
Abstract
Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), a TNF superfamily member, induces damage of the epithelial cells (ECs) and production of inflammatory mediaters through its receptor Fn14 in a model of acute colitis. In our current study of chronic colitis induced by repeated rectal injection of a hapten, we found that inflammation, fibrosis, and T helper 2 (Th2)-type immunity were significantly reduced in Fn14 gene knockout (KO) mice when compared with wild-type (WT) control mice. Expression of thymic stromal lymphopoietin (TSLP) was lower in Fn14 KO colon ECs than in WT ECs. TWEAK potentiates the induction of TSLP by interleukin-13 (IL-13) in colon explants from WT but not in Fn14 KO tissue. TSLP receptor KO mice exhibit milder chronic colitis, similar to that in Fn14 KO mice. TWEAK and IL-13 synergistically promote fibroblast proliferation. Thus we propose an IL-13-TWEAK/Fn14-TSLP axis as a key mechanism underlying chronic colitis with fibrosis.
Similar content being viewed by others


Introduction
Crohn’s disease and ulcerative colitis are chronic relapsing inflammatory diseases of gastrointestinal tract. In Crohn’s disease, which involves both the small and large intestines, relapsing acute inflammatory illness causes significant tissue remodeling and deformity of the intestine, such as intestinal fibrotic stricture. These legions start as inflammation involving all layers of the intestinal wall, frequently associated with cleft ulcers and fistula, followed by severe fibrosis and stenosis even after inflammation has subdued. However, the mechanism of the pathogenic intestinal fibrosis has just recently been recognized as an important target for the treatment.1, 2
Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) is a cytokine of the TNF ligand superfamily that is constitutively expressed by many immune cell types, including monocytes/macrophages, dendritic cells (DC), neutrophils, natural killer (NK) cells, and also T cells,3, 4, 5, 6 although TWEAK expression has also been reported in some non-hematopoietic cell types, including renal cells.7 The only known signaling receptor for TWEAK, FGF (fibroblast growth factor)-inducible molecule 14 (Fn14), is a highly inducible molecule in epithelial cells (ECs), endothelial cells, and other mesenchymal cell types wherein TWEAK signals through Fn14 to promote the canonical and noncanonical nuclear factor (NF)-κB, mitogen-activated protein kinase, and potentially other pathways.8, 9 Fn14 is induced by various growth factors and proinflammatory cytokines9 and as well as by the bacterial component CpG-DNA10 and cytokines TNF-α and interleukin-13 (IL-13) in the intestinal ECs.11 Fn14 expression appears to be universally upregulated in contexts of tissue injury and disease (reviewed in Winkles,9 Burkly and Dohi,12 and Zheng and Burkly13), including in intestinal epithelium after hapten-induced injury and γ−irradiation injury.10 TWEAK significantly contributes to driving tissue pathology, likely by acting on Fn14-expressing tissue cell types to directly promote tissue damage, potentiate inflammation locally in the disease target tissue, including the infiltration of innate and adaptive immune cell types, and pathological remodeling, including fibrogenic responses.8, 12 However, the contribution of TWEAK/Fn14 signaling to systemic acquired immunity seems to be minimal. In the model of collagen-induced arthritis,14, 15 hapten-induced acute colitis,10 and autoantibody-mediated nephritis induced by chronic graft-versus-host disease,16 TWEAK/Fn14 pathway blocking or deficiency ameliorated inflammation and tissue damage locally within the disease target tissue, with no effect on systemic antigen-specific humoral or cellular immune responses. Consistent with this, Fn14 expression is absent in T and B cells and immune responses to systemic neo-antigen immunization are intact in TWEAK or Fn14 gene–deficient mice (Burkly, unpublished).
We recently found that there is crosstalk between the IL-13, TWEAK/Fn14 and TNF-α signaling pathways in mediating intestinal EC death.11 Therefore, we hypothesized that in disease models where IL-13 has major role, TWEAK/Fn14 may also be significantly involved. IL-13 is a pathogenic profibrotic cytokine in the respiratory tract17 and the major effector of lung tissue remodeling. IL-13 stimulates the proliferation of lung fibroblasts from human subjects with asthma, by induction of platelet-derived growth factor (PDGF)-AA and PDGF-CC.18 IL-13 also mediates fibrosis in a model of chronic intestinal damage induced by the weekly intracolonic injection of hapten, trinitrobenzene sulfonic acid (TNBS) with ethanol.19, 20 This model features increased collagen and tissue architecture remodeling of the colon along with increased expression of transforming growth factor (TGF)-β1, collagen (Col1a2), matrix metalloproteinases (MMPs), and tissue inhibitor of metalloproteinases TIMP-1.21 These pathological changes were partially dependent on NF-κB.20 Further, this model induced in T helper type 2 (Th2)-prone BALB/c mice was shown to be mediated primarily by IL-13 followed by the induction of TGF-β.19 In this study, we used this model to investigate the potential contribution of the TWEAK/Fn14 pathway to the pathology of the Th2-type chronic intestinal inflammation and fibrosis and interplay with IL-13 in mediating its effects.
