were cleared within 5 days in the kidneys, significant numbers of bacteria were still found in the bladder as late as 1 month after infection. They demonstrated that this prolonged bacterial survival was due to production of IL-10, and the absence of significant levels of E. coli-specific antibodies, in the bladder.317 There is evidence that mouse MCs can represent an important source of IL-10 during inflammation141, 317 and that MC-derived IL-10 can: (1) limit inflammation during contact hypersensitivity141 (although these findings have been recently challenged by Dudeck et al.168) as well as (2) diminish the severity of experimental graft-versus-host disease.318 In line with these findings, Chan et al.317 demonstrated that MC-derived IL-10 contributed importantly to the suppression of E. coli-specific antibody production during experimental urinary tract infection in mice and accounted, at least in part, for the persistence of E. coli in the bladder. Therefore, MCs may have a dual role during E. coli infections in the bladder, first reducing numbers of bacteria by promoting aspects of the initial response to infection but later limiting the antibody response to E. coli by producing IL-10.317
Because c-kit mutant MC-deficient mice have many c-kit-related phenotypic abnormalities that may influence the responses of such animals to infection (including those which do or do not affect MC numbers or functions),152, 158, 175 it will be of great interest to continue to evaluate the roles of MCs in infection models using some of the newer, c-kit-independent, models of MC deficiency. For example, Rönnberg et al.319 recently reported that peritoneal MCs are activated by Staphylococcus aureus in vitro; however, the authors used c-kit-independent MC-deficient Mcpt5-Cre; DTA+ mice to demonstrate that MCs do not influence the in vivo manifestations of one model of i.p. S. aureus infection. Such work will help to clarify which roles of MCs are variably redundant with those of other cell types (including neutrophils or macrophages, among others) and which MC roles—whether to enhance and/or suppress aspects of these innate or acquired immune responses—may be nonredundant.
Viral infections
MCs have been implicated in the defense against certain viruses, although there have been relatively few studies in this area.79 Sendai virus can induce histamine release from rat peritoneal MCs ex vivo320 and infection of rats with Sendai virus results in increased numbers of MCs in the lung.321, 322 Kulka et al. showed that human peripheral blood-derived cultured MCs and two lines of human MCs (LAD and HMC-1), as well as mouse BMCMCs, can respond to stimulation with dsRNA (poly(I:C)) by producing type I IFN-α through TLR3. They also found that human peripheral blood-derived cultured MCs can produce IFN-α when stimulated with live respiratory syncytial virus, reovirus type 1, or ultraviolet-inactivated influenza virus.323 It has been reported, based on studies in MC knock-in KitW/W-v mice, that MCs can promote the recruitment of CD8+ T cells following i.p. injection of poly(I:C).324
Several reports suggest that MCs can contribute to the pathology induced by some viruses in vivo. Both MC-deficient KitW/W-v mice and KitlSl/Sl-d mice exhibited reduced myocardial inflammation and necrosis as well as increased survival as compared with the respective littermate WT mice after i.p. infection with encephalomyocarditis virus.325 Furthermore, adoptive transfer of BMCMCs into KitW/W-v mice or repeated subcutaneous treatment of KitlSl/Sl-d mice with recombinant SCF (which can induce the appearance of both CTMCs and MMCs in these mice63, 326) significantly increased the histopathological severity of the myocardial lesions induced by the virus (albeit not to levels observed in WT mice).325 By contrast, studies in KitW-sh/W-sh mice, including engraftment of these mice with BMCMCs, showed that MCs can participate in host defense against vaccinia virus, and MC production of the antimicrobial peptide cathelicidin was implicated as a key defense mechanism against this virus.327, 328
In humans, infection with dengue virus leads to increased levels of MC chymase in the serum,329 and chymase levels are significantly higher in patients with severe dengue fever (also known as dengue hemorrhagic fever) as compared with patients with dengue fever.329, 330 Using the MC knock-in model in KitW-sh/W-sh mice, St John et al.329, 330, 331 demonstrated that MCs can promote the recruitment of NK (natural killer) and NK T cells during dengue virus infection in mice. Ebert et al.332 recently used a similar approach to demonstrate that MCs can contribute to clearance of pulmonary murine cytomegalovirus in the lung by enhancing the recruitment of CD8+ T cells to the infection site.
There is evidence that MCs may have roles in human immunodeficiency virus infection. In vitro experiments show that the human immunodeficiency virus gp120 envelope protein can promote production of Th2 cytokines (IL-4 and IL-5) in human MCs.323 MCs and their progenitors might also serve as a reservoir for latent virus, a role which would be detrimental to the host.333, 334, 335, 336
In line with the potential of MCs to help in orchestrating protective adaptive responses at mucosal sites, McLachlan et al.337 demonstrated that certain small molecules (“MC activators”) are potent mucosal adjuvants, and provided evidence that these agents mediate such functions in a largely MC-dependent manner. The so-called “MC activators” comprise a family of structurally diverse cationic peptides and polymeric compounds that can induce strong MC degranulation;338, 339 such agents include compound 48/80 (c48/80),340, 341, 342 and a variety of peptide toxins, such as MC-degranulating peptide, which is found in honeybee and bumblebee venoms.343 Using the MC knock-in system in c-kit mutant mice, McLachlan et al. demonstrated that compound 48/80 (which promotes MC degranulation, but also has other effects) can act as a potent mucosal adjuvant when coadministered in the footpad with recombinant anthrax protective antigen, and that this adjuvant effect largely depends on MCs and MC-derived TNF. Importantly, vaccination with c48/80 coadministered with the vaccinia virus antigen B5R intranasally conferred protection against intranasal challenge with a normally lethal dose of vaccinia virus.337
Conclusions
We are in the midst of an interesting period in MC research. For many years, an increasing understanding of the diversity of MC products, signaling mechanisms, and interactions with other cell types has led to the generation of many attractive hypotheses about the diverse potential effector and immunoregulatory roles of MCs in the biology and pathology of mucosal tissues (and in other settings). Increasingly, these hypotheses are being tested in ways that permit us to accrue definitive evidence regarding the nature, and the importance, of such proposed MC roles. In addition to long-established mouse model systems, including “MC knock-in c-kit mutant mice” and various MC protease-deficient mice, there are now many promising new models of constitutive or inducible MC deficiency, as well as many new models for achieving the targeted deletion of individual products in MCs. On the basis of the results obtained so far with both the older and newer models for MC research, we think that the most robust conclusions about the nature and importance of the roles of MCs in various biological responses in vivo, in mucosal tissues and other sites, probably will be derived from studies employing multiple informative model systems.152 Taken together, such approaches offer many opportunities to obtain increasingly solid evidence to support (or discard) notions about how MCs might influence the development, physiology, homeostasis, immunology, and pathology of mucosal tissues.
It hardly needs mentioning that findings in mice do not prove that the same processes occur in humans, and there are likely to be multiple differences in the details of immune responses and disease pathogenesis in the two species, not just differences in the roles of MCs in such settings. However, preclinical studies using models in which individual cells or products can be manipulated offer the promise of revealing pathways that, with luck, might be exploited to provide benefit to those suffering from any of the diverse mucosal pathologies in which MCs have been implicated. Time will tell to what extent this hope will be realized.
References
Mekori, Y.A. & Metcalfe, D.D. Mast cells in innate immunity. Immunol. Rev. 173, 131–140 (2000).
Metz, M., Grimbaldeston, M.A., Nakae, S., Piliponsky, A.M., Tsai, M. & Galli, S.J. Mast cells in the promotion and limitation of chronic inflammation. Immunol. Rev. 217, 304–328 (2007).
Galli, S.J., Grimbaldeston, M. & Tsai, M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat. Rev. Immunol. 8, 478–486 (2008).
Moon, T.C. et al. Advances in mast cell biology: new understanding of heterogeneity and function. Mucosal Immunol. 3, 111–128 (2010).
Gurish, M.F. & Austen, K.F. Developmental origin and functional specialization of mast cell subsets. Immunity 37, 25–33 (2012).
Kalesnikoff, J. & Galli, S.J. New developments in mast cell biology. Nat. Immunol. 9, 1215–1223 (2008).
Galli, S.J., Nakae, S. & Tsai, M. Mast cells in the development of adaptive immune responses. Nat. Immunol. 6, 135–142 (2005).
Galli, S.J., Kalesnikoff, J., Grimbaldeston, M.A., Piliponsky, A.M., Williams, C.M. & Tsai, M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786 (2005).
Kitamura, Y., Shimada, M., Hatanaka, K. & Miyano, Y. Development of mast cells from grafted bone marrow cells in irradiated mice. Nature 268, 442–443 (1977).
Kitamura, Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu. Rev. Immunol. 7, 59–76 (1989).
Douaiher, J. et al. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv. Immunol. 122, 211–252 (2014).
Oliveira, S.H. & Lukacs, N.W. Stem cell factor: a hemopoietic cytokine with important targets in asthma. Curr. Drug Targets Inflamm. Allergy 2, 313–318 (2003).
Reber, L., Da Silva, C.A. & Frossard, N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur. J. Pharmacol. 533, 327–340 (2006).
Galli, S.J., Zsebo, K.M. & Geissler, E.N. The kit ligand, stem cell factor. Adv. Immunol. 55, 1–96 (1994).
Razin, E. et al. Interleukin 3: a differentiation and growth factor for the mouse mast cell that contains chondroitin sulfate E proteoglycan. J. Immunol. 132, 1479–1486 (1984).
Toru, H., Ra, C., Nonoyama, S., Suzuki, K., Yata, J. & Nakahata, T. Induction of the high-affinity IgE receptor (FcɛRI) on human mast cells by IL-4. Int. Immunol. 8, 1367–1373 (1996).
