
Abstract
Changes in macrophage phenotype have been implicated in apoptotic cell-mediated immune modulation via induction of peroxisome proliferator-activated receptor-γ (PPARγ). In this study, we characterized PPARγ induction by apoptotic cell instillation over the course of bleomycin-induced lung injury in C57BL/6 mice. Next, the role of PPARγ activation in resolving lung inflammation and fibrosis was investigated. Our data demonstrate that apoptotic cell instillation after bleomycin results in immediate and prolonged enhancement of PPARγ mRNA and protein in alveolar macrophages and lung. Moreover, PPARγ activity and expression of its target molecules, including CD36, macrophage mannose receptor, and arginase 1, were persistently enhanced following apoptotic cell instillation. Coadministration of the PPARγ antagonist, GW9662, reversed the enhanced efferocytosis, and the reduced proinflammatory cytokine expression, neutrophil recruitment, myeloperoxidase activity, hydroxyproline contents, and fibrosis markers, including type 1 collagen α2, fibronectin and α-smooth muscle actin (α-SMA), in the lung by apoptotic cell instillation. In addition, inhibition of PPARγ activity reversed the expression of transforming growth factor-β (TGF-β), interleukin (IL)-10, and hepatocyte growth factor (HGF). These findings indicate that one-time apoptotic cell instillation contributes to anti-inflammatory and antifibrotic responses via upregulation of PPARγ expression and subsequent activation, leading to regulation of efferocytosis and production of proresolving cytokines.
Similar content being viewed by others


INTRODUCTION
Pulmonary fibrosis is a potentially fatal disease characterized by continuous alveolar epithelial injury and dysregulated repair, leading to myofibroblast accumulation and excessive deposition of extracellular matrix and connective tissue. Previous studies have indicated that efficient alveolar epithelial cell repair is critical for normal healing without fibrosis.1, 2 Because apoptotic cell recognition and clearance result in growth factor release and activation of signaling molecules involved in maintaining the epithelium and endothelium, the maintenance of alveolar integrity has been proposed to be critically dependent on intact mechanisms of apoptotic cell clearance.3
Apoptotic cell recognition and efferocytosis by macrophages greatly influences the resolution of inflammation by affecting secretion of the anti-inflammatory cytokines, transforming growth factor-β (TGF-β) and interleukin (IL)-10, that inhibit inflammatory mediator production.4, 5 In addition, the ability of macrophages to clear apoptotic cells involved in resolving inflammation is dependent on their programming to the alternative M2 phenotype. These macrophages exhibit increased expression and activity of macrophage-specific peroxisome proliferator-activated receptor-γ (PPARγ) that is essential to their alternative activation.6, 7, 8 One mechanism by which apoptotic cells enhance efferocytic programming is through phosphatidylserine-dependent induction of IL-4 signaling that upregulates PPARγ activation through production of 12/15-lipoxygenase-dependent 15-hyrdoxyoctadecadienoic acid (15-HETE).9
PPARγ is a member of the nuclear hormone receptor family that modulates gene expression upon ligand binding. Activation of PPARγ has been linked to glucose homeostasis,10 cellular differentiation,11 apoptosis,12 and anti-inflammatory responses.13 Within the lung, PPARγ is expressed by the epithelium, smooth muscle cells, fibroblasts, endothelium, macrophages, eosinophils, and dendritic cells.14 Several studies have demonstrated that PPARs play important roles in regulating processes related to fibrogenesis, including cellular differentiation, inflammation, and wound healing in vitro and in vivo.15, 16 In particular, PPARγ synthetic ligands interfered with Smad-dependent and -independent TGF-β signaling, thereby opposing the profibrotic effect of TGF-β.17, 18 Administration of PPARγ agonists markedly reduced expression of nitrotyrosine, poly(ADP ribose) polymerase activation, inducible nitric oxide synthase formation, and lung damage induced by bleomycin in mice.19 On the other hand, PPARγ agonists induced expression of the antifibrotic mediator, hepatocyte growth factor (HGF), via a peroxisome proliferator response element within the HGF promoter.20 Moreover, these agonists upregulate the Smad corepressor TG-interacting factor (TGIF), leading to inhibition of α-smooth muscle actin (α-SMA) and fibronectin expression.6 However, it remains unclear how this multipotent molecule PPARγ prevents excessive fibrosis, thus leading to successful tissue repair, in the context of enhanced apoptotic cell recognition and clearance from the lesion.
Recently, the rodent bleomycin model of lung fibrosis allows the use of molecular tools to dissect the cellular and subcellular processes leading to fibrosis.21 Interestingly, a single bleomycin instillation effectively replicates several of the specific pathogenic molecular changes associated with idiopathic pulmonary fibrosis, and may be best used as a model for patient with active disease.22, 23 Nonetheless, in truth, the bleomycin model has never been promoted as an experimental equivalent of human idiopathic pulmonary fibrosis, a progressive interstitial lung disorder.
Here, we hypothesized that increased PPARγ activation by apoptotic cell recognition and clearance programming reinforces the efferocytic ability of phagocytes, such as alveolar macrophages, thus hastening the resolution of pulmonary inflammation and fibrosis. To explore this hypothesis, we first characterized PPARγ expression and activation in lung and alveolar macrophages following apoptotic cell instillation over the course of bleomycin-induced lung injury. Next, using a pharmacological approach involving a PPARγ antagonist GW9662, we investigated the role of enhanced PPARγ activation following apoptotic cell instillation in altering inflammatory and fibrogenic programs in alveolar macrophages and the lung, including the effects of PPARγ on TGF-β, IL-10, and HGF, that are induced by apoptotic cells in vivo and in vitro.24, 25, 26
RESULTS
Immediate and sustained enhancement of PPARγ expression following apoptotic cell instillation into bleomycin-stimulated lungs
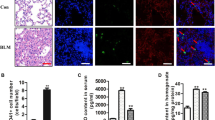
Although PPARγ mRNA and protein expression appeared to increase slightly in alveolar macrophages and lung tissue at 2 days after bleomycin treatment, these increases were not significant compared with the saline control (Figure 1a–c). Apoptotic cell instillation further increased PPARγ mRNA and protein expression relative to the bleomycin+saline group. In contrast, viable cell instillation had no effect. In addition, confocal microscopy revealed an enhancement of nuclear PPARγ staining in alveolar macrophages in bleomycin+apoptotic cells compared with bleomycin+saline or viable cells (Figure 1d). In comparison, exposure to apoptotic cells did not affect the low levels of PPARγ mRNA and protein present in lungs from naive mice (Figure 1a–c).

Apoptotic cell instillation induces immediate and persistent enhancement of peroxisome proliferator-activated receptor-γ (PPARγ) expression in bleomycin (BLM)-stimulated lungs. After intratracheal instillation of saline alone (Sal), viable Jurkat cells (ViaJ), or apoptotic Jurkat cells (ApoJ) at 2 days after BLM treatment, lavage supernatant was collected at 2 h after ApoJ instillation and on days 7 (7D), 14 (14D), and 21 (21D) after BLM treatment. PPARγ mRNA was analyzed by real-time PCR in alveolar macrophages (AM) and lung tissue at 2 h after ApoJ instillation (a and b) and on days 7, 14, and 21 after BLM treatment (e). Western blot analysis of PPARγ protein in lung tissue homogenates at 2 h after ApoJ instillation (c) and on days 7, 14, and 21 after BLM treatment (f). The relative densitometric intensity was determined for each band and normalized to α-tubulin. (d) Immunofluorescence staining (red) of PPARγ in alveolar macrophages at 2 h after ApoJ instillation. Images were captured at × 800 magnification. Representative results from five mice per group are shown. (a–c, e, f) Values represent the mean±s.e.m. from five mice in each group. *P<0.05 compared with saline control, +P<0.05 for BLM+ApoJ vs. BLM+Sal or BLM+ViaJ.
To demonstrate that apoptotic cell induction is not restricted to T cells, we introduced apoptotic HeLa epithelial cells into bleomycin-stimulated lungs and then assessed PPARγ mRNA and protein expression levels. Apoptotic HeLa epithelial cells exerted similar effects on PPARγ mRNA and protein expression in lung tissue (Supplementary Figure S1a,b online). Likely, no significant differences were observed with human Jurkat T cells and mouse thymocytes (Supplementary Figure S1c,d). These findings suggest that apoptotic cell-induced PPARγ expression is not dependent on cell type and the heterologous nature of the target apoptotic cells.