Results
The TWEAK/Fn14 pathway promotes chronic colitis with fibrosis in chronic TNBS-induced colitis in mice
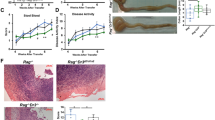
Chronic colitis was induced by rectal injection of TNBS with ethanol weekly for 6 consecutive weeks in wild-type (WT) and Fn14 or TWEAK knockout (KO) BALB/c mice. Colons from naive WT and KO mice were comparable, as previously reported.10
There were no macroscopic open ulcers in the WT mouse colon after chronic TNBS administration, in contrast to that frequently seen in the acute model. However, histologically, the crypts were lost and replaced by leukocyte infiltration along with fibroblasts and connective tissue. Accumulated collagen was detected in the mucosal as well as in the submucosal layer as previously reported in this model (Figure 1a). The degree of crypt loss and accumulation of collagen was much reduced in Fn14 KO as compared with WT mice (Figure 1a). Weight gain of WT mice with colitis was reduced, whereas the Fn14 KO mice gained weight normally (Figure 1b). In agreement with histological findings, when the ratio of collagen/non-collagen protein was measured in colon sections, we found an increased ratio in WT mice with chronic colitis, with a significantly lower value in Fn14 KO (Figure 1c). Total histological scores as well as scores for the degree of crypt loss were significantly higher in WT as compared with that in Fn14 KO mice (Figure 1d). Infiltration of CD45+ leukocytes, F4/80+ macrophages, Gr1+ granulocytes, and CD11c+ DC was observed and infiltration of these cell types was attenuated in Fn14 KO as compared with that in WT mice (Figure 2a). Lymphocyte infiltration in the LP was not as obvious (data not shown); however, enlargement of colonic patches and lymphoid follicles were more severe in WT mice than in Fn14 KO mice (Figure 2a). In addition, infiltration of eosinophils was seen in WT mice but was not seen much in Fn14 KO mice, likely reflecting the reduction in granulocytes (Figure 2a). Total leukocyte infiltration area was significantly reduced in the Fn14 KO colon (Figure 2b). These results indicate that Fn14 deficiency ameliorated colonic damage, inflammation, and fibrosis in the chronic colitis induced by repeated injection of TNBS.

Preserved epithelial cells and less fibrosis in Fn14 (FGF-inducible molecule 14) knockout (KO) mice in chronic colitis. (a) After 6 weekly injections of trinitrobenzene sulfonic acid (TNBS), formalin-fixed paraffin-embedded colon sections were prepared for hematoxylin and eosin (H&E) staining (left column) and detection of collagen with Sirius red and non-collagen proteins with fast green (right column). Naive wild-type (WT) is also representative of both naive WT and KO mice. Representative WT and KO with TNBS-induced colitis are shown. Bars =100 μm. (b) Body weight curves of WT or Fn14 KO mice after induction of colitis (WT:TNBS, Fn14 KO:TNBS, N=9, respectively). Naive mice (N=4) are shown with dotted lines. Data are shown as mean with s.e.m. Difference between WT:TNBS and Fn14 KO:TNBS was statistically significant (P<0.001 by two-way analysis of variance analysis). There was no difference between naive WT and FN14 KO mice. (c) Ratio of collagen/non-collagen proteins extracted from formalin-fixed paraffin-embedded colon sections prepared from naive or inflamed colon of WT or Fn14 KO mice. (d) Scores for total histology and loss of crypts.In c and d, each dot represents an individual mouse.