Valent, P. et al. Interleukin 4 promotes expression of mast cell ICAM-1 antigen. Proc. Natl. Acad. Sci. USA 88, 3339–3342 (1991).
Sillaber, C. et al. IL-4 regulates c-kit proto-oncogene product expression in human mast and myeloid progenitor cells. J. Immunol. 147, 4224–4228 (1991).
Toru, H., Eguchi, M., Matsumoto, R., Yanagida, M., Yata, J. & Nakahata, T. Interleukin-4 promotes the development of tryptase and chymase double-positive human mast cells accompanied by cell maturation. Blood 91, 187–195 (1998).
Matsuzawa, S., Sakashita, K., Kinoshita, T., Ito, S., Yamashita, T. & Koike, K. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J. Immunol. 170, 3461–3467 (2003).
Mwamtemi, H.H. et al. An increase in circulating mast cell colony-forming cells in asthma. J. Immunol. 166, 4672–4677 (2001).
Bailey, D.P., Kashyap, M., Bouton, L.A., Murray, P.J. & Ryan, J.J. Interleukin-10 induces apoptosis in developing mast cells and macrophages. J. Leukoc. Biol. 80, 581–589 (2006).
Royer, B., Varadaradjalou, S., Saas, P., Guillosson, J.J., Kantelip, J.P. & Arock, M. Inhibition of IgE-induced activation of human mast cells by IL-10. Clin. Exp. Allergy 31, 694–704 (2001).
Yeatman, C.F. II et al. Combined stimulation with the T helper cell type 2 cytokines interleukin (IL)-4 and IL-10 induces mouse mast cell apoptosis. J. Exp. Med. 192, 1093–1103 (2000).
Ho, L.H. et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcɛRI signals. J. Leukoc. Biol. 82, 1481–1490 (2007).
Iikura, M. et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab. Invest. 87, 971–978 (2007).
Allakhverdi, Z., Smith, D.E., Comeau, M.R. & Delespesse, G. Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 179, 2051–2054 (2007).
Lin, T.J., Issekutz, T.B. & Marshall, J.S. Human mast cells transmigrate through human umbilical vein endothelial monolayers and selectively produce IL-8 in response to stromal cell-derived factor-1α. J. Immunol. 165, 211–220 (2000).
Godot, V. et al. H4 histamine receptor mediates optimal migration of mast cell precursors to CXCL12. J. Allergy Clin. Immunol. 120, 827–834 (2007).
Macey, M.R. et al. IL-4 and TGF-β1 counterbalance one another while regulating mast cell homeostasis. J. Immunol. 184, 4688–4695 (2010).
Matsuda, H. et al. Nerve growth factor induces development of connective tissue-type mast cells in vitro from murine bone marrow cells. J. Exp. Med. 174, 7–14 (1991).
Ruitenberg, E.J. & Elgersma, A. Absence of intestinal mast cell response in congenitally athymic mice during Trichinella spiralis infection. Nature 264, 258–260 (1976).
Friend, D.S., Ghildyal, N., Austen, K.F., Gurish, M.F., Matsumoto, R. & Stevens, R.L. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J. Cell Biol. 135, 279–290 (1996).
Friend, D.S. et al. Reversible expression of tryptases and chymases in the jejunal mast cells of mice infected with Trichinella spiralis. J. Immunol. 160, 5537–5545 (1998).
Hunt, J.E., Stevens, R.L., Austen, K.F., Zhang, J., Xia, Z. & Ghildyal, N. Natural disruption of the mouse mast cell protease 7 gene in the C57BL/6 mouse. J. Biol. Chem. 271, 2851–2855 (1996).
Jippo, T., Lee, Y.M., Ge, Y., Kim, D.K., Okabe, M. & Kitamura, Y. Tissue-dependent alteration of protease expression phenotype in murine peritoneal mast cells that were genetically labeled with green fluorescent protein. Am. J. Pathol. 158, 1695–1701 (2001).
Galli, S.J., Borregaard, N. & Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat. Immunol. 12, 1035–1044 (2011).
Irani, A.M., Bradford, T.R., Kepley, C.L., Schechter, N.M. & Schwartz, L.B. Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti-tryptase and anti-chymase antibodies. J. Histochem. Cytochem. 37, 1509–1515 (1989).
Li, L., Meng, X.W. & Krilis, S.A. Mast cells expressing chymase but not tryptase can be derived by culturing human progenitors in conditioned medium obtained from a human mastocytosis cell strain with c-kit ligand. J. Immunol. 156, 4839–4844 (1996).
Weidner, N. & Austen, K.F. Heterogeneity of mast cells at multiple body sites. Fluorescent determination of avidin binding and immunofluorescent determination of chymase, tryptase, and carboxypeptidase content. Pathol. Res. Pract. 189, 156–162 (1993).
Irani, A.M., Craig, S.S., DeBlois, G., Elson, C.O., Schechter, N.M. & Schwartz, L.B. Deficiency of the tryptase-positive, chymase-negative mast cell type in gastrointestinal mucosa of patients with defective T lymphocyte function. J. Immunol. 138, 4381–4386 (1987).
Irani, A.A., Schechter, N.M., Craig, S.S., DeBlois, G. & Schwartz, L.B. Two types of human mast cells that have distinct neutral protease compositions. Proc. Natl. Acad. Sci. USA 83, 4464–4468 (1986).
Matin, R., Tam, E.K., Nadel, J.A. & Caughey, G.H. Distribution of chymase-containing mast cells in human bronchi. J. Histochem. Cytochem. 40, 781–786 (1992).
Bischoff, S.C. Physiological and pathophysiological functions of intestinal mast cells. Semin. Immunopathol. 31, 185–205 (2009).
Bischoff, S.C. et al. Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology 28, 1–13 (1996).
Raithel, M., Winterkamp, S., Pacurar, A., Ulrich, P., Hochberger, J. & Hahn, E.G. Release of mast cell tryptase from human colorectal mucosa in inflammatory bowel disease. Scand. J. Gastroenterol. 36, 174–179 (2001).
Cho, E.Y. et al. Nafamostat mesilate attenuates colonic inflammation and mast cell infiltration in the experimental colitis. Int. Immunopharmacol. 11, 412–417 (2011).
Brandt, E.B. et al. Mast cells are required for experimental oral allergen-induced diarrhea. J. Clin. Invest. 112, 1666–1677 (2003).
Hagel, A.F. et al. Mast cell tryptase levels in gut mucosa in patients with gastrointestinal symptoms caused by food allergy. Int. Arch. Allergy Immunol. 160, 350–355 (2013).
Betts, C.J. & Else, K.J. Mast cells, eosinophils and antibody-mediated cellular cytotoxicity are not critical in resistance to Trichuris muris. Parasite Immunol. 21, 45–52 (1999).
Lantz, C.S. et al. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature 392, 90–93 (1998).
Marone, G., Triggiani, M. & de Paulis, A. Mast cells and basophils: friends as well as foes in bronchial asthma? Trends Immunol. 26, 25–31 (2005).
Brightling, C.E., Bradding, P., Symon, F.A., Holgate, S.T., Wardlaw, A.J. & Pavord, I.D. Mast-cell infiltration of airway smooth muscle in asthma. N Engl. J. Med. 346, 1699–1705 (2002).
Brightling, C.E., Symon, F.A., Birring, S.S., Bradding, P., Wardlaw, A.J. & Pavord, I.D. Comparison of airway immunopathology of eosinophilic bronchitis and asthma. Thorax 58, 528–532 (2003).
Yu, M., Tsai, M., Tam, S.Y., Jones, C., Zehnder, J. & Galli, S.J. Mast cells can promote the development of multiple features of chronic asthma in mice. J. Clin. Invest. 116, 1633–1641 (2006).
Yu, M. et al. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J. Clin. Invest. 121, 3133–3143 (2011).
Kawanami, O., Ferrans, V.J., Fulmer, J.D. & Crystal, R.G. Ultrastructure of pulmonary mast cells in patients with fibrotic lung disorders. Lab. Invest. 40, 717–734 (1979).
Edwards, S.T. et al. c-Kit immunophenotyping and metalloproteinase expression profiles of mast cells in interstitial lung diseases. J. Pathol. 206, 279–290 (2005).
Yoshizaki, A. et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. Am. J. Pathol. 172, 1650–1663 (2008).
Reber, L.L., Daubeuf, F., Pejler, G., Abrink, M. & Frossard, N. Mast cells contribute to bleomycin-induced lung inflammation and injury in mice through a chymase/mast cell protease 4-dependent mechanism. J. Immunol. 192, 1847–1854 (2014).
Sonoda, T. et al. Proliferation of peritoneal mast cells in the skin of W/Wv mice that genetically lack mast cells. J. Exp. Med. 160, 138–151 (1984).
Kanakura, Y. et al. Multiple bidirectional alterations of phenotype and changes in proliferative potential during the in vitro and in vivo passage of clonal mast cell populations derived from mouse peritoneal mast cells. Blood 72, 877–885 (1988).
Tsai, M. et al. The rat c-kit ligand, stem cell factor, induces the development of connective tissue-type and mucosal mast cells in vivo. Analysis by anatomical distribution, histochemistry, and protease phenotype. J. Exp. Med. 174, 125–131 (1991).
Dvorak, A.M., Mihm, M.C. Jr . & Dvorak, H.F. Morphology of delayed-type hypersensitivity reactions in man. II. Ultrastructural alterations affecting the microvasculature and the tissue mast cells. Lab. Invest. 34, 179–191 (1976).
Dahlin, J.S. & Hallgren, J. Mast cell progenitors: origin, development and migration to tissues. Mol. Immunol. 63, 9–17 (2014).