The kinetics of apoptotic cell-induced PPARγ expression were also examined at days 7–21 after bleomycin treatment. PPARγ mRNA in alveolar macrophages and lung tissue gradually increased up to 21 days after bleomycin treatment (Figure 1e and Supplementary Figure S2a). However, the apoptotic cell instillation further enhanced PPARγ mRNA expression in alveolar macrophages and lung tissue each day after bleomycin treatment. Similarly, PPARγ protein levels in lung tissue were further enhanced by apoptotic cells, reaching peak levels at day 21 (Figure 1f). As PPARγ mRNA and protein levels followed similar kinetics after bleomycin treatment and apoptotic cell instillation, PPARγ protein expression is likely regulated at the level of transcription.
As expected, lung PPARγ mRNA and protein expressions were not statistically different between the bleomycin+viable cells and bleomycin+saline groups at days 7 and 14 after bleomycin treatment (Supplementary Figure S2b,c). In addition, as all 10 mice treated with viable cells died within 21 days after bleomycin treatment, the effects between the two groups could not be compared.
Activation of PPARγ and its target molecules are enhanced following apoptotic cell instillation into bleomycin-stimulated lungs
Next, we examined whether nuclear PPARγ activity was also enhanced by apoptotic cell instillation of lung tissue. PPARγ activity was further enhanced in a similar temporal pattern by apoptotic cell instillation following bleomycin-induced lung injury (Figure 2a,b). As expected, lung PPARγ activity was not statistically different between the bleomycin+viable cells and bleomycin+saline groups on days 2, 7, and 14 after bleomycin treatment (Supplementary Figure S2d).

Apoptotic cell instillation induces enhancement of peroxisome proliferator-activated receptor-γ (PPARγ) activity and its target molecule expression. After intratracheal instillation of saline alone (Sal) or apoptotic Jurkat cells (ApoJ) at 2 days after bleomycin (BLM) treatment, lavage supernatant was collected at 2 h after ApoJ instillation and on days 7, 14, and 21 after BLM treatment. Where indicated, the first dose of GW9662 (1 mg kg−1, intraperitoneally (i.p.)) was administered simultaneously with ApoJ instillation into BLM-stimulated lungs (2 days) and every day thereafter. PPARγ activity in nuclear extracts from lung tissue was analyzed at (a) 2 h after ApoJ instillation and on (b) days 7, 14, and 21 after BLM treatment as described in Methods. The mRNA level of PPARγ target molecules, including CD36 and arginase1 (Arg1), was analyzed by real-time PCR at (c and d) 2 h after ApoJ instillation and on (f and g) days 7 (7D), 14 (14D), and 21 (21D) after BLM treatment. Western blot analysis of CD36 and Arg1 protein in lung tissue homogenates at (e) 2 h after ApoJ instillation and on (h) days 7, 14, and 21 after BLM treatment. The relative densitometric intensity was determined for each band and normalized to α-tubulin. AM, alveolar macrophage. Values represent the mean±s.e.m. from five mice in each group. *P<0.05 compared with saline control, +P<0.05 for BLM+ApoJ vs. BLM+Sal, #P<0.05 for BLM+ApoJ vs. BLM+ApoJ+GW.
As an indication of PPARγ functional activity, we examined the expression of CD36, macrophage mannose receptor (MMR), and arginase 1 (Arg1), all of which are upregulated by PPARγ and characteristic of alternative macrophage programming.6, 27, 28 Our data demonstrate that these PPARγ targets increased at the levels of mRNA and protein in alveolar macrophages and lung tissue at 2 h following apoptotic cell instillation (Figure 2c–e and Supplementary Figure S3a–c) and throughout bleomycin treatment (Figure 2f–h and Supplementary Figure S3d,e). Notably, the expression of these markers paralleled the pattern of PPARγ expression in the bleomycin+saline or apoptotic cell groups. To confirm that these target molecules are dependent on apoptotic cell-enhanced PPARγ activity, the PPARγ antagonist GW9662 was coadministered with apoptotic cells into the bleomycin-stimulated lungs. The enhanced activity of PPARγ, as well as expression of these target molecules in alveolar macrophages and lung tissue, induced by introduction of apoptotic cells after bleomycin treatment was reduced by GW9662 treatment each day after bleomycin treatment. The inhibitor administered with buffer had no effects.
Cellular source of PPARγ in the lung
Immunohistochemistry was performed on lung tissue using an anti-PPARγ monoclonal antibody or an immunoglobulin G (IgG) isotype control to determine which cells express PPARγ in this organ. PPARγ expression was mainly localized to alveolar macrophages and, to some extent, lung parenchymal cells on days 7 and 14 after bleomycin treatment with saline (Supplementary Figure S4b,c). Strong PPARγ expression was observed in these cells following introduction of bleomycin+apoptotic cells at these time points (Supplementary Figure S4e,f). In contrast, PPARγ staining was very weak in lung sections of unstimulated saline control animals (Supplementary Figure S4a). No staining was observed in lung sections stained with the isotype control (Supplementary Figure S4d). Similarly, double immunofluorescence staining demonstrated increased PPARγ expression following apoptotic cell instillation primarily in macrophages and, to some extent, fibroblasts on days 7 (Supplementary Figure S5a,b) and 14 after bleomycin treatment (Figure 3a,b).

Peroxisome proliferator-activated receptor-γ (PPARγ)-positive staining in alveolar macrophages and lung fibroblasts after apoptotic cell instillation. At 2 days after bleomycin (BLM) treatment, lungs were instilled with saline alone (Sal) or apoptotic Jurkat cells (ApoJ) intratracheally. (a and b) Immunofluorescence staining for PPARγ (red) in macrophage-specific marker (Mac3, green) or fibroblast-specific marker (S100A4, green) was performed in lung sections on day 14 (14D) following BLM treatment. Arrowheads indicate colocalization of PPARγ in alveolar macrophages or lung fibroblasts. The imaging medium was Vectashield fluorescence mounting medium containing 4',6-diamidino-2-phenylindole (DAPI; not shown). Original magnification × 800. Representative images were obtained from three mice in each group.
PPARγ activation enhances the efferocytic ability of alveolar macrophages
Endogenous apoptotic cells are generated and removed during an inflammatory reaction. Apoptotic neutrophils are likely the dominant cells being removed endogenously and alveolar macrophages may be globally activated after bleomycin treatment.25 Similar to the findings of our previous studies,24 the phagocytic indices in alveolar macrophages from bleomycin+apoptotic cells were significantly higher over the course of lung injury compared with treatment with bleomycin+saline.24 In this study, we evaluated the role of PPARγ activation in increasing the efferocytic ability of alveolar macrophages. Coadministration of GW9662 with apoptotic cells significantly reversed the enhanced efferocytic ability of alveolar macrophages on days 2 (Figure 4a,c) and throughout days 7–21 after bleomycin treatment (Figure 4b). Treatment with the inhibitor and buffers had no effect.

Inhibition of peroxisome proliferator-activated receptor-γ (PPARγ) restores the basal efferocytic ability of alveolar macrophages following apoptotic cell instillation into bleomycin (BLM)-stimulated lungs. Where indicated, 1 mg kg−1 GW9662 (GW, intraperitoneally (i.p.)), NS398 (NS; 3 mg kg−1, intraosseously (i.o.)), or AH6809 (AH; 5 mg kg−1, i.p.) was administered simultaneously with apoptotic cell (ApoJ) instillation 2 days after bleomycin (BLM) treatment and every 12 h (NS398) or every day (GW9662, AH6809) thereafter. Mice were killed on (a) day 2 and on (b) days 7 (7D), 14 (14D), and 21 (21D) following BLM treatment. (a and b) Phagocytic indices were measured in bronchoalveolar lavage (BAL) alveolar macrophages. (c) Representative photomicrographs showing cytospin-stained BAL cells at 2 h after apoptotic cell instillation into the BLM-stimulated lung. Arrowheads indicate alveolar macrophages with engulfed apoptotic cells or fragments. Original magnification × 400. Values represent the mean±s.e.m. of results from five mice in each group. *P<0.05 compared with saline control, +P<0.05 for BLM+ApoJ vs. BLM+Sal, #P<0.05 for BLM+ApoJ vs. BLM+ApoJ+inhibitor.