Infiltrating cells in chronic trinitrobenzene sulfonic acid (TNBS)-induced colitis. (a) Frozen colon sections from wild-type (WT) and Fn14 (FGF-inducible molecule 14) knockout (KO) mice after 6 weekly injections of TNBS stained with anti-CD45, anti-Gr1, anti-CD11c, or anti-F4/80, to visualize leukocytes, neutrophils, dendritic cells (DC), and macrophages, respectively, show infiltration in both the mucosal and submucosal layers. CD11c+ DCs were mainly in the mucosal layer. All types of cells were localized in the areas of crypt loss and were more abundant in WT colon than in KO tissue. Panels with hematoxylin and eosin (H&E) staining demonstrate the enlargement of colonic patches and eosinophil infiltration at a higher extent in WT mice than Fn14 KO mice. White bars=100 μm, thick black bars=500 μm, and thin black bars=50 μm. Colonic patches and some typical eosinophils infiltrating in lamina propria are indicated with arrows and arrowheads, repectively. (b) Total leukocyte cell infiltration determined as CD45+ area. Images as shown in a were subjected to image analysis to measure CD45 positive area. Each group included values of three fields from each of six mice.
Differential gene expression in chronic TNBS-induced colitis in WT and Fn14 KO mice
In order to elucidate the mechanisms underlying protection by Fn14 deficiency in chronic TNBS-induced colitis, gene expression profiling was conducted using whole-colon tissue after 6 weekly TNBS administrations (Figure 3). In WT colon, mast cell and eosinophil-related molecules are highly increased in mice with colitis as compared with the naive condition, reflecting the local infiltration of these cell types. In addition, molecules expressed in macrophages and DC, including the expression of innate sensors and associated molecules, are upregulated, and these cells are apparently also activated because MHC (major histocompatibility complex) class II molecules are upregulated. Interestingly, we also found strong upregulation of signature molecules of alternatively activated macrophages, which are induced by Th2-type cytokines,22 namely, arginase 1 (arg1), chitinase 3-like 3 (chi3l3 also known as YM1), and dectin-1 (Clec7a). Intelectin (Itlna), which was reported to be induced in ECs by parasite infection23 and IL-13,24 is also upregulated. In addition, genes related to NK cells and neutrophils and B-cell- and T-cell-related molecules were increased in the inflamed WT colon. An array of chemokines that mediate leukocyte recruitment, as well as a variety of effector cytokines, MMPs, and inflammation-related adhesion molecules, were also upregulated. Also, of note, TGF-β and IL-13Rα2, both of which are molecular mediators of the fibrotic changes in this colitis model,19 were upregulated. Importantly, collagen and matrix molecule expression were upregulated matching the result of the histology.

Gene expression in chronic trinitrobenzene sulfonic acid (TNBS)-induced colitis in wild-type (WT) and Fn14 (FGF-inducible molecule 14) knockout (KO) colon. Total RNA from whole-colon tissue was harvested from 4–7 mice per experimental group and subjected to microarray analysis. Genes with >two fold change in expression with TNBS as compared with naive tissue are shown for WT and Fn14 KO, with fold change corresponding to the color key, and genes, manually classified according to their gene ontology and cell type expression. Asterisk indicates statistically significant difference between WT and Fn14 KO mice. DC, dendriticells; MHC, major histocompatibility complex; MMP, matrix metalloproteinases; NK, natural killer.
By contrast, in the Fn14 KO colon, mast cell and eosinophil-related molecules were upregulated to a lesser degree than in WT mice. Macrophage and DC-related molecules as well as MHC molecules, which were highly induced WT mice, mostly did not change from the naive condition in Fn14 KO mice. These findings may reflect the lower cell infiltration in Fn14 KO mice. Interestingly, the expression pattern of genes related to B and T cells suggests that a different type of adaptive response is induced in WT and in Fn14 KO mice. For example, immunoglobulin G1 (Igh-4), which is characteristic of Th2-type responses, is increased in WT colon, whereas immunoglobulin G2a (Igh-1a), which characterizes Th1-type responses, is increased in Fn14 KO mice. In addition, the increase in ICOS (inducible costimulator on activated T cells), a hallmark of Th2 cells, and CD69 indicates activated T-cell infiltrates in WT mice but not in KO colons. Most cytokines and chemokines and their receptors, including TGF-β and IL-13Rα2, and connective tissue components and MMPs that were upregulated in WT did not change in Fn14 KO mice as compared with the naive condition, matching the histologically decreased fibrosis and cell infiltration in Fn14 KO mice.
Overall the gene expression profiles support a more prominent Th2-type response and more fibrotic changes with severe inflammation in WT as compared with Fn14 KO mice, mediated by proinflammatory cytokines and chemokines, matrix remodeling enzymes, TGF-β, and IL-13Rα2.