Arinobu, Y. et al. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc. Natl. Acad. Sci. USA 102, 18105–18110 (2005).
Jones, T.G. et al. Antigen-induced increases in pulmonary mast cell progenitor numbers depend on IL-9 and CD1d-restricted NKT cells. J. Immunol. 183, 5251–5260 (2009).
Abramson, J. & Pecht, I. Regulation of the mast cell response to the type 1 Fcɛ receptor. Immunol. Rev. 217, 231–254 (2007).
Galli, S.J. & Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 18, 693–704 (2012).
Reber, L.L. & Frossard, N. Targeting mast cells in inflammatory diseases. Pharmacol. Ther. 142, 416–435 (2014).
Turner, H. & Kinet, J.P. Signalling through the high-affinity IgE receptor FcɛRI. Nature 402, B24–B30 (1999).
Rivera, J., Fierro, N.A., Olivera, A. & Suzuki, R. New insights on mast cell activation via the high affinity receptor for IgE. Adv. Immunol. 98, 85–120 (2008).
Cheng, L.E., Hartmann, K., Roers, A., Krummel, M.F. & Locksley, R.M. Perivascular mast cells dynamically probe cutaneous blood vessels to capture immunoglobulin E. Immunity 38, 166–175 (2013).
Suzuki, R. et al. Molecular editing of cellular responses by the high-affinity receptor for IgE. Science 343, 1021–1025 (2014).
Marichal, T. et al. A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity 39, 963–975 (2013).
Palm, N.W., Rosenstein, R.K., Yu, S., Schenten, D.D., Florsheim, E. & Medzhitov, R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity 39, 976–985 (2013).
Palm, N.W., Rosenstein, R.K. & Medzhitov, R. Allergic host defences. Nature 484, 465–472 (2012).
Profet, M. The function of allergy: immunological defense against toxins. Q Rev. Biol. 66, 23–62 (1991).
Abraham, S.N. & St John, A.L. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10, 440–452 (2010).
Supajatura, V. et al. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Invest. 109, 1351–1359 (2002).
Metz, M. et al. Mast cells can enhance resistance to snake and honeybee venoms. Science 313, 526–530 (2006).
Akahoshi, M. et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J. Clin. Invest. 121, 4180–4191 (2011).
Schneider, L.A., Schlenner, S.M., Feyerabend, T.B., Wunderlin, M. & Rodewald, H.R. Molecular mechanism of mast cell mediated innate defense against endothelin and snake venom sarafotoxin. J. Exp. Med. 204, 2629–2639 (2007).
Ali, H. Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol. Lett. 128, 36–45 (2010).
Schafer, B. et al. Mast cell anaphylatoxin receptor expression can enhance IgE-dependent skin inflammation in mice. J. Allergy Clin. Immunol. 131, 541–548 e1-9 (2013).
Kajiwara, N. et al. Activation of human mast cells through the platelet-activating factor receptor. J. Allergy Clin. Immunol. 125, 1137–1145 e1136 (2010).
Metz, M., Gilles, S., Geldmacher, A., Behrendt, H., Traidl-Hoffmann, C. & Maurer, M. Evidence for non-allergic mast cell activation in pollen-associated inflammation. J. Invest. Dermatol. 131, 987–990 (2011).
Cirino, G. et al. Factor Xa as an interface between coagulation and inflammation. Molecular mimicry of factor Xa association with effector cell protease receptor-1 induces acute inflammation in vivo. J. Clin. Invest. 99, 2446–2451 (1997).
Cirino, G., Cicala, C., Bucci, M.R., Sorrentino, L., Maraganore, J.M. & Stone, S.R. Thrombin functions as an inflammatory mediator through activation of its receptor. J. Exp. Med. 183, 821–827 (1996).
Enoksson, M., Lyberg, K., Moller-Westerberg, C., Fallon, P.G., Nilsson, G. & Lunderius-Andersson, C. Mast cells as sensors of cell injury through IL-33 recognition. J. Immunol. 186, 2523–2528 (2011).
Lunderius-Andersson, C., Enoksson, M. & Nilsson, G. Mast cells respond to cell injury through the recognition of IL-33. Front. Immunol. 3, 82 (2012).
Kaur, D. et al. Mast cell-airway smooth muscle crosstalk: the role of thymic stromal lymphopoietin. Chest 142, 76–85 (2011).
Zhou, Y. et al. Aryl hydrocarbon receptor controls murine mast cell homeostasis. Blood 121, 3195–3204 (2013).
Sibilano, R. et al. The aryl hydrocarbon receptor modulates acute and late mast cell responses. J. Immunol. 189, 120–127 (2012).
Pawankar, R., Okuda, M., Yssel, H., Okumura, K. & Ra, C. Nasal mast cells in perennial allergic rhinitics exhibit increased expression of the FcɛRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J. Clin. Invest. 99, 1492–1499 (1997).
Nakae, S. et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 176, 2238–2248 (2006).
Kashiwakura, J., Yokoi, H., Saito, H. & Okayama, Y. T cell proliferation by direct cross-talk between OX40 ligand on human mast cells and OX40 on human T cells: comparison of gene expression profiles between human tonsillar and lung-cultured mast cells. J. Immunol. 173, 5247–5257 (2004).
Sibilano, R. et al. Modulation of FcɛRI-dependent mast cell response by OX40L via Fyn, PI3K, and RhoA. J. Allergy Clin. Immunol. 130, 751–760 e752 (2012).
Lee, H. et al. Activation of human synovial mast cells from rheumatoid arthritis or osteoarthritis patients in response to aggregated IgG through Fcγ receptor I and Fcγ receptor II. Arthritis Rheum. 65, 109–119 (2013).
Malbec, O. & Daeron, M. The mast cell IgG receptors and their roles in tissue inflammation. Immunol. Rev. 217, 206–221 (2007).
Fantozzi, R. et al. Mast cell and neutrophil interactions: a role for superoxide anion and histamine. Agents Actions 16, 260–264 (1985).
Gri, G. et al. CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 29, 771–781 (2008).
Gaudenzio, N., Espagnolle, N., Mars, L.T., Liblau, R., Valitutti, S. & Espinosa, E. Cell-cell cooperation at the T helper cell/mast cell immunological synapse. Blood 114, 4979–4988 (2009).
Karra, L. & Levi-Schaffer, F. Down-regulation of mast cell responses through ITIM containing inhibitory receptors. Adv. Exp. Med. Biol. 716, 143–159 (2011).
Katz, H.R. Inhibitory receptors and allergy. Curr. Opin. Immunol. 14, 698–704 (2002).
Ott, V.L. & Cambier, J.C. Activating and inhibitory signaling in mast cells: new opportunities for therapeutic intervention? J. Allergy Clin. Immunol. 106, 429–440 (2000).
Hill, P.B., MacDonald, A.J., Thornton, E.M., Newlands, G.F., Galli, S.J. & Miller, H.R. Stem cell factor enhances immunoglobulin E-dependent mediator release from cultured rat bone marrow-derived mast cells: activation of previously unresponsive cells demonstrated by a novel ELISPOT assay. Immunology 87, 326–333 (1996).
Ito, T. et al. Stem cell factor programs the mast cell activation phenotype. J. Immunol. 188, 5428–5437 (2012).
Komai-Koma, M. et al. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naive mice. Allergy 67, 1118–1126 (2012).
Okayama, Y., Kirshenbaum, A.S. & Metcalfe, D.D. Expression of a functional high-affinity IgG receptor, FcγRI, on human mast cells: up-regulation by IFN-γ. J. Immunol. 164, 4332–4339 (2000).
Riley, J.F. Histamine in tissue mast cells. Science 118, 332 (1953).
Razin, E. et al. IgE-mediated release of leukotriene C4, chondroitin sulfate E proteoglycan, β-hexosaminidase, and histamine from cultured bone marrow-derived mouse mast cells. J. Exp. Med. 157, 189–201 (1983).
Windelborg Nielsen, B., Engberg, T.M., Herlin, T., Bjerke, T. & Schiotz, P.O. Histamine release from cord blood basophils. Int. Arch. Allergy Appl. Immunol. 93, 314–322 (1990).
Ghosh, A.K., Hirasawa, N., Ohtsu, H., Watanabe, T. & Ohuchi, K. Defective angiogenesis in the inflammatory granulation tissue in histidine decarboxylase-deficient mice but not in mast cell-deficient mice. J. Exp. Med. 195, 973–982 (2002).
Xu, X. et al. Neutrophil histamine contributes to inflammation in mycoplasma pneumonia. J. Exp. Med. 203, 2907–2917 (2006).
Caughey, G.H. Mast cell proteases as protective and inflammatory mediators. Adv. Exp. Med. Biol. 716, 212–234 (2011).
Pejler, G., Ronnberg, E., Waern, I. & Wernersson, S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood 115, 4981–4990 (2010).
Pejler, G., Abrink, M., Ringvall, M. & Wernersson, S. Mast cell proteases. Adv. Immunol. 95, 167–255 (2007).
Huang, C. et al. Induction of a selective and persistent extravasation of neutrophils into the peritoneal cavity by tryptase mouse mast cell protease 6. J. Immunol. 160, 1910–1919 (1998).
Compton, S.J., Cairns, J.A., Holgate, S.T. & Walls, A.F. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1β and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J. Immunol. 161, 1939–1946 (1998).
Algermissen, B., Laubscher, J.C., Bauer, F. & Henz, B.M. Purification of mast cell proteases from murine skin. Exp. Dermatol. 8, 413–418 (1999).
Compton, S.J., Cairns, J.A., Holgate, S.T. & Walls, A.F. Human mast cell tryptase stimulates the release of an IL-8-dependent neutrophil chemotactic activity from human umbilical vein endothelial cells (HUVEC). Clin. Exp. Immunol. 121, 31–36 (2000).