Cyclooxygenase-2 (COX-2)-induced production of prostaglandin E2 (PGE2), which is the endogenous activator of Rac1, results in increased efferocytic capacity in vitro.29 Thus, we examined whether the COX-2/PGE2 pathway is involved in the apoptotic cell-induced efferocytic ability of alveolar macrophages. Coadministration of the COX-2 selective inhibitor NS-398, or the selective PGE2 receptor EP2 inhibitor AH6809, did not alter efferocytosis of alveolar macrophages at 2 h after apoptotic cell instillation and on days 7–21 after bleomycin treatment (Figure 4a,b).
PPARγ activation mediates the anti-inflammatory response induced by apoptotic cell instillation
Studies have established PPARγ as an inhibitor of activator protein-1, specific protein 1, and nuclear factor-κB-driven proinflammatory cytokine transcription.6, 30 Thus, we examined whether inhibition of apoptotic cell-enhanced PPARγ activity affects tumor necrosis factor-α and macrophage inflammatory protein-2 expression in the lung. We also examined the effects of this inhibition on the inflammatory response, and this we investigated by assessing neutrophil numbers, total protein levels in bronchoalveolar lavage (BAL) fluid, and myeloperoxidase (MPO) activity in lung tissue. Coadministration of GW9662 reversed the reduction of tumor necrosis factor-α and macrophage inflammatory protein-2 mRNA in alveolar macrophages and lung tissue at 2 h after apoptotic cell instillation and 3 days after bleomycin treatment (Figure 5a,b,d,e and Supplementary Figure S6a,b,d,e). GW9662 also restored the protein levels of these cytokines in BAL fluid on days 2 and 3 after bleomycin treatment (Figure 5c,f and Supplementary Figure S6c,f). Inhibition of PPARγ signaling also reversed the apoptotic cell-induced reduction of neutrophil numbers and total protein levels in BAL fluid, as well as MPO activity in lung tissue over the course of lung injury after bleomycin treatment (Figure 5g–i).

Inhibition of peroxisome proliferator-activated receptor-γ (PPARγ) abrogates the anti-inflammatory effects of apoptotic cell instillation. Where indicated, 1 mg kg−1 GW9662 (GW) was administered intraperitoneally (i.p.) simultaneously with apoptotic cell (ApoJ) instillation 2 days after bleomycin (BLM) treatment and every day thereafter. (a–c) Mice were killed at 2 h after ApoJ instillation and on (d–f) days 7, 14, and 21 following BLM treatment. 3D, 3 days; 7D, 7 days; Sal, saline alone. (a, b, d and e) Tumor necrosis factor-α (TNF-α) mRNA levels in alveolar macrophages (AM) and lung homogenates were analyzed by real-time PCR. (c and f) TNF-α protein levels in the BAL fluid were quantified by enzyme-linked immunosorbent assay (ELISA). (g) Neutrophil numbers and (h) total protein levels in the bronchoalveolar lavage (BAL) fluid and (i) myeloperoxidase (MPO) activity in lung tissue. Values represent the mean±s.e.m. from (a, b, d and e) 5 or (c and f, g–i) 10 mice in each group. +P<0.05 for BLM+ApoJ vs. BLM+Sal, #P<0.05 for BLM+ApoJ vs. BLM+ApoJ+GW. *P<0.05 compared with saline control.
PPARγ activation regulates proresolving cytokines after apoptotic cell instillation
The proresolution cytokines, TGF-β, IL-10, and HGF, are induced by apoptotic cell recognition and clearance. We have demonstrated that the presence of apoptotic cells upregulates TGF-β production during the early phase (days 2 and 3 after bleomycin treatment), but downregulates this cytokine during the fibrotic phase after bleomycin treatment.19 In this study, we used an antagonist of PPARγ to determine its role in the expression of these proresolving cytokines over time following apoptotic cell instillation. Coadministration of GW9662 reversed the enhancement of TGF-β mRNA in alveolar macrophages and lung tissue and the protein expression levels in BAL fluid following apoptotic cell instillation during the early phase (Figure 6a–f). Moreover, the reduced levels of TGF-β mRNA and protein following apoptotic cell instillation were partially restored by GW9662 during the late fibrotic phase (days 14 or 21) (Figure 6d–f). These data suggest that PPARγ signaling is involved in apoptotic cell-induced TGF-β production at the transcriptional level during the early inflammatory phase and transcriptional repression during the late fibrotic phase following bleomycin treatment.

Peroxisome proliferator-activated receptor-γ (PPARγ) activation following apoptotic cell instillation mediates changes of transforming growth factor-β (TGF-β) and interleukin-10 (IL-10) expression over the course of bleomycin (BLM)-stimulated lung injury. Where indicated, 1 mg kg−1 GW9662 (GW) was administered intraperitoneally (i.p.) at the same time with apoptotic cell (ApoJ) instillation 2 days after BLM treatment and every day thereafter. (a–c, g–i) Mice were killed at 2 h after ApoJ instillation and on (d–f, j–l) days 7, 14, and 21 after BLM treatment. Sal, saline alone. TGF-β and IL-10 mRNA levels in (a, d, g and j) alveolar macrophages (AM) and (b, e, h and k) lung homogenates were analyzed by real-time PCR. (c, f, i and l) TGF-β1 and IL-10 protein levels in bronchoalveolar lavage fluid (BALF) were quantified by enzyme-linked immunosorbent assay (ELISA). Values represent the mean±s.e.m. from (a, b, d, e, g, h, j and k) 5 or (c, f, i and l) 10 mice in each group. +P<0.05 for BLM+ApoJ vs. BLM+Sal, #P<0.05 for BLM+ApoJ vs. BLM+ApoJ+GW. *P<0.05 compared with saline control.
In comparison, the levels of IL-10 mRNA in alveolar macrophages and lung tissue, as well as protein levels in BAL fluid, were significantly increased at 2 days after bleomycin treatment. IL-10 expression peaked at day 7 before gradually declining until day 21. The expression of IL-10 remained higher than the saline-treated control group (Figure 6g–l). The apoptotic cell instillation increased IL-10 mRNA expression in alveolar macrophages and lung to levels higher than that of the bleomycin+saline group up to day 14. However, the protein levels in BAL fluid were significantly different on days 2, 3, and 7 after bleomycin treatment. Apoptotic cell-induced IL-10 mRNA and protein levels peaked 7 days after bleomycin treatment. In contrast, instillation of viable cells did not alter baseline IL-10 protein levels (Supplementary Figure S2e). Coadministration of GW9662 reversed apoptotic cell-enhanced IL-10 mRNA and protein production on days 2, 3, and 7 after bleomycin treatment, reverting close to the levels exhibited by mice treated with bleomycin only. No significant difference between bleomycin+apoptotic cells with or without GW9662 was observed during the late fibrotic phase.
Previously, we demonstrated that apoptotic cell instillation resulted in persistent induction of HGF mRNA and protein expression during bleomycin-induced lung fibrosis.24 Moreover, inhibition of HGF signaling reversed the anti-inflammatory and antifibrotic effects of apoptotic cell instillation.24, 31 Data from other studies indicated that PPARγ agonists induced PPARγ binding to the peroxisome proliferator response element in the HGF promoter region.32 Thus, we examined the association between PPARγ activity and HGF induction following apoptotic cell instillation. When GW9662 was coadministered with apoptotic cell instillation, the immediate and prolonged enhancement of HGF mRNA and protein were reversed in alveolar macrophages and the lung on day 2 (Figure 7a,b and Supplementary Figure S6g) and throughout days 7–21 (Figure 7d and Supplementary Figure S6h). This treatment also reversed the enhancement of HGF protein in BAL fluid following apoptotic cell instillation on day 2 (Figure 7c) and throughout days 7–21 (Figure 7e). However, coadministration of GW9662 with buffer had no effect throughout the time period examined.