Gallwitz, M., Reimer, J.M. & Hellman, L. Expansion of the mast cell chymase locus over the past 200 million years of mammalian evolution. Immunogenetics 58, 655–669 (2006).
Tchougounova, E., Pejler, G. & Abrink, M. The chymase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. A role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J. Exp. Med. 198, 423–431 (2003).
Andersson, M.K., Karlson, U. & Hellman, L. The extended cleavage specificity of the rodent β-chymases rMCP-1 and mMCP-4 reveal major functional similarities to the human mast cell chymase. Mol. Immunol. 45, 766–775 (2008).
Walsh, L.J., Trinchieri, G., Waldorf, H.A., Whitaker, D. & Murphy, G.F. Human dermal mast cells contain and release tumor necrosis factor α, which induces endothelial leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. USA 88, 4220–4224 (1991).
Frangogiannis, N.G. et al. Resident cardiac mast cells degranulate and release preformed TNF-α, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 98, 699–710 (1998).
Gordon, J.R. & Galli, S.J. Mast cells as a source of both preformed and immunologically inducible TNF-α/cachectin. Nature 346, 274–276 (1990).
Kunder, C.A. et al. Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J. Exp. Med. 206, 2455–2467 (2009).
Austen, K.F. The mast cell and the cysteinyl leukotrienes. Novartis Found. Symp. 271, 166–175 discussion 176-168, 198-169 (2005).
Boyce, J.A. Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol. Rev. 217, 168–185 (2007).
Dreskin, S.C. & Abraham, S.N. Production of TNF-α by murine bone marrow derived mast cells activated by the bacterial fimbrial protein, FimH. Clin. Immunol. 90, 420–424 (1999).
Malaviya, R., Ikeda, T., Ross, E. & Abraham, S.N. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature 381, 77–80 (1996).
Gordon, J.R. & Galli, S.J. Promotion of mouse fibroblast collagen gene expression by mast cells stimulated via the FcɛRI. Role for mast cell-derived transforming growth factor β and tumor necrosis factor α. J. Exp. Med. 180, 2027–2037 (1994).
Burd, P.R. et al. Interleukin 3-dependent and -independent mast cells stimulated with IgE and antigen express multiple cytokines. J. Exp. Med. 170, 245–257 (1989).
Hagaman, D.D., Okayama, Y., D’Ambrosio, C., Prussin, C., Gilfillan, A.M. & Metcalfe, D.D. Secretion of interleukin-1 receptor antagonist from human mast cells after immunoglobulin E-mediated activation and after segmental antigen challenge. Am. J. Respir. Cell Mol. Biol. 25, 685–691 (2001).
Lin, T.J., Garduno, R., Boudreau, R.T. & Issekutz, A.C. Pseudomonas aeruginosa activates human mast cells to induce neutrophil transendothelial migration via mast cell-derived IL-1 α and β. J. Immunol. 169, 4522–4530 (2002).
Oldford, S.A., Haidl, I.D., Howatt, M.A., Leiva, C.A., Johnston, B. & Marshall, J.S. A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. J. Immunol. 185, 7067–7076 (2010).
Liu, J. et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 15, 940–945 (2009).
Ishizuka, T., Okayama, Y., Kobayashi, H. & Mori, M. Interleukin-10 is localized to and released by human lung mast cells. Clin. Exp. Allergy 29, 1424–1432 (1999).
Grimbaldeston, M.A., Nakae, S., Kalesnikoff, J., Tsai, M. & Galli, S.J. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat. Immunol. 8, 1095–1104 (2007).
Song, C., Zhang, Q., Liu, X. & Shan, Y. IL-12 and IL-10 production are differentially regulated by phosphatidylinositol 3-kinase in mast cells. Scand. J. Immunol. 75, 266–272 (2012).
Mrabet-Dahbi, S., Metz, M., Dudeck, A., Zuberbier, T. & Maurer, M. Murine mast cells secrete a unique profile of cytokines and prostaglandins in response to distinct TLR2 ligands. Exp. Dermatol. 18, 437–444 (2009).
Buckland, J. New role for mast cells as IL-17-expressing effector cells in established RA. Nat. Rev. Rheumatol. 6, 243 (2010).
Hueber, A.J. et al. Mast cells express IL-17A in rheumatoid arthritis synovium. J. Immunol. 184, 3336–3340 (2010).
Boesiger, J. et al. Mast cells can secrete vascular permeability factor/vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of Fcɛ receptor I expression. J. Exp. Med. 188, 1135–1145 (1998).
Grützkau, A. et al. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF206 . Mol. Biol. Cell 9, 875–884 (1998).
Detoraki, A. et al. Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J. Allergy Clin. Immunol. 123, 1142–1149 1149 e1141-1145 (2009).
Zhang, S. et al. Human mast cells express stem cell factor. J. Pathol. 186, 59–66 (1998).
de Paulis, A. et al. Stem cell factor is localized in, released from, and cleaved by human mast cells. J. Immunol. 163, 2799–2808 (1999).
Sibilano, R., Frossi, B. & Pucillo, C.E. Mast cell activation: a complex interplay of positive and negative signaling pathways. Eur. J. Immunol. 44, 2558–2566 (2014).
Reber, L.L., Marichal, T. & Galli, S.J. New models for analyzing mast cell functions in vivo. Trends Immunol. 33, 613–625 (2012).
Oka, T., Kalesnikoff, J., Starkl, P., Tsai, M. & Galli, S.J. Evidence questioning cromolyn’s effectiveness and selectivity as a 'mast cell stabilizer' in mice. Lab. Invest. 92, 1472–1482 (2012).
Dawicki, W. & Marshall, J.S. New and emerging roles for mast cells in host defence. Curr. Opin. Immunol. 19, 31–38 (2007).
Grimbaldeston, M.A., Chen, C.C., Piliponsky, A.M., Tsai, M., Tam, S.Y. & Galli, S.J. Mast cell-deficient W-sash c-kit mutant KitW-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 167, 835–848 (2005).
Grimbaldeston, M.A., Metz, M., Yu, M., Tsai, M. & Galli, S.J. Effector and potential immunoregulatory roles of mast cells in IgE-associated acquired immune responses. Curr. Opin. Immunol. 18, 751–760 (2006).
Zhou, J.S., Xing, W., Friend, D.S., Austen, K.F. & Katz, H.R. Mast cell deficiency in Kit(W-sh mice does not impair antibody-mediated arthritis. J. Exp. Med. 204, 2797–2802 (2007).
Piliponsky, A.M. et al. Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am. J. Pathol. 176, 926–938 (2010).
Nigrovic, P.A. et al. Genetic inversion in mast cell-deficient Wsh mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 173, 1693–1701 (2008).
Chervenick, P.A. & Boggs, D.R. Decreased neutrophils and megakaryocytes in anemic mice of genotype W/Wv. J. Cell. Physiol. 73, 25–30 (1969).
Nagle, D.L., Kozak, C.A., Mano, H., Chapman, V.M. & Bucan, M. Physical mapping of the Tec and Gabrb1 loci reveals that the Wsh mutation on mouse chromosome 5 is associated with an inversion. Hum. Mol. Genet. 4, 2073–2079 (1995).
Lyon, M.F. & Glenister, P.H. A new allele sash (Wsh) at the W-locus and a spontaneous recessive lethal in mice. Genet. Res. 39, 315–322 (1982).
Wolters, P.J. et al. Tissue-selective mast cell reconstitution and differential lung gene expression in mast cell-deficient KitW-sh/KitW-sh sash mice. Clin. Exp. Allergy 35, 82–88 (2005).
Huizinga, J.D., Thuneberg, L., Kluppel, M., Malysz, J., Mikkelsen, H.B. & Bernstein, A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 373, 347–349 (1995).
Nakano, T. et al. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J. Exp. Med. 162, 1025–1043 (1985).
Galli, S.J. & Kitamura, Y. Genetically mast-cell-deficient W/Wvand Sl/Sldmice. Their value for the analysis of the roles of mast cells in biologic responses in vivo. Am. J. Pathol. 127, 191–198 (1987).
Lilla, J.N. et al. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 118, 6930–6938 (2011).
Dudeck, A. et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984 (2011).
Scholten, J. et al. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 17, 307–315 (2008).
Musch, W., Wege, A.K., Mannel, D.N. & Hehlgans, T. Generation and characterization of alpha-chymase-Cre transgenic mice. Genesis 46, 163–166 (2008).
Otsuka, A. et al. Requirement of interaction between mast cells and skin dendritic cells to establish contact hypersensitivity. PLoS One 6, e25538 (2011).
Feyerabend, T.B. et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 35, 832–844 (2011).
Furumoto, Y. et al. PTEN deficiency in mast cells causes a mastocytosis-like proliferative disease that heightens allergic responses and vascular permeability. Blood 118, 5466–5475 (2011).
Rodewald, H.R. & Feyerabend, T.B. Widespread immunological functions of mast cells: fact or fiction? Immunity 37, 13–24 (2012).
Galli, S.J., Tsai, M., Marichal, T., Chugunova, E., Reber, L.L. & Pejler, G. Approaches for analyzing the roles of mast cells and their proteases in vivo. Adv. Immunol. (in press).
Wernersson, S. & Pejler, G. Mast cell secretory granules: armed for battle. Nat. Rev. Immunol. 14, 478–494 (2014).
Groschwitz, K.R. et al. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc. Natl. Acad. Sci. USA 106, 22381–22386 (2009).
Berkes, J., Viswanathan, V.K., Savkovic, S.D. & Hecht, G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut 52, 439–451 (2003).