Peroxisome proliferator-activated receptor-γ (PPARγ) activation following apoptotic cell instillation alters hepatocyte growth factor (HGF), interleukin (IL)-4, IL-13, and interferon-γ (IFN-γ) over the course of bleomycin (BLM)-stimulated lung injury. Where indicated, 1 mg kg−1 GW9662 (GW) was administered intraperitoneally (i.p.) simultaneously with apoptotic cell (ApoJ) instillation 2 days after BLM treatment and every day thereafter. (a–c) Mice were killed at 2 h after ApoJ instillation and on (d and e, f–h) days 7, 14, and 21 after BLM treatment. Sal, saline alone. HGF mRNA levels in (a and d, left) alveolar macrophages (AM) and (b and d, right) lung homogenates were analyzed by quantitative real-time PCR. The levels of (c and e) HGF, (f) IL-4, (g) IL-13, and (h) IFN-γ in bronchoalveolar lavage fluid (BALF) were quantified by enzyme-linked immunosorbent assay (ELISA). Values represent the mean±s.e.m. from (a, b and d) 5 or (c and e) 10 mice in each group. +P<0.05 for BLM+ApoJ vs. BLM+Sal, #P<0.05 for BLM+ApoJ vs. BLM+ApoJ+GW. *P<0.05 compared with saline control.
Helper T cell type 2 (Th2) cytokines, such as IL-4 and IL-13, may play an important role in the pathogenesis of pulmonary fibrosis.33 Helper T cell type 1 (Th1) cytokine interferon-γ (IFN-γ) is thought to be an inhibitory modulator.34, 35 We therefore assessed whether apoptotic cell-enhanced PPARγ activity affects Th1/Th2 cytokine levels in BAL fluid. IL-4 and IL-13 protein levels increased significantly at 7 days and declined until 21 days after bleomycin treatment (Figure 7f,g), consistent with the previous investigation.36 Apoptotic cell instillation reduced bleomycin-induced increases in IL-4 and IL-13 levels on day 7 after bleomycin treatment. However, these cytokine levels were not significantly different on days 14 and 21 compared with the bleomycin+saline group. Coadministration of GW9662 reversed partially the reduction of IL-4 and IL-13 in BAL fluid on day 7. In comparison, IFN-γ levels in BAL fluid decreased slightly at 7, 14, and 21 days after bleomycin treatment compared with the saline group (Figure 7h). Apoptotic cell instillation significantly enhanced the IFN-γ levels on day 7 after bleomycin treatment. GW9662 reversed the enhanced IFN-γ levels by apoptotic cell instillation at this time point.
PPARγ activation mediates the antiapoptotic and antifibrotic effects of apoptotic cell instillation
Several studies have demonstrated that PPARγ agonists prevent fibrogenesis in vitro and in vivo.15, 16 Because our data demonstrate that apoptotic cell-enhanced PPARγ signaling mediates persistent HGF production and biphasic TGF-β regulation, we next examined the role of PPARγ in promoting the anti-apoptotic and antifibrotic effects of apoptotic cell instillation by assessing apoptotic activity and several fibrosis markers in experimental lung samples. In our previous study, we assessed DNA damage and apoptosis levels in lungs at day 7 after bleomycin treatment using TUNEL (TdT-mediated dNTP nick end labeling) staining. Apoptotic cell instillation reduced the number of TUNEL-positive alveolar epithelial cells.24 In this study, the decrease in apoptosis levels was confirmed by measure of activities of caspase-3 and -9 and western blot analysis for the cleaved form of caspase-3 (Figure 8a–c). Coadministration of GW9662 reversed reduction of activities of caspase-3 and -9 and cleaved caspase-3 expression at 7 days after bleomycin treatment. Moreover, the reduction of matrix markers of fibroproliferation, such as type 1 collagen α2 and fibronectin, and a marker of myofibroblast differentiation, α-SMA, at days 14 and 21 after bleomycin treatment was also reversed by GW9662 (Figure 8d). Hydroxyproline, a marker of collagen content in lung tissue, was also restored by this inhibitor (Figure 8e). Inhibitor alone had no effect. Furthermore, trichrome staining revealed that the reduction in collagen-stained interstitial areas with damaged alveolar structures by apoptotic cell instillation at day 21 after bleomycin treatment was reversed by long-term PPARγ inhibition (Figure 8f).

Inhibition of peroxisome proliferator-activated receptor-γ (PPARγ) activation abrogates the antiapoptotic and antifibrotic effects of apoptotic cell instillation into bleomycin (BLM)-stimulated lungs. Where indicated, 1 mg kg−1 GW9662 (GW) was administered intraperitoneally (i.p.) simultaneously with apoptotic cell (ApoJ) instillation 2 days after BLM treatment and every day thereafter. Mice were killed on days 7, 14, and 21 following BLM treatment. (a) Caspase-3, (b) cleaved caspase-3 expression, and (c) caspase-9 activities in lung tissue on day 7. (b and d) Western blots probed with anticleaved caspase-3, anti-type 1 collagen α1 (Col1), or anti-fibronectin (Fn) antibody were employed to monitor the cleaved caspase-3 fragments, Col1, or Fn in lung tissue homogenates. The relative densitometric intensity was determined for each band and normalized to α-tubulin. (e) Collagen deposition in the whole lung was determined by measuring hydroxyproline content on days 14 (14D) and 21 (21D). Values represent the mean±s.e.m. from five mice in each group. *P<0.05 compared with saline control, +P<0.05 for BLM+ApoJ vs. BLM+Sal, #P<0.05 for BLM+ApoJ vs. BLM+ApoJ+GW. (f) Lung sections were visualized with Masson’s trichrome staining on day 21 (original magnification × 200). Representative results from five mice per group are shown.
PPARγ activation enhances survival rate following apoptotic cell instillation
Of the mice treated with bleomycin, 50% and 85% died within 14 and 21 days, respectively (Figure 9). However, instillation of apoptotic cells reduced the death rate (15% and 20% mortality, respectively). However, coadministration of GW9662 with apoptotic cell instillation reversed reduction of the death rate (32% and 63% mortality, respectively).

Inhibition of peroxisome proliferator-activated receptor-γ (PPARγ) activation reduces the survival rate following apoptotic cell instillation. Where indicated, 1 mg kg−1 GW9662 (GW) was administered intraperitoneally (i.p.) simultaneously with apoptotic cell (ApoJ) instillation 2 days after bleomycin (BLM) treatment and every day thereafter. The percentage of surviving mice was plotted over a 21-day period. There were 20 mice in each group.
DISCUSSION
The feasibility of cellular therapy based on the immunomodulatory properties of apoptotic cells has already been evaluated in different experimental models of acute and chronic inflammation to restore or induce immune tolerance. Indeed, the administration of apoptotic cells has been shown to attenuate lipopolysaccharide-induced acute lung injury or sepsis.25, 37, 38 Moreover, apoptotic cell injection has also been used to reduce both the acute and chronic phases of inflammatory arthritis,39 and insulitis in mice type-1 diabetes.40 These beneficial effects have been attributed to the release of anti-inflammatory cytokines, such as TGF-β and IL-10, by macrophages upon apoptotic cell recognition and clearance. However, TGF-β1 production in alternatively activated macrophages has been linked to the development of pulmonary fibrosis.41, 42, 43 Several studies have demonstrated that PPARγ plays important roles in regulating processes related to fibrogenesis in vitro and in vivo.14, 15 Thus, important questions are raised regarding whether PPARγ activation is enhanced by in vivo exposure to apoptotic cells over the course of lung injury, thereby leading to altered alveolar macrophage programming and, ultimately, hastening the resolution of both lung inflammation and fibrosis. Inflammatory responses have been associated with the initial stage of idiopathic pulmonary fibrosis.44 Therefore, an effective treatment strategy for pulmonary fibrosis should be administrated during the early state.45, 46 Indeed, in our preliminary studies, early exposure of apoptotic cells 2 days after bleomycin treatment provided significant effects on inflammatory responses, including neutrophil recruitment and total protein contents, and HGF production in BAL fluid, when examined on days 7 and 14 after bleomycin treatment (Supplementary Figure S7a–c). However, little or no changes in these parameters were shown when apoptotic cells were exposed at 6 or 13 days after bleomycin treatment. Thus, based on this observation, apoptotic cells were instilled 2 days after bleomycin treatment not adding them at the late phase of inflammation and fibrosis induction.
We first characterized changes in PPARγ expression and activation following apoptotic cell instillation over the course of bleomycin-induced lung injury. Genovese et al.19 have demonstrated enhanced PPARγ staining in lung sections obtained from bleomycin-treated mice. Interestingly, Standiford et al.47 found higher PPARγ mRNA levels and transcriptional activity in alveolar macrophages isolated from patients with acute lung injury compared with healthy control subjects. The administration of apoptotic cells has been shown to attenuate lipopolysaccharide-induced acute lung injury in mice.25 Thus, it might be an important question of whether administration of apoptotic cells to patients with acute lung injury enhances the induction and activity of PPARγ, then consequently resulting in resolution of inflammation and prevention of lung injury. In this study, we show that PPARγ mRNA and protein levels in alveolar macrophages and lung tissue were immediately enhanced following instillation of apoptotic cells relative to bleomycin with or without viable cells. Moreover, the levels of PPARγ mRNA and protein following early exposure to apoptotic cells increased gradually for up to 21 days after bleomycin treatment. However, the presence of viable Jurkat cells had no effect. These data demonstrate the specificity of PPARγ induction for apoptotic cell recognition systems.
The factors that lead to upregulation of PPARγ in the setting of bleomycin-induced lung injury are beyond the scope of this project, but a variety of endogenous ligands for PPARγ such as fatty acids, PGD2-derived 15-deoxy-12,14 PGJ2 (15d-PGJ2), and lysophosphatidic acid can activate PPARγ in a variety of cell types.19, 47 Indeed, levels of PPARγ and PPARγ ligands, such as PGD2 and 15-hyrdoxyoctadecadienoic acid (15-HETE) were elevated in alveolar macrophages isolated from patients with acute lung injury.47 Similarly, in this study, we found that 15d-PGJ2 in BAL fluid increased up to 14 days and slightly declined at 21 days after bleomycin treatment (Supplementary Figure S8a,b). Many cell types in the lung are capable of expressing PPARγ. Based on our results, alveolar macrophages are a primary source of PPARγ following in vivo exposure to apoptotic cells. Moreover, immunohistochemistry and double immunofluorescence staining studies indicate that apoptotic cell-induced PPARγ expression was observed mainly in macrophages and, to some extent, fibroblasts on days 7 and 14 after bleomycin treatment.
Concomitant with PPARγ expression, immediate and prolonged enhancement of PPARγ activity following apoptotic cell instillation was demonstrated in bleomycin-stimulated lung tissue. In support of this, enhanced expression of direct PPARγ targets, including CD36, MMR, and Arg1, in alveolar macrophages and lung tissue following apoptotic cell instillation was greater than that observed following bleomycin with or without viable cells. Interestingly, all of these genes are known to be upregulated by PPARγ activation and are characteristic of alternative macrophage programming.6, 27, 28 Indeed, inhibition of PPARγ activity by treatment with its antagonist GW9662 reversed apoptotic cell-enhanced mRNA and protein expression of these targets. Notably, the enhancement of phagocytic indices in alveolar macrophages after 2 h of in vivo exposure to apoptotic cells and up to 21 days after bleomycin treatment was restored to the levels seen in the bleomycin-only-treated group. However, inhibition of other signaling pathway, including the COX-2/PGE2 pathway, did not reverse the enhanced phagocytic ability of alveolar macrophages. These data suggest that enhanced PPARγ activity plays a critical role in inducing efferocytosis by alveolar macrophages following in vivo exposure to apoptotic cells. Consistent with this, previous studies have shown that macrophage-specific deletion of PPARγ in mice also resulted in decreased apoptotic cell uptake.6 GW9662 could also reverse the observed augmentation of efferocytosis in human macrophages induced by glucocorticoids. Thus, persistently enhanced PPARγ activity following early in vivo exposure to apoptotic cells might strengthen apoptotic cell recognition and clearance system and prevent a defect in the ability of macrophages to clear them during bleomycin-induced lung injury. Notably, in human pulmonary fibrosis, Morimoto et al.48 showed that, like other chronic lung diseases, there is defective efferocytic ability and an increase in the number of apoptotic cells. However, the rate of neutrophil apoptosis as well as secondary necrosis along with efferocytic ability in the progress of human pulmonary fibrosis has not been reported. Nevertheless, the therapeutic use of apoptotic cells needs to be carefully considered in cases in which the capacity for apoptotic cell clearance is reduced during lung injury, and in which administered cells may progress into secondary necrosis that could exacerbate inflammation or injury.49
The mechanisms by which PPARγ activation mediates enhanced efferocytosis require further investigation. However, this biological process likely involves ligand or signaling-dependent transactivation of efferocytic surface receptors and markers of alternative macrophage activation, such as CD36, MMR, and Arg1.6, 27, 28 These data support an important and emerging concept that macrophage recognition of apoptotic cells can reinforce signaling pathways that shift their programming toward enhanced efferocytic ability in a feedback loop.50, 51 One mechanism by which apoptotic cells enhance efferocytic programming is through phosphatidylserine-dependent induction of IL-4 signaling that upregulates PPARγ.9 Autocrine stimulation by TGF-β produced in response to apoptotic cell recognition may similarly enhance PPARγ expression.26 These proposed mechanisms need to be verified in our in vivo experimental setting. Nevertheless, these hypotheses may not explain the effects observed during the late fibrotic phase as TGF-β and PPARγ are regulated reciprocally after apoptotic cell instillation. In addition, it is possible that PPARγ ligands, such as 15d-PGJ2 and 15-HETE, might act as other factors to enhance PPARγ expression following apoptotic cell instillation. However, the significant enhancement of 15d-PGJ2 following apoptotic cell instillation was not observed during late inflammatory and fibrotic phases after bleomycin treatment (Supplementary Figure 8b).
GW9662 restored the levels of proinflammatory mediators, including tumor necrosis factor-α and macrophage inflammatory protein-2, in alveolar macrophages, lung, and BAL fluid. Moreover, GW9662 inhibited apoptotic cell-induced neutrophil recruitment and enhancement of total protein in BAL fluid and MPO activity in the lung during early inflammatory phase. These data suggest that induction of the inflammatory response may be prevented by enhanced PPARγ expression and activity following apoptotic cell instillation. The effects of PPARγ activation may be mediated by several mechanisms. The anti-inflammatory action of PPARγ agonism is reportedly mediated via its transrepression of proinflammatory mediator transcription.6, 31 In addition, significant indirect effect via enhanced apoptotic cell recognition that then leads to the production of suppressive mediators, such as TGF-β and IL-10, might be even more significant.25, 26, 52 Our study supports this concept as coadministration of GW9662 reversed apoptotic cell-induced TGF-β and IL-10 expression during early inflammatory phase after bleomycin treatment. These data suggest that enhanced PPARγ activity upregulates efferocytosis in a positive feedback loop, leading to increased TGF-β and IL-10 gene expression and production that may be critical for regulating the activity of these anti-inflammatory mediators. However, further investigation is required to understand why IL-10 was upregulated for longer periods than TGF-β following apoptotic cell instillation in early inflammatory phase (peak response at 7 days for IL-10 vs. 3 days for TGF-β after bleomycin treatment). These results highlight the complexity of the effect of apoptotic cells as well as the possible involvement of various cross-talking networks that act on transcription and/or translation to produce these cytokines.
The fibroblast is now recognized as the key effector cell in the normal wound-healing process as well as the development of fibrosis.53 Improper repair may result in dysregulated proliferation and activation of fibroblasts with increased expression of collagen and other matrix proteins. Myofibroblasts are recruited from a variety of sources including local mesenchymal cells, bone marrow progenitors (called fibrocytes), and via a process called epithelial–mesenchymal transition. Recent studies employing in vitro and in vivo approaches have revealed an entirely novel role for PPARγ in fibrogenesis. Remarkably, both natural (15d-PGJ2) and synthetic PPARγ agonists inhibited TGF-β-driven myofibroblast differentiation and type I collagen protein production.54, 55 In particular, Milam et al.18 demonstrated that treatment of lung fibroblasts obtained from idiopathic pulmonary fibrosis patients or normal human controls with PPARγ agonists results in inhibition of proliferative responses and induces cell cycle arrest. The PPARγ agonist inhibited fibrosis, as assessed by measurement of lung hydroxyproline and collagen and by trichrome staining in the bleomycin-induced murine model. Thus, in our study, we could hypothesize that enhanced PPARγ expression and activation following apoptotic cell instillation contributes to the prevention of excessive fibrosis, leading to successful tissue repair because of enhanced apoptotic cell recognition and clearance from the lesion. Indeed, coadministration of GW9662 reversed reduction in apoptotic activity, hydroxyproline contents, type 1 collagen α2, fibronectin, and SMA expression in lung tissue. The antifibrotic effect of apoptotic cell instillation via enhanced PPARγ activity was confirmed by lung histology using trichrome staining of lung sections.