Perdue, M.H., Masson, S., Wershil, B.K. & Galli, S.J. Role of mast cells in ion transport abnormalities associated with intestinal anaphylaxis. Correction of the diminished secretory response in genetically mast cell-deficient W/Wv mice by bone marrow transplantation. J. Clin. Invest. 87, 687–693 (1991).
Groschwitz, K.R. & Hogan, S.P. Intestinal barrier function: molecular regulation and disease pathogenesis. J. Allergy Clin. Immunol. 124, 3–20 quiz 21-22 (2009).
McDermott, J.R., Bartram, R.E., Knight, P.A., Miller, H.R., Garrod, D.R. & Grencis, R.K. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc. Natl. Acad. Sci. USA 100, 7761–7766 (2003).
Chabot, B., Stephenson, D.A., Chapman, V.M., Besmer, P. & Bernstein, A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 335, 88–89 (1988).
Kapur, R., Cooper, R., Xiao, X., Weiss, M.J., Donovan, P. & Williams, D.A. The presence of novel amino acids in the cytoplasmic domain of stem cell factor results in hematopoietic defects in Steel17H mice. Blood 94, 1915–1925 (1999).
Wang, L., Stanisz, A.M., Wershil, B.K., Galli, S.J. & Perdue, M.H. Substance P induces ion secretion in mouse small intestine through effects on enteric nerves and mast cells. Am. J. Physiol. 269, G85–G92 (1995).
Bousquet, J., Jeffery, P.K., Busse, W.W., Johnson, M. & Vignola, A.M. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med. 161, 1720–1745 (2000).
Fish, J.E. & Peters, S.P. Airway remodeling and persistent airway obstruction in asthma. J. Allergy Clin. Immunol. 104, 509–516 (1999).
Wills-Karp, M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu. Rev. Immunol. 17, 255–281 (1999).
Busse, W.W. & Lemanske, R.F. Asthma. N Engl. J. Med. 344, 350–362 (2001).
Holt, P.G. & Sly, P.D. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat. Med. 18, 726–735 (2012).
Wenzel, S.E. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat. Med. 18, 716–725 (2012).
Slager, R.E., Hawkins, G.A., Li, X., Postma, D.S., Meyers, D.A. & Bleecker, E.R. Genetics of asthma susceptibility and severity. Clin. Chest Med. 33, 431–443 (2012).
Meyers, D.A., Bleecker, E.R., Holloway, J.W. & Holgate, S.T. Asthma genetics and personalised medicine. Lancet Respir. Med. 2, 405–415 (2014).
Dougherty, R.H. et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in TH2-high asthma. J. Allergy Clin. Immunol. 125, 1046–1053 e1048 (2010).
Balzar, S. et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 183, 299–309 (2011).
Carroll, N.G., Mutavdzic, S. & James, A.L. Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur. Respir. J. 19, 879–885 (2002).
Bradley, B.L. et al. Eosinophils, T-lymphocytes, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects with asthma: comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. J. Allergy Clin. Immunol. 88, 661–674 (1991).
Liesker, J.J.W., Ten Hacken, N.H.T., Rutgers, S.R., Zeinstra-Smith, M., Postma, D.S. & Timens, W. Mast cell numbers in airway smooth muscle and PC20AMP in asthma and COPD. Respir. Med. 101, 882–887 (2007).
Lamb, D. & Lumsden, A. Intra-epithelial mast cells in human airway epithelium: evidence for smoking-induced changes in their frequency. Thorax 37, 334–342 (1982).
Holgate, S.T., Lackie, P.M., Davies, D.E., Roche, W.R. & Walls, A.F. The bronchial epithelium as a key regulator of airway inflammation and remodelling in asthma. Clin. Exp. Allergy. 29 (Suppl 2), 90–95 (1999).
Wilson, J.W. & Bamford, T.L. Assessing the evidence for remodelling of the airway in asthma. Pulm. Pharmacol. Ther. 14, 229–247 (2001).
Altin, J., Shen, C. & Liston, A. Understanding the genetic regulation of IgE production. Blood Rev. 24, 163–169 (2010).
Takhar, P. et al. Class switch recombination to IgE in the bronchial mucosa of atopic and nonatopic patients with asthma. J. Allergy Clin. Immunol. 119, 213–218 (2007).
Williams, C.M. & Galli, S.J. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J. Exp. Med. 192, 455–462 (2000).
Taube, C. et al. Mast cells, FcɛRI, and IL-13 are required for development of airway hyperresponsiveness after aerosolized allergen exposure in the absence of adjuvant. J. Immunol. 172, 6398–6406 (2004).
Nakae, S. et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J. Allergy Clin. Immunol. 120, 48–55 (2007).
Reuter, S. et al. Mast cell-derived tumour necrosis factor is essential for allergic airway disease. Eur. Respir. J. 31, 773–782 (2008).
Kung, T.T. et al. Mast cells modulate allergic pulmonary eosinophilia in mice. Am. J. Respir. Cell Mol. Biol. 12, 404–409 (1995).
Kobayashi, T. et al. An essential role of mast cells in the development of airway hyperresponsiveness in a murine asthma model. J. Immunol. 164, 3855–3861 (2000).
Nogami, M. et al. Experimental pulmonary eosinophilia in mice by Ascaris suum extract. Am. Rev. Respir. Dis. 141, 1289–1295 (1990).
Okudaira, H. et al. T-cell-dependent accumulation of eosinophils in the lung and its inhibition by monoclonal anti-interleukin-5. Int. Arch. Allergy Appl. Immunol. 94, 171–173 (1991).
Brusselle, G.G. et al. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin. Exp. Allergy 24, 73–80 (1994).
Takeda, K. et al. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J. Exp. Med. 186, 449–454 (1997).
Becker, M. et al. Genetic variation determines mast cell functions in experimental asthma. J. Immunol. 186, 7225–7231 (2011).
Yang, M., Kumar, R.K. & Foster, P.S. Pathogenesis of steroid-resistant airway hyperresponsiveness: interaction between IFN-γ and TLR4/MyD88 pathways. J. Immunol. 182, 5107–5115 (2009).
Li, J.J. et al. IL-27/IFN-γ induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J. Immunol. 185, 4401–4409 (2010).
Hunter, C.A. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 5, 521–531 (2005).
Bullens, D.M.A. et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir. Res. 7, 135 (2006).
Iwakura, Y., Ishigame, H., Saijo, S. & Nakae, S. Functional specialization of interleukin-17 family members. Immunity 34, 149–162 (2011).
Tan, B.B., Weald, D., Strickland, I. & Friedmann, P.S. Double-blind controlled trial of effect of housedust-mite allergen avoidance on atopic dermatitis. Lancet 347, 15–18 (1996).
He, R., Oyoshi, M.K., Jin, H. & Geha, R.S. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc. Natl. Acad. Sci. USA 104, 15817–15822 (2007).
Hellings, P.W. et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am. J. Respir. Cell Mol. Biol. 28, 42–50 (2003).
Lin, A.M. et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 187, 490–500 (2011).
Suurmond, J. et al. Mast cells are the main interleukin 17-positive cells in anticitrullinated protein antibody-positive and -negative rheumatoid arthritis and osteoarthritis synovium. Arthritis Res. Ther. 13, R150 (2011).
Préfontaine, D. et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 125, 752–754 (2010).
Préfontaine, D. et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J. Immunol. 183, 5094–5103 (2009).
Gudbjartsson, D.F. et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 41, 342–347 (2009).
Moffatt, M.F. et al. A large-scale, consortium-based genomewide association study of asthma. N Engl. J. Med. 363, 1211–1221 (2010).
Torgerson, D.G. et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 43, 887–892 (2011).
Pecaric-Petkovic, T., Didichenko, S.A., Kaempfer, S., Spiegl, N. & Dahinden, C.A. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood 113, 1526–1534 (2009).
Yagami, A. et al. IL-33 mediates inflammatory responses in human lung tissue cells. J. Immunol. 185, 5743–5750 (2010).
Junttila, I.S. et al. Efficient cytokine-induced IL-13 production by mast cells requires both IL-33 and IL-3. J. Allergy Clin. Immunol. 132, 704–712 e710 (2013).
Liew, F.Y., Pitman, N.I. & McInnes, I.B. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat. Rev. Immunol. 10, 103–110 (2010).
Jung, M.-Y. et al. IL-33 induces a hyporesponsive phenotype in human and mouse mast cells. J. Immunol. 190, 531–538 (2013).
Waern, I. et al. Mouse mast cell protease 4 is the major chymase in murine airways and has a protective role in allergic airway inflammation. J. Immunol. 183, 6369–6376 (2009).
Waern, I., Lundequist, A., Pejler, G. & Wernersson, S. Mast cell chymase modulates IL-33 levels and controls allergic sensitization in dust-mite induced airway inflammation. Mucosal Immunol. 6, 911–920 (2013).
Sicherer, S.H. & Sampson, H.A. Food allergy: recent advances in pathophysiology and treatment. Annu. Rev. Med. 60, 261–277 (2009).
Bock, S.A., Munoz-Furlong, A. & Sampson, H.A. Fatalities due to anaphylactic reactions to foods. J. Allergy Clin. Immunol. 107, 191–193 (2001).
Bock, S.A., Munoz-Furlong, A. & Sampson, H.A. Further fatalities caused by anaphylactic reactions to food, 2001-2006. J. Allergy Clin. Immunol. 119, 1016–1018 (2007).
Finkelman, F.D. Anaphylaxis: lessons from mouse models. J. Allergy Clin. Immunol. 120, 506–515 quiz 516-507 (2007).
Tsujimura, Y. et al. Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 28, 581–589 (2008).
Jonsson, F. et al. Mouse and human neutrophils induce anaphylaxis. J. Clin. Invest. 121, 1484–1496 (2011).
Sun, J. et al. Impact of CD40 ligand, B cells, and mast cells in peanut-induced anaphylactic responses. J. Immunol. 179, 6696–6703 (2007).
Reber, L.L. et al. Selective ablation of mast cells or basophils reduces peanut-induced anaphylaxis in mice. J. Allergy Clin. Immunol. 132, 881–888 e1-11 (2013).
Arias, K. et al. Distinct immune effector pathways contribute to the full expression of peanut-induced anaphylactic reactions in mice. J. Allergy Clin. Immunol. 127, 1552–1561 e1551 (2011).
Smit, J.J. et al. Contribution of classic and alternative effector pathways in peanut-induced anaphylactic responses. PLoS One 6, e28917 (2011).
Khodoun, M. et al. Peanuts can contribute to anaphylactic shock by activating complement. J. Allergy Clin. Immunol. 123, 342–351 (2009).
Ghosh, S., Hoselton, S.A. & Schuh, J.M. μ-Chain-deficient mice possess B-1 cells and produce IgG and IgE, but not IgA, following systemic sensitization and inhalational challenge in a fungal asthma model. J. Immunol. 189, 1322–1329 (2012).
Orinska, Z. et al. Novel B cell population producing functional IgG in the absence of membrane IgM expression. Eur. J. Immunol. 32, 3472–3480 (2002).
Perona-Wright, G. et al. Cutting edge: Helminth infection induces IgE in the absence of mu- or delta-chain expression. J. Immunol. 181, 6697–6701 (2008).
Simons, F.E. Anaphylaxis: recent advances in assessment and treatment. J. Allergy Clin. Immunol. 124, 625–636 quiz 637-628 (2009).
Vadas, P. et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl. J. Med. 358, 28–35 (2008).
Vadas, P., Perelman, B. & Liss, G. Platelet-activating factor, histamine, and tryptase levels in human anaphylaxis. J. Allergy Clin. Immunol. 131, 144–149 (2013).
Cheifetz, A. et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am. J. Gastroenterol. 98, 1315–1324 (2003).
Sampson, H.A. et al. Second symposium on the definition and management of anaphylaxis: summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. J. Allergy Clin. Immunol. 117, 391–397 (2006).
Bischoff, S. & Crowe, S.E. Gastrointestinal food allergy: new insights into pathophysiology and clinical perspectives. Gastroenterology 128, 1089–1113 (2005).
Hogan, S.P., Wang, Y.H., Strait, R. & Finkelman, F.D. Food-induced anaphylaxis: mast cells as modulators of anaphylactic severity. Semin. Immunopathol. 34, 643–653 (2012).
Caffarelli, C., Romanini, E., Caruana, P., Street, M.E. & de’ Angelis, G. Clinical food hypersensitivity: the relevance of duodenal immunoglobulin E-positive cells. Pediatr. Res. 44, 485–490 (1998).
Nolte, H., Schiotz, P.O., Kruse, A. & Stahl Skov, P. Comparison of intestinal mast cell and basophil histamine release in children with food allergic reactions. Allergy 44, 554–565 (1989).
Wang, M. et al. Peanut-induced intestinal allergy is mediated through a mast cell-IgE-FcɛRI-IL-13 pathway. J. Allergy Clin. Immunol. 126, 306–316 316 e301-312 (2010).
Forbes, E.E. et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J. Exp. Med. 205, 897–913 (2008).
Boyce, J.A. et al. Guidelines for the diagnosis and management of food allergy in the United States: summary of the NIAID-sponsored expert panel report. Nutr. Res. 31, 61–75 (2011).
Rance, F. Food allergy in children suffering from atopic eczema. Pediatr. Allergy Immunol. 19, 279–284 quiz 285 (2008).
Noti, M. et al. Exposure to food allergens through inflamed skin promotes intestinal food allergy through the thymic stromal lymphopoietin-basophil axis. J. Allergy Clin. Immunol. 133, 1390–1399 1399 e1391-1396 (2014).
Burton, O.T. et al. Immunoglobulin E signal inhibition during allergen ingestion leads to reversal of established food allergy and induction of regulatory T cells. Immunity 41, 141–151 (2014).
Choi, H.W. & Abraham, S.N. Mast cell mediator responses and their suppression by pathogenic and commensal microorganisms. Mol. Immunol. 63, 74–79 (2015).
Chan, C.Y., St John, A.L. & Abraham, S.N. Plasticity in mast cell responses during bacterial infections. Curr. Opin. Microbiol. 15, 78–84 (2012).
Kumar, V. & Sharma, A. Mast cells: emerging sentinel innate immune cells with diverse role in immunity. Mol. Immunol. 48, 14–25 (2010).
Galli, S.J. & Tsai, M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur. J. Immunol. 40, 1843–1851 (2010).
Woodbury, R.G., Miller, H.R., Huntley, J.F., Newlands, G.F., Palliser, A.C. & Wakelin, D. Mucosal mast cells are functionally active during spontaneous expulsion of intestinal nematode infections in rat. Nature 312, 450–452 (1984).
Askenase, P.W. Immune inflammatory responses to parasites: the role of basophils, mast cells and vasoactive amines. Am. J. Trop. Med. Hyg. 26, 96–103 (1977).
Knight, P.A., Wright, S.H., Lawrence, C.E., Paterson, Y.Y. & Miller, H.R. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J. Exp. Med. 192, 1849–1856 (2000).
Barrett, K.E. et al. The immune response to nematode parasites: modulation of mast cell numbers and function during Strongyloides stercoralis infections in nonhuman primates. Am. J. Trop. Med. Hyg. 38, 574–581 (1988).
Gustowska, L., Ruitenberg, E.J., Elgersma, A. & Kociecka, W. Increase of mucosal mast cells in the jejunum of patients infected with Trichinella spiralis. Int. Arch. Allergy Appl. Immunol. 71, 304–308 (1983).
Allen, J.E. & Maizels, R.M. Diversity and dialogue in immunity to helminths. Nat. Rev. Immunol. 11, 375–388 (2011).
Grencis, R.K., Humphreys, N.E. & Bancroft, A.J. Immunity to gastrointestinal nematodes: mechanisms and myths. Immunol. Rev. 260, 183–205 (2014).
Maizels, R.M., Pearce, E.J., Artis, D., Yazdanbakhsh, M. & Wynn, T.A. Regulation of pathogenesis and immunity in helminth infections. J. Exp. Med. 206, 2059–2066 (2009).
Taylor, M.D., van der Werf, N. & Maizels, R.M. T cells in helminth infection: the regulators and the regulated. Trends Immunol. 33, 181–189 (2012).
Ohnmacht, C. & Voehringer, D. Basophils protect against reinfection with hookworms independently of mast cells and memory Th2 cells. J. Immunol. 184, 344–350 (2010).
Crowle, P.K. Mucosal mast cell reconstitution and Nippostrongylus brasiliensis rejection by W/Wv mice. J. Parasitol. 69, 66–69 (1983).
Abe, T. & Nawa, Y. Reconstitution of mucosal mast cells in W/WV mice by adoptive transfer of bone marrow-derived cultured mast cells and its effects on the protective capacity to Strongyloides ratti-infection. Parasite Immunol. 9, 31–38 (1987).
Sasaki, Y. et al. IL-18 with IL-2 protects against Strongyloides venezuelensis infection by activating mucosal mast cell-dependent type 2 innate immunity. J. Exp. Med. 202, 607–616 (2005).
Khan, A.I., Horii, Y. & Nawa, Y. Defective mucosal immunity and normal systemic immunity of Mongolian gerbils, Meriones unguiculatus, to reinfection with Strongyloides venezuelensis. Parasite Immunol. 15, 565–571 (1993).
Ha, T.Y., Reed, N.D. & Crowle, P.K. Delayed expulsion of adult Trichinella spiralis by mast cell-deficient W/Wv mice. Infect. Immun. 41, 445–447 (1983).
Oku, Y., Itayama, H. & Kamiya, M. Expulsion of Trichinella spiralis from the intestine of W/Wv mice reconstituted with haematopoietic and lymphopoietic cells and origin of mucosal mast cells. Immunology 53, 337–344 (1984).
Koyama, K. & Ito, Y. Mucosal mast cell responses are not required for protection against infection with the murine nematode parasite Trichuris muris. Parasite Immunol. 22, 13–20 (2000).
Hepworth, M.R. et al. Mast cells orchestrate type 2 immunity to helminths through regulation of tissue-derived cytokines. Proc. Natl. Acad. Sci. USA 109, 6644–6649 (2012).
Abe, T. & Nawa, Y. Localization of mucosal mast cells in W/Wv mice after reconstitution with bone marrow cells or cultured mast cells, and its relation to the protective capacity to Strongyloides ratti infection. Parasite Immunol. 9, 477–485 (1987).
Wedemeyer, J. & Galli, S.J. Decreased susceptibility of mast cell-deficient KitW/KitW-v mice to the development of 1, 2-dimethylhydrazine-induced intestinal tumors. Lab. Invest. 85, 388–396 (2005).
Shin, K. et al. Mouse mast cell tryptase mMCP-6 is a critical link between adaptive and innate immunity in the chronic phase of Trichinella spiralis infection. J. Immunol. 180, 4885–4891 (2008).
Blankenhaus, B. et al. Foxp3+ regulatory T cells delay expulsion of intestinal nematodes by suppression of IL-9-driven mast cell activation in BALB/c but not in C57BL/6 mice. PLoS Pathog. 10, e1003913 (2014).