PPARγ agonists have been shown to induce the expression of HGF via a peroxisome proliferator response element in the HGF promoter20, 32 and repress TGF-β-mediated myofibroblast activation.56 Similarly, our data demonstrate that inhibition of PPARγ activity by GW9662 reversed the immediate and prolonged increase in HGF mRNA and protein following apoptotic cell instillation. These findings emphasize that persistent amplification of HGF production after exposure to apoptotic cells is strengthened through additional PPARγ signaling. In comparison, long-term inhibition of PPARγ signaling partially reversed the reduction of TGF-β mRNA and protein levels induced by apoptotic cell instillation during late fibrotic phase after bleomycin treatment. However, administration of GW9662 with buffer alone had no effect throughout the time period investigated. These findings indicate that enhanced PPARγ activity mimics the apoptotic cell-mediated effects on HGF and TGF-β production after bleomycin treatment. The mechanisms by which PPARγ activity modulates reduced TGF-β expression by apoptotic cell instillation during fibrosis remain poorly understood in the context of enhanced efferocytosis. Nevertheless, it may be because of synergy of the transrepressive effects on TGF-β30 and upregulation of HGF by greater and prolonged enhanced PPARγ activity. Indeed, studies have reported that HGF decreases TGF-β mRNA and protein to promote epithelial cell survival57 and inhibit epithelial–mesenchymal transition.58, 59 Previously, we found that long-term inhibition of HGF signaling reversed the decrease in TGF-β production induced by apoptotic cell instillation 21 days after bleomycin treatment.31 On the other hand, as few effects on IL-10 levels were observed following apoptotic cell instillation with or without GW9662 at days 14 and 21 after bleomycin treatment, it remains possible that persistent, apoptotic cell-enhanced PPARγ activity might contribute little to no changes in IL-10 expression during late fibrotic phase, even though this proresolving cytokine has been demonstrated to attenuate bleomycin-induced pulmonary fibrosis.60
Th1/Th2 imbalance is proposed to be one of the hypothesis related to developing pulmonary fibrosis.33, 36 We found that coadministration of GW9662 reversed the apoptotic cell-induced reduction of IL-4 and IL-13 levels and the enhancement of IFN-γ levels in BAL fluid on day 7. These results suggest that enhanced PPARγ activity contributes, at least in part, to improving the Th1/Th2 balance. Thus, apoptotic cell-induced PPARγ activity may suppress initiation of a fibrotic response by regulating Th1/Th2 balance. In addition, oxidative stress may play an important role in the development of bleomycin-induced fibrosis. Ligand-activated PPARγ promotes the expression of antioxidant genes, such as manganese superoxide dismutase, glutathione peroxidase, heme-oxygenase 1, endothelial nitric oxide synthase, and the mitochondrial uncoupling protein 2.61 Therefore, further studies are needed to determine the effects of enhanced PPARγ activity by apoptotic cells on these antioxidant genes during bleomycin-induced pulmonary inflammation and fibrosis.
Our data demonstrated that apoptotic cell instillation after bleomycin treatment resulted in immediate and prolonged enhancement of PPARγ mRNA and protein expression, accompanied by increased PPARγ activity above the high basal levels found in bleomycin-stimulated lungs. The functional consequence of enhanced PPARγ expression and activity, in turn, regulates the macrophage program of alternative activation with increased efferocytic surface receptors, including the PPARγ target molecules, CD36, MMR, and Arg1. These effects reinforce the enhanced efferocytic ability of alveolar macrophages while reducing lung proinflammatory cytokine expression, neutrophil recruitment, total protein levels, and MPO activity. The additional complex molecular events provided by enhanced PPARγ activity mediate the biphasic regulation of TGF-β during early and late phases, as well as upregulate IL-10 during the early inflammatory phase and HGF during both early and late phases following apoptotic cell instillation. A mechanism diagram summarizing and integrating the effects of enhanced PPARγ activity following apoptotic cell instillation in bleomycin-induced lung fibrosis is shown in Figure 10. Our study supports the concept that administration of apoptotic cells may be a potential tool with great therapeutic efficacy due to induction of PPARγ shifting alternatively programmed alveolar macrophages associated with enhanced efferocytic ability and coordinated regulation of TGF-β, IL-10, and HGF in favor of resolving inflammation and preventing excessive, fibrotic outcomes.

A mechanism diagram summarizing and integrating the effects of enhanced peroxisome proliferator-activated receptor-γ (PPARγ) activity following apoptotic cell instillation in bleomycin-induced lung fibrosis. Arg1, arginase 1; Col1, type 1 collagen α2; ECM, extracellular matrix; Fn, fibronectin; HGF, hepatocyte growth factor; IL-10, interleukin-10; MIP-2, macrophage inflammatory protein-2; MMR, macrophage mannose receptor; α-SMA, α-smooth muscle actin; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; Th1/Th2, helper T cell type 1/2.
METHODS
Reagents. Bleomycin and paraformaldehyde were purchased from Sigma-Aldrich (St Louis, MO). GW9662, NS398, and AH6809 (Cayman Chemical, Ann Arbor, MI) were used as supplied. The antibodies used for western blotting were as follows: anti-PPARγ, anti-CD36, anti-MMR, anti-Arg1 (Cayman Chemical), anti-HGF (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-α-tubulin (Sigma-Aldrich).
Animal protocols. Specific pathogen-free male C57BL/6 mice (Orient Bio, Sungnam, Korea) weighing 20–25 g were used in all experiments. The Animal Care Committee of the Ewha Medical Research Institute approved the experimental protocol. Mouse pharyngeal aspiration was used to administer the test solution containing bleomycin (5 U kg−1 body weight in 30 μl).24, 62 At 2 days after bleomycin treatment, saline alone or 10 × 106 apoptotic or viable Jurkat cells in 50 μl saline were administered intratracheally through pharyngeal aspiration.25, 49 Mice were killed 2 h after apoptotic cell instillation, as well as on days 7, 14, and 21 following bleomycin treatment (n=10 per group). Animals given apoptotic or viable human HeLa epithelial cells or mouse thymocytes were killed 2 h after apoptotic cell instillation (n=10 per group).
For the immediate inhibition experiments, the selective PPARγ antagonist GW9662 (1 mg kg−1, intraperitoneally)63 was administered simultaneously with intratracheal instillation of 10 × 106 apoptotic cells 2 days after bleomycin treatment (n=10 per group). Mice were euthanized at 2 h after apoptotic cell instillation. For the long-term inhibition experiments, after the first dose of GW9662, the inhibitor was administrated once a day, and mice euthanized on days 7, 14, and 21 after bleomycin treatment.
In efferocytosis assay experiments, the COX-2 selective inhibitor NS398 (3 mg kg−1, intraosseously) or the selective PGE2 receptor EP2 antagonist AH6809 (5 mg kg−1, intraperitoneally) were administered simultaneously with instillation of 10 × 106 apoptotic Jurkat cells 2 days after bleomycin treatment (n=10 per group) and mice were killed 2 h later. In long-term inhibition experiments, after the first dose, the inhibitor was administered twice a day (NS-398) or once a day (AH6809), and mice were killed on days 7, 14, and 21 after bleomycin treatment.
BAL cells, lung tissue, and cell counts. BAL was performed and cell counts were determined using an electronic Coulter Counter fitted with a cell sizing analyzer (Coulter Model ZBI with a channelizer 256; Coulter Electronics, Bedfordshire, UK). BAL cells were isolated and cytospun to assess phagocytic indices.24, 25 The phagocytic index was calculated using the following formula: (number of apoptotic bodies)/(200 total macrophages) × 100. After BAL, lungs were removed, immediately frozen in liquid nitrogen, and stored at −70 °C.
Induction of apoptosis. Human T lymphocyte Jurkat cells and HeLa epithelial cells were obtained from the American Type Culture Collection (Rockville, MD). Murine thymocytes were isolated from thymi of 4-week-old C57BL/6 mice by homogenizing the tissue through a 40-μm strainer to separate individual cells. Apoptosis was induced by ultraviolet irradiation at 254 nm for 10 min. The cells were then incubated for 2.5 h before use. They were ∼70% apoptotic by evaluation of nuclear morphology by light microscopy.64
Alveolar macrophage culture. Suspended alveolar macrophages from mice were over 95% viable as determined by Trypan blue exclusion. Alveolar macrophages were isolated by adhesion (60 min).