Newlands, G.F., Miller, H.R., MacKellar, A. & Galli, S.J. Stem cell factor contributes to intestinal mucosal mast cell hyperplasia in rats infected with Nippostrongylus brasiliensis or Trichinella spiralis, but anti-stem cell factor treatment decreases parasite egg production during N brasiliensis infection. Blood 86, 1968–1976 (1995).
Arizono, N. et al. Infection of Nippostrongylus brasiliensis induces development of mucosal-type but not connective tissue-type mast cells in genetically mast cell-deficient Ws/Ws rats. Blood 81, 2572–2578 (1993).
Townsend, J.M., Fallon, G.P., Matthews, J.D., Smith, P., Jolin, E.H. & McKenzie, N.A. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity 13, 573–583 (2000).
Harris, N. & Gause, W.C. To B or not to B: B cells and the Th2-type immune response to helminths. Trends Immunol. 32, 80–88 (2011).
Pritchard, D.I. Immunity to helminths: is too much IgE parasite—rather than host-protective? Parasite Immunol. 15, 5–9 (1993).
Wakelin, D. Immunity to intestinal parasites. Nature 273, 617–620 (1978).
Zhu, D., Kepley, C.L., Zhang, M., Zhang, K. & Saxon, A. A novel human immunoglobulin FcγFcε bifunctional fusion protein inhibits FcεRI-mediated degranulation. Nat. Med. 8, 518–521 (2002).
Zhu, D., Kepley, C.L., Zhang, K., Terada, T., Yamada, T. & Saxon, A. A chimeric human-cat fusion protein blocks cat-induced allergy. Nat. Med. 11, 446–449 (2005).
Echtenacher, B., Mannel, D.N. & Hultner, L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature 381, 75–77 (1996).
Maurer, M. et al. The c-kit ligand, stem cell factor, can enhance innate immunity through effects on mast cells. J. Exp. Med. 188, 2343–2348 (1998).
Maurer, M. et al. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 432, 512–516 (2004).
Mallen-St Clair, J., Pham, C.T., Villalta, S.A., Caughey, G.H. & Wolters, P.J. Mast cell dipeptidyl peptidase I mediates survival from sepsis. J. Clin. Invest. 113, 628–634 (2004).
Piliponsky, A.M. et al. Neurotensin increases mortality and mast cells reduce neurotensin levels in a mouse model of sepsis. Nat. Med. 14, 392–398 (2008).
Sutherland, R.E., Olsen, J.S., McKinstry, A., Villalta, S.A. & Wolters, P.J. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J. Immunol. 181, 5598–5605 (2008).
Piliponsky, A.M. et al. The chymase mouse mast cell protease 4 degrades TNF, limits inflammation, and promotes survival in a model of sepsis. Am. J. Pathol. 181, 875–886 (2012).
Nakano, N. et al. Involvement of mast cells in IL-12/23 p40 production is essential for survival from polymicrobial infections. Blood 109, 4846–4855 (2007).
Kanamaru, Y., Sumiyoshi, K., Ushio, H., Ogawa, H., Okumura, K. & Nakao, A. Smad3 deficiency in mast cells provides efficient host protection against acute septic peritonitis. J. Immunol. 174, 4193–4197 (2005).
Orinska, Z. et al. IL-15 constrains mast cell-dependent antibacterial defenses by suppressing chymase activities. Nat. Med. 13, 927–934 (2007).
Huang, C. et al. Evaluation of the substrate specificity of human mast cell tryptase βI and demonstration of its importance in bacterial infections of the lung. J. Biol. Chem. 276, 26276–26284 (2001).
Thakurdas, S.M. et al. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J. Biol. Chem. 282, 20809–20815 (2007).
Xu, X. et al. Mast cells protect mice from Mycoplasma pneumonia. Am. J. Respir. Crit. Care Med. 173, 219–225 (2006).
Wershil, B.K., Castagliuolo, I. & Pothoulakis, C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology 114, 956–964 (1998).
Malaviya, R., Navara, C. & Uckun, F.M. Role of Janus kinase 3 in mast cell-mediated innate immunity against gram-negative bacteria. Immunity 15, 313–321 (2001).
Anderson, G.G., Dodson, K.W., Hooton, T.M. & Hultgren, S.J. Intracellular bacterial communities of uropathogenic Escherichia coli in urinary tract pathogenesis. Trends Microbiol. 12, 424–430 (2004).
Shelburne, C.P. et al. Mast cells augment adaptive immunity by orchestrating dendritic cell trafficking through infected tissues. Cell Host Microbe 6, 331–342 (2009).
Malaviya, R., Ikeda, T., Abraham, S.N. & Malaviya, R. Contribution of mast cells to bacterial clearance and their proliferation during experimental cystitis induced by type 1 fimbriated E. coli. Immunol. Lett. 91, 103–111 (2004).
Chan, C.Y., St John, A.L. & Abraham, S.N. Mast cell interleukin-10 drives localized tolerance in chronic bladder infection. Immunity 38, 349–359 (2013).
Leveson-Gower, D.B. et al. Mast cells suppress murine GVHD in a mechanism independent of CD4+CD25+ regulatory T cells. Blood 122, 3659–3665 (2013).
Ronnberg, E. et al. Mast cells are activated by Staphylococcus aureus in vitro but do not influence the outcome of intraperitoneal S. aureus infection in vivo. Immunology 143, 155–163 (2014).
Sugiyama, K. Histamine release from rat mast cells induced by Sendai virus. Nature 270, 614–615 (1977).
Castleman, W.L., Sorkness, R.L., Lemanske, R.F. Jr . & McAllister, P.K. Viral bronchiolitis during early life induces increased numbers of bronchiolar mast cells and airway hyperresponsiveness. Am. J. Pathol. 137, 821–831 (1990).
Sorden, S.D. & Castleman, W.L. Virus-induced increases in bronchiolar mast cells in Brown Norway rats are associated with both local mast cell proliferation and increases in blood mast cell precursors. Lab. Invest. 73, 197–204 (1995).
Kulka, M., Alexopoulou, L., Flavell, R.A. & Metcalfe, D.D. Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. J. Allergy Clin. Immunol. 114, 174–182 (2004).
Orinska, Z., Bulanova, E., Budagian, V., Metz, M., Maurer, M. & Bulfone-Paus, S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood 106, 978–987 (2005).
Higuchi, H. et al. Mast cells play a critical role in the pathogenesis of viral myocarditis. Circulation 118, 363–372 (2008).
Zsebo, K.M. et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell 63, 213–224 (1990).
Wang, Z. et al. Skin mast cells protect mice against vaccinia virus by triggering mast cell receptor S1PR2 and releasing antimicrobial peptides. J. Immunol. 188, 345–357 (2012).
Wang, Z., MacLeod, D.T. & Di Nardo, A. Commensal bacteria lipoteichoic acid increases skin mast cell antimicrobial activity against vaccinia viruses. J. Immunol. 189, 1551–1558 (2012).
St John, A.L., Rathore, A.P., Raghavan, B., Ng, M.L. & Abraham, S.N. Contributions of mast cells and vasoactive products, leukotrienes and chymase, to dengue virus-induced vascular leakage. Elife 2, e00481 (2013).
St John, A.L. Influence of mast cells on dengue protective immunity and immune pathology. PLoS Pathog. 9, e1003783 (2013).
St John, A.L. et al. Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc. Natl. Acad. Sci. USA 108, 9190–9195 (2011).
Ebert, S. et al. Mast cells expedite control of pulmonary murine cytomegalovirus infection by enhancing the recruitment of protective CD8 T cells to the lungs. PLoS Pathog. 10, e1004100 (2014).
Sundstrom, J.B. et al. Human tissue mast cells are an inducible reservoir of persistent HIV infection. Blood 109, 5293–5300 (2007).
Bannert, N. et al. Human mast cell progenitors can be infected by macrophagetropic human immunodeficiency virus type 1 and retain virus with maturation in vitro. J. Virol. 75, 10808–10814 (2001).
Sundstrom, J.B., Little, D.M., Villinger, F., Ellis, J.E. & Ansari, A.A. Signaling through Toll-like receptors triggers HIV-1 replication in latently infected mast cells. J. Immunol. 172, 4391–4401 (2004).
Li, Y. et al. Mast cells/basophils in the peripheral blood of allergic individuals who are HIV-1 susceptible due to their surface expression of CD4 and the chemokine receptors CCR3, CCR5, and CXCR4. Blood 97, 3484–3490 (2001).
McLachlan, J.B. et al. Mast cell activators: a new class of highly effective vaccine adjuvants. Nat. Med. 14, 536–541 (2008).
Ferry, X., Brehin, S., Kamel, R. & Landry, Y. G protein-dependent activation of mast cell by peptides and basic secretagogues. Peptides 23, 1507–1515 (2002).
Aridor, M., Rajmilevich, G., Beaven, M.A. & Sagi-Eisenberg, R. Activation of exocytosis by the heterotrimeric G protein Gi3 . Science 262, 1569–1572 (1993).
Paton, W.D. Compound 48/80: a potent histamine liberator. Br. J. Pharmacol. Chemother. 6, 499–508 (1951).
Fawcett, D.W. Cytological and pharmacological observations on the release of histamine by mast cells. J. Exp. Med. 100, 217–224 (1954).
Rothschild, A.M. Mechanisms of histamine release by compound 48-80. Br. J. Pharmacol. 38, 253–262 (1970).
Gushchin, I.S., Miroshnikov, A.I., Martynov, V.I. & Sviridov, V.V. Histamine releasing and anti-inflammatory activities of MCD-peptide and its modified forms. Agents Actions 11, 69–71 (1981).