Quantitative real-time PCR. Gene expression was analyzed using quantitative real-time PCR on a StepOnePlus System (Applied Biosystems, Life Technologies, Carlsbad, CA). For each quantitative real-time PCR assay, a total of 50 ng of complementary DNA was used. Primer sets (Supplementary Table S1) for PCR amplifications were designed using Primer Express software (Applied Biosystems, Foster City, CA). All data were normalized to glyceraldehyde 3-phosphate dehydrogenase52 and are reported as fold change in expression over the control group.
Semiquantitative reverse transcription–PCR. Total RNA was isolated from cultured cells using Invitrogen reagent solution (iNtRon Biotechnology, Seoul, Korea). The concentrations and purities of the RNA samples were evaluated by spectrophotometry. Reverse transcription was conducted for 60 min at 42 °C with 2 μg of total RNA using M-MLV reverse transcriptase (Enzynomics, Seoul, Korea). The primer sequences are listed in Supplementary Table S2. Amplification was carried out using a GeneAmp PCR System 2400 (PerkinElmer, Waltham, MA). Samples were visualized on 1–2% agarose gels stained with ethidium bromide. The relative amounts, as compared with β-actin, were determined by densitometry.
Western blot analysis. Lung tissue homogenate samples were separated on 10% sodium dodecyl sulfate–polyacrylamide gels. Separated proteins were electrophoretically transferred onto nitrocellulose paper. The membranes were probed with specific antibodies to PPARγ, CD36, MMR, Arg1, HGF, type 1 collagen α2, fibronectin, α-SMA, or α-tubulin and visualized by chemiluminescence.
Measurement of PPARγ activity. PPARγ activity was determined in nuclear extracts (8 μg) from lung tissue prepared using a kit and TRANSAM assay (Activ Motif, Carlsbad, CA) according to the manufacturer’s instructions.
Caspase-3 and caspase-9 activities. The bioactivity of caspase-3 and caspase-9 was measured with a Fluorometric Assay Kit (ABcam, Cambridge, UK). In brief, lung homogenate samples were incubated with caspase-3 substrate DEVD-AFC or caspase-9 substrate LEHD-AFC. The fluorescence of the cleaved substrates was determined at an excitation wave length of 400 nm and an emission wave length of 505 nm.
Measurement of total protein concentration. Protein concentrations of the BAL samples were used as indicators of blood-pulmonary epithelial cell barrier integrity. Total protein content in BAL fluid was measured according to the manufacture’s protocols.
Enzyme-linked immunosorbent assay (ELISA). BAL fluid samples were assayed using tumor necrosis factor-α, macrophage inflammatory protein-2, IL-4, IL-13, IFN-γ, TGF-β1, IL-10, and HGF ELISA kits as per the manufacturer’s instructions (R&D Systems, Minneapolis, MN). The activity of MPO in lung homogenates was estimated by using a specific ELISA kit.
Immunocytochemistry. PPARγ protein expression was evaluated in cytospin preparations from freshly isolated alveolar macrophages by immunocytochemistry. Cells were fixed with 4% paraformaldehyde, permeabilized with Triton X-100, and stained with mouse monoclonal anti-PPARγ antibody (Santa Cruz) overnight at 4 °C. Subsequently, cells were washed with phosphate-buffered saline three times and incubated with fluorescent isothiocyanate-conjugated donkey anti-rabbit IgG (Jackson ImmunoReseach, West Grove, PA). The slides were mounted in Vectashield mounting medium with 4',6-diamidino-2-phenylindole (Vector Laboratories, Youngstown, OH). All slides were imaged using a confocal microscope (LSM5 PASCAL; Carl Zeiss, Jena, Germany) equipped with a filter set with excitation at 488 and 543 nm.
Immunohistochemistry. From formalin-fixed, paraffin-embedded tissues, 4 μm sections were obtained. Slides were deparaffinized twice in xylene and rehydrated through graded ethanol solutions to distilled water. Sections were incubated with primary antibody against PPARγ or control rabbit IgG at room temperature for 30 min, incubated with polymeric horseradish peroxidase-linker antibody conjugates as secondary antibodies, and then visualized using 3,3′-diaminobenzidine. To evaluate expression of PPARγ on macrophages and fibroblasts, immunofluorescent staining for the macrophage-specific marker Mac3 (BD Pharmingen, San Jose, CA), fibroblast-specific marker S100A4 (Thermo Fisher Scientific, Fremont, CA), and PPARγ was performed. The sections were incubated with Texas Red-conjugated anti-mouse IgG and fluorescein isothiocyanate-conjugated goat anti-rat IgG (Vector Laboratories). The sections were washed with Tris-buffered saline between all steps. The sections were mounted in Vectashield mounting medium with 4',6-diamidino-2-phenylindole. All slides were imaged using a confocal microscope.
Measurement of hydroxyproline. Lung hydroxyproline content was measured using a hydroxyproline assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) as per the manufacturer’s instructions.
Lung histology. Lung tissue was fixed with 10% buffered formalin and embedded in paraffin. Masson’s trichrome was used to stain 4 μm sections to evaluate inflammation and collagen deposition.
Statistical analysis. Values are expressed as the mean±s.e.m. Analysis of variance was applied for multiple comparisons and Tukey’s post hoc test was applied where appropriate. Student’s t-tests were used for comparisons of two sample means. A P-value of <0.05 was considered statistically significant. All data were analyzed using JMP software (SAS Institute, Cary, NC).
References
Thannickal, V.J., Toews, G.B., White, E.S., Lynch, J.P. III & Martinez, F.J. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 5, 395–417 (2004).
Harari, S. & Caminati, A. IPF: new insight on pathogenesis and treatment. Allergy 5, 537–553 (2010).
Vandivier, R.W., Henson, P.M. & Douglas, I.S. Burying the dead. The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 129, 1673–1682 (2006).
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R & Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 390, 350–351 (1997).
Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L. & Henson, P. M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216 (1992).
Majai, G., Sarang, Z., Csomós, K., Zahuczky, G. & Fésüs, L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur. J. Immunol. 37, 1343–1354 (2007).
Asada, K., Sasaki, S., Suda, T., Chida, K. & Nakamura, H. Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am. J. Respir. Crit. Care Med. 169, 195–200 (2004).
Odegaard, J.I. et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007).
Fernandez-Boyanapalli, R.F. et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine-dependent production of IL-4. Blood 113, 2047–2055 (2009).
Nolan, J.J., Ludvik, B., Beerdsen, P., Joyce, M. & Olefsky, J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 331, 1188–1193 (1994).
Tontonoz, P. et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc. Natl. Acad. Sci. USA 94, 237–241 (1997).
Chang, T.H. & Szabo, E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor gamma in non-small cell lung cancer. Cancer Res. 60, 1129–1138 (2000).
Ricote, M., Li, A.C., Willson, T.M., Kelly, C.J. & Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79–82 (1998).
Simon, D.M. et al. Epithelial cell PPARγ is an endogenous regulator of normal lung maturation and maintenance. Proc. Am. Thorac. Soc. 3, 510–511 (2006).
Uto, H. et al. The peroxisome proliferator-activated receptor-γ agonist, pioglitazone, inhibits fat accumulation and fibrosis in the livers of rats fed a choline-deficient, L-amino acid-defined diet. Hepatol. Res. 32, 235–242 (2005).
Calkin, A.C., Giunti, S., Jandeleit-Dahm, K.A., Allen, T.J., Cooper, M.E. & Thomas, M.C. PPAR-α and -γ agonists attenuate diabetic kidney disease in the apolipoprotein E knockout mouse. Nephrol. Dial. Transplant. 21, 2399–2405 (2006).
Zhao, C., Chen, W., Yang, L., Chen, L., Stimpson, S.A. & Diehl, A.M. PPARγ agonists prevent TGFβ1/Smad3-signaling in human hepatic stellate cells. Biochem. Biophys. Res. Commun. 350, 385–391 (2006).
Milam, J.E. et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 294, L891–L901 (2008).
Genovese, T. et al. Effect of rosiglitazone and 15-deoxy-Delta12,14-prostaglandin J2 on bleomycin-induced lung injury. Eur. Respir. J. 25, 225–234 (2005).
Li, Y., Wen, X., Spataro, B.C., Hu, K., Dai, C. & Liu, Y. Hepatocyte growth factor is a downstream effector that mediates the antifibrotic action of peroxisome proliferator-activated receptor-γ agonists. J. Am. Soc. Nephrol. 17, 54–65 (2006).