Galli, S.J., Wershil, B.K., Bose, R., Walker, P.A. & Szabo, S. Ethanol-induced acute gastric injury in mast cell-deficient and congenic normal mice. Evidence that mast cells can augment the area of damage. Am. J. Pathol. 128, 131–140 (1987).
Pae, S., Cho, J.Y., Dayan, S., Miller, M., Pemberton, A.D. & Broide, D.H. Chronic allergen challenge induces bronchial mast cell accumulation in BALB/c but not C57BL/6 mice and is independent of IL-9. Immunogenetics 62, 499–506 (2010).
Xing, W., Austen, K.F., Gurish, M.F. & Jones, T.G. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc. Natl. Acad. Sci. USA 108, 14210–14215 (2011).
Forsberg, E. et al. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature 400, 773–776 (1999).
Humphries, D.E. et al. Heparin is essential for the storage of specific granule proteases in mast cells. Nature 400, 769–772 (1999).
Rönnberg, E., Melo, F.R. & Pejler, G. Mast cell proteoglycans. J. Histochem. Cytochem. 60, 950–962 (2012).
Schwartz, L.B. Mast cells: function and contents. Curr. Opin. Immunol. 6, 91–97 (1994).
Craig, S.S., Irani, A.M., Metcalfe, D.D. & Schwartz, L.B. Ultrastructural localization of heparin to human mast cells of the MCTC and MCT types by labeling with antithrombin III-gold. Lab. Invest. 69, 552–561 (1993).
Wernersson, S. & Pejler, G. Mast cell secretory granules: armed for battle. Nat. Rev. Immunol. 14, 478–494 (2014).
Wong, G.W. et al. Identification of a new member of the tryptase family of mouse and human mast cell proteases which possesses a novel COOH-terminal hydrophobic extension. J. Biol. Chem. 274, 30784–30793 (1999).
Xia, H.Z., Kepley, C.L., Sakai, K., Chelliah, J., Irani, A.M. & Schwartz, L.B. Quantitation of tryptase, chymase, FcɛRIα, and FcɛRIγ mRNAs in human mast cells and basophils by competitive reverse transcription-polymerase chain reaction. J. Immunol. 154, 5472–5480 (1995).
Caughey, G.H. et al. Characterization of human γ-tryptases, novel members of the chromosome 16p mast cell tryptase and prostasin gene families. J. Immunol. 164, 6566–6575 (2000).
Schechter, N.M., Irani, A.M., Sprows, J.L., Abernethy, J., Wintroub, B. & Schwartz, L.B. Identification of a cathepsin G-like proteinase in the MCTC type of human mast cell. J. Immunol. 145, 2652–2661 (1990).
Irani, A.M., Goldstein, S.M., Wintroub, B.U., Bradford, T. & Schwartz, L.B. Human mast cell carboxypeptidase. Selective localization to MCTC cells. J. Immunol. 147, 247–253 (1991).
Vanderslice, P., Ballinger, S.M., Tam, E.K., Goldstein, S.M., Craik, C.S. & Caughey, G.H. Human mast cell tryptase: multiple cDNAs and genes reveal a multigene serine protease family. Proc. Natl. Acad. Sci. USA 87, 3811–3815 (1990).
Katz, H.R., Stevens, R.L. & Austen, K.F. Heterogeneity of mammalian mast cells differentiated in vivo and in vitro. J. Allergy Clin. Immunol. 76, 250–259 (1985).
Li, H.J. et al. Distinct cellular origins for serotonin-expressing and enterochromaffin-like cells in the gastric corpus. Gastroenterology 146, 754–764 e753 (2014).
von Stebut, E., Metz, M., Milon, G., Knop, J. & Maurer, M. Early macrophage influx to sites of cutaneous granuloma formation is dependent on MIP-1α/β released from neutrophils recruited by mast cell-derived TNFα. Blood 101, 210–215 (2003).
Kushnir-Sukhov, N.M., Brown, J.M., Wu, Y., Kirshenbaum, A. & Metcalfe, D.D. Human mast cells are capable of serotonin synthesis and release. J. Allergy Clin. Immunol. 119, 498–499 (2007).
Lowman, M.A., Rees, P.H., Benyon, R.C. & Church, M.K. Human mast cell heterogeneity: histamine release from mast cells dispersed from skin, lung, adenoids, tonsils, and colon in response to IgE-dependent and nonimmunologic stimuli. J. Allergy Clin. Immunol. 81, 590–597 (1988).
Rudich, N., Ravid, K. & Sagi-Eisenberg, R. Mast cell adenosine receptors function: a focus on the A3 adenosine receptor and inflammation. Front. Immunol. 3, 134 (2012).
Zhong, H. et al. Activation of murine lung mast cells by the adenosine A3 receptor. J. Immunol. 171, 338–345 (2003).
Tilley, S.L., Wagoner, V.A., Salvatore, C.A., Jacobson, M.A. & Koller, B.H. Adenosine and inosine increase cutaneous vasopermeability by activating A3 receptors on mast cells. J. Clin. Invest. 105, 361–367 (2000).
Sereda, M.J., Bradding, P. & Vial, C. Adenosine potentiates human lung mast cell tissue plasminogen activator activity. J. Immunol. 186, 1209–1217 (2011).
Gomez, G., Zhao, W. & Schwartz, L.B. Disparity in FcɛRI-induced degranulation of primary human lung and skin mast cells exposed to adenosine. J. Clin. Immunol. 31, 479–487 (2011).
Ujike, A. et al. Modulation of immunoglobulin (Ig)E-mediated systemic anaphylaxis by low-affinity Fc receptors for IgG. J. Exp. Med. 189, 1573–1579 (1999).
Takai, T., Ono, M., Hikida, M., Ohmori, H. & Ravetch, J.V. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature 379, 346–349 (1996).
Hazenbos, W.L. et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in FcγRIII (CD16) deficient mice. Immunity 5, 181–188 (1996).
Dombrowicz, D., Flamand, V., Miyajima, I., Ravetch, J.V., Galli, S.J. & Kinet, J.P. Absence of FcɛRI α chain results in upregulation of FcγRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between FcɛRI and FcγRIII for limiting amounts of FcR β and γ chains. J. Clin. Invest. 99, 915–925 (1997).
Coleman, J.W. & Godfrey, R.C. The number and affinity of IgE receptors on dispersed human lung mast cells. Immunology 44, 859–863 (1981).
Sellge, G. et al. Interferon-γ regulates growth and controls Fcγ receptor expression and activation in human intestinal mast cells. BMC Immunol. 15, 27 (2014).
Helm, B., Marsh, P., Vercelli, D., Padlan, E., Gould, H. & Geha, R. The mast cell binding site on human immunoglobulin E. Nature 331, 180–183 (1988).
Tkaczyk, C., Okayama, Y., Woolhiser, M.R., Hagaman, D.D., Gilfillan, A.M. & Metcalfe, D.D. Activation of human mast cells through the high affinity IgG receptor. Mol. Immunol. 38, 1289–1293 (2002).
Zhao, W., Kepley, C.L., Morel, P.A., Okumoto, L.M., Fukuoka, Y. & Schwartz, L.B. FcγRIIa, not FcγRIIb, is constitutively and functionally expressed on skin-derived human mast cells. J. Immunol. 177, 694–701 (2006).
Trivedi, N.N., Tamraz, B., Chu, C., Kwok, P.Y. & Caughey, G.H. Human subjects are protected from mast cell tryptase deficiency despite frequent inheritance of loss-of-function mutations. J. Allergy Clin. Immunol. 124, 1099–1105 e1091-1094 (2009).
Befus, A.D., Pearce, F.L., Gauldie, J., Horsewood, P. & Bienenstock, J. Mucosal mast cells. I. Isolation and functional characteristics of rat intestinal mast cells. J. Immunol. 128, 2475–2480 (1982).
Katz, H.R. & Lobell, R.B. Expression and function of FcγR in mouse mast cells. Int. Arch. Allergy Immunol. 107, 76–78 (1995).
Acknowledgements
We thank the members of the Galli lab and our collaborators and colleagues for their contributions to some of the work reviewed herein and we apologize to the many contributors to this field whose work was not cited because of space limitations. L.L.R. is the recipient of fellowships from the French “Fondation pour la Recherche Médicale FRM” and the Stanford Pediatric Research Fund and grants from the Arthritis National Research Foundation and National Institutes of Health (K99AI110645); R.S. was supported by the Lucile Packard Foundation for Children’s Health and the Stanford NIH/NCRR CTSA award number UL1 RR025744; K.M. was supported in part by a Postdoctoral Fellowship for Research Abroad of the Japan Society for the Promotion of Science; S.J.G. acknowledges support from the National Institutes of Health grants U19 AI104209, NS 080062 and from Tobacco-Related Disease Research Program at University of California.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article

Cite this article
Reber, L., Sibilano, R., Mukai, K. et al. Potential effector and immunoregulatory functions of mast cells in mucosal immunity. Mucosal Immunol 8, 444–463 (2015). https://doi.org/10.1038/mi.2014.131
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.131
This article is cited by
-
The combined use of photobiomodulation and curcumin-loaded iron oxide nanoparticles significantly improved wound healing in diabetic rats compared to either treatment alone
Lasers in Medical Science (2022)
-
GM-CSF drives dysregulated hematopoietic stem cell activity and pathogenic extramedullary myelopoiesis in experimental spondyloarthritis
Nature Communications (2020)
-
Pike intestinal reaction to Acanthocephalus lucii (Acanthocephala): immunohistochemical and ultrastructural surveys
Parasites & Vectors (2018)
-
The role of extracellular vesicles when innate meets adaptive
Seminars in Immunopathology (2018)
-
The involvement of mast cells in the irinotecan-induced enteric neurons loss and reactive gliosis
Journal of Neuroinflammation (2017)