Shimabukuro, D.W., Sawa, T. & Gropper, M.A. Injury and repair in lung and airways. Crit. Care Med. 31, S524–S531 (2003).
Peng, R. et al. Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for ‘active’ disease. PLoS One 8, e59348 (2013).
Chung, M.P., Monick, M.M., Hamzeh, N.Y., Butler, N.S., Powers, L.S. & Hunninghake, G.W. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am. J. Respir. Cell Mol. Biol. 29, 375–380 (2003).
Lee, Y.J. et al. Apoptotic cell instillation after bleomycin attenuates lung injury through hepatocyte growth factor induction. Eur. Respir. J. 40, 424–435 (2012).
Huynh, M.L., Fadok, V.A. & Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 109, 41–50 (2002).
Freire-de-Lima, C.G., Xiao, Y.Q., Gardai, S.J., Bratton, D.L., Schiemann, W.P. & Henson, PM Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 281, 38376–38384 (2006).
Coste, A. et al. PPARgamma promotes mannose receptor gene expression in murine macrophages and contributes to the induction of this receptor by IL-13. Immunity 19, 329–339 (2003).
Kiss, R.S., Elliott, M.R., Ma, Z., Marcel, Y.L. & Ravichandran, K.S. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr. Biol. 16, 2252–2258 (2006).
Frasch, S.C. et al. Signaling via macrophage G2A enhances efferocytosis of dying neutrophils by augmentation of Rac activity. J. Biol. Chem. 286, 12108–12122 (2011).
Ricote, M. & Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 1771, 926–935 (2007).
Yoon, Y.S., Lee, Y.J., Choi, J.Y., Cho, M.S. & Kang, J.L. Coordinated induction of cyclooxygenase-2/prostaglandin E2 and hepatocyte growth factor by apoptotic cells prevents lung fibrosis. J. Leukoc. Biol. 94, 1037–1049 (2013).
Jiang, J.G., Johnson, C. & Zarnegar, R. PPAR gamma-mediated transcriptional upregulation of the hepatocyte growth factor gene promoter via a novel composite cis-acting element. J. Biol. Chem. 276, 25049–25056 (2001).
Kikuchi, N. et al. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 11, 31–42 (2010).
Segel, M.J. et al. Role of interferon-gamma in the evolution of murine bleomycin lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 285, L1255–L1262 (2003).
Ziesche, R., Hofbauer, E., Wittmann, K., Petkov, V. & Block, L.H. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 341, 1264–1269 (1999).
Izumo, T., Kondo, M. & Nagai, A. Effects of a leukotriene B4 receptor antagonist on bleomycin-induced pulmonary fibrosis. Eur. Respir. J. 34, 1444–1451 (2009).
Ren, Y. et al. Apoptotic cells protect mice against lipopolysaccharide-induced shock. J. Immunol. 180, 4978–4985 (2008).
Kleinclauss, F. et al. Intravenous apoptotic spleen cell infusion induces a TGF-β-dependent regulatory T-cell expansion. Cell Death Differ. 13, 41–52 (2006).
Perruche, S., Saas, P. & Chen, W. Apoptotic cell-mediated suppression of streptococcal cell wall-induced arthritis is associated with alteration of macrophage function and local regulatory T-cell increase: a potential cell-based therapy? Arthritis Res. Ther. 11, R104–R111 (2009).
Xia, C.Q., Peng, R., Qiu, Y., Annamalai, M., Gordon, D. & Clare-Salzler, M.J. Transfusion of apoptotic beta-cells induces immune tolerance to beta-cell antigens and prevents type 1 diabetes in NOD mice. Diabetes 56, 2116–2123 (2007).
Wynn, T.A. & Barron, L. Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257 (2010).
Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 (2003).
Gibbons, M.A. et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 569–581 (2011).
Pociask, D.A., Chen, K., Choi, S.M., Oury, T.D., Steele, C. & Kolls, J.K. γδ T cells attenuate bleomycin-induced fibrosis through the production of CXCL10. Am. J. Pathol. 178, 1167–1176 (2011).
Cao, H. et al. Hydrogen sulfide protects against bleomycin-induced pulmonary fibrosis in rats by inhibiting NF-κB expression and regulating Th1/Th2 balance. Toxicol. Lett. 224, 387–394 (2014).
Cooper, J.A.D. Pulmonary fibrosis pathways are slowly coming into light. Am. J. Respir. Cell Mol. Biol. 22, 520–523 (2000).
Standiford, T.J., Keshamouni, V.G. & Reddy, R.C. Peroxisome proliferator-activated receptor-{gamma} as a regulator of lung inflammation and repair. Proc. Am. Thorac. Soc. 2, 226–231 (2005).
Morimoto, K., Janssen, W.J. & Terada, M. Defective efferocytosis by alveolar macrophages in IPF patients. Respir. Med. 106, 1800–1803 (2012).
Poon, I.K., Lucas, C.D., Rossi, A.G. & Ravichandran, K.S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180 (2014).
Mukundan, L. et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med. 15, 1266–1272 (2009).
Roszer, T. et al. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 186, 621–631 (2011).
Brown, J.R., Goldblatt, D., Buddle, J., Morton, L. & Thrasher, A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J. Leukoc. Biol. 73, 591–599 (2003).
White, E.S., Lazar, M.H. & Thannickal, V.J. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J. Pathol. 201, 343–354 (2003).
Tan, X., Dagher, H., Hutton, C.A. & Bourke, J.E. Effects of PPAR gamma ligands on TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respir. Res. 11, 21–33 (2010).
Burgess, H.A. et al. PPARgamma agonists inhibit TGF-beta induced pulmonary myofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 288, L1146–L1153 (2005).
Ghosh, A.K., Bhattacharyya, S., Lakos, G., Chen, S.J., Mori, Y. & Varga, J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 50, 1305–1318 (2004).
Fan, S. et al. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3′ kinase. Oncogene 19, 2212–2223 (2000).
Zhang, A., Wang, M.H., Dong, Z. & Yang, T. Prostaglandin E2 is a potent inhibitor of epithelial-to-mesenchymal transition: interaction with hepatocyte growth factor. Am. J. Physiol. Renal Physiol. 291, F1323–F1331 (2006).
Shukla, M.N., Rose, J.L., Ray, R., Lathrop, K.L., Ray, A. & Ray, P. Hepatocyte growth factor inhibits epithelial to myofibroblast transition in lung cells via SMAD7. Am. J. Respir. Cell Mol. Biol. 40, 643–653 (2009).
Nakagome, K., Dohi, M., Okunishi, K., Tanaka, R., Miyazaki, J. & Yamamoto, K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 61, 886–894 (2006).
Polvani, S., Tarocchi, M. & Galli, A. PPARγ and oxidative stress: Con(β) catenating NRF2 and FOXO. PPAR Res. 2012, 641087 (2012).
Rao, G.V.S. et al. Efficacy of a technique for exposing the mouse lung to particles aspirated from the pharynx. J. Toxicol. Environ. Health A 66, 1441–1452 (2003).
Collin, M., Patel, N.S., Dugo, L. & Thiemermann, C. Role of peroxisome proliferator-activated receptor-gamma in the protection afforded by 15-deoxydelta12,14 prostaglandin J2 against the multiple organ failure caused by endotoxin. Crit. Care Med. 32, 826–831 (2004).
Hoffman, P.R. et al. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 155, 649–659 (2001).
Acknowledgements
This work was supported by a National Research Foundation (NRF) grant funded by the Korean government (MSIP; grant 2010-0029353).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Yoon, YS., Kim, SY., Kim, MJ. et al. PPARγ activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol 8, 1031–1046 (2015). https://doi.org/10.1038/mi.2014.130
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.130
This article is cited by
-
Intestinal microbe-dependent ω3 lipid metabolite αKetoA prevents inflammatory diseases in mice and cynomolgus macaques
Mucosal Immunology (2022)
-
The Role of PPAR-γ in Allergic Disease
Current Allergy and Asthma Reports (2021)
-
Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases
Nature Reviews Rheumatology (2020)
-
Impact of fatty acid binding protein 5‐deficiency on COPD exacerbations and cigarette smoke‐induced inflammatory response to bacterial infection
Clinical and Translational Medicine (2019)
-
Programming of macrophages by UV-irradiated apoptotic cancer cells inhibits cancer progression and lung metastasis
Cellular & Molecular Immunology (2019)