Trinitrophenyl (TNP)-specific immune response was attenuated in Fn14 KO mice with chronic TNBS-induced colitis
As activated T and B cells were involved in the severe inflammation in the WT colon with TNBS-induced colitis and as the expression of genes associated with these adaptive immune cell types altered in KO colon, reflecting reduced Th2-type infiltrates in the KO colon, we investigated the basis for this phenotype. Sacral lymph node (SLN), the draining lymph node of the colon, was swollen after induction of chronic colitis in WT mice, the number of harvested cells was significantly increased in WT as compared with Fn14 KO mice (Figure 4a), and a TNP-specific proliferative response was seen in WT but not in Fn14 KO SLN cells (Figure 4b). This was surprising as Fn14 is not expressed by T or B cells, and TWEAK/Fn14 pathway deficiency has not previously been shown to affect adaptive immunity.13

Decreased antigen-specific responses in sacral lymph node (SLN) of Fn14 (FGF-inducible molecule 14) knockout (KO) mice with chronic trinitrobenzene sulfonic acid (TNBS)-induced colitis. (a) Number of cells freshly harvested from SLN. (b) Trinitrophenyl (TNP)-specific SLN cell proliferation. SLN cells were cultured with or without TNBS and cultured for 2 days. Proliferation response was shown as the ratio of cell number after the culture with TNBS/without TNBS. (c) Cytokine mRNA levels of freshly harvested SLN from wild-type (WT) or Fn14 KO (N=4 for each group). The quantitative reverse transcriptase–PCR (qRT-PCR) results for interleukin (IL)-13 and interferon (IFN)-γ each expressed relative to WT value defined as one. (d) TNP-specific cytokine production in SLN cells. SLN cells were cultured with (+) or without (−) TNBS treatment and culture supernatant was subjected to multiplex cytokine assay. (e) TNP-specific cytokine mRNA induction in SLN cells. SLN cells were cultured as in d and harvested cells subjected to qRT-PCR for IL-13 or IFN-γ. Each dot represents an individual mouse. Statistically significant P values (P<0.05) are shown. TNF, tumor necrosis factor.
When cytokine mRNA was measured in pooled fresh SLN cells from WT and Fn14 KO mice with chronic colitis, SLN cells from Fn14 KO mice expressed lower levels of IL-13 transcripts than those of WT mice (Figure 4c). No difference was seen in the levels of interferon (IFN)-γ. We also measured the TNP-specific cytokine release by SLN cells and found that IL-2, IL-4, IL-5, IL-6, IL-10, IL-13, and TNF-α were secreted in a TNP-specific manner in WT SLN and were higher in WT than in Fn14 KO mice. IL-12 and IFN-γ were detected without TNBS stimuli at comparable levels in SLN cultures from WT and Fn14 KO mice, and statistically significant induction of IL-12 and IFN-γ was not seen in either WT or in KO cultures (Figure 4d). IL-17 and IL-25 were also measured but not detected in most of the samples. We further confirmed the cytokine expression of TNP-stimulated SLN cells by quantitative reverse transcriptase–PCR. The level of IL-13 was significantly lower in Fn14 KO mice, while there was no difference in the level of IFN-γ transcripts (Figure 4e). These results indicate that a Th2-type TNP-specific immune response with higher production of IL-13 was the major event in WT mice, and it was much attenuated in Fn14 KO mice.
To identify the IL-13-producing cells, we did intracellular staining of IL-13 in mesenteric lymph node (MLN) from WT mice with chronic colitis. Given the relative paucity of cells in SLN, we used MLN cells for the assay after confirming they exhibited a similar cytokine gene expression pattern as reported for SLN in Figure 4c (data not shown). Without stimulation, IL-13-producing cells were not detected. With anti-CD3 and anti-CD28 antibody stimulation, IL-13-producing cells were detected within the CD3+ and T-cell receptor-β+ (TCRβ+) cells but not in TCRγδ+ cells or NKG2D+ NK-type cells (Supplementary Figure S2). Recently, new IL-13-producing innate cell types were identified.25, 26, 27 To address the possibility that these cell types may produce IL-13, we also stimulated MLN cells with phorbol 12-myristate 13-acetate plus ionomycin and examined IL-13+ cells in the Lin−ST2+ IL-7R+ or IL-7R− subsets. We detected IL-13+ cells but found them to be very rare (2.73% of the total IL-13+ cells). Therefore, we conclude that major source of IL-13 in this model is CD3+ T cells.
In order to address whether there was an intrinsic functional defect in IL-13 production as a result of Fn14 deficiency, the MLN cells from WT or Fn14 KO mice were stimulated with anti-CD3/CD28 antibodies. The frequency of IL-13+ cells was comparable in KO and WT mice (Figure 5a) as were the levels of IL-13 released (Figure 5b), indicating no such intrinsic defect in Fn14 KO MLN cells. This was also true in case of lamina propria mononuclear cells (LPMC; data not shown). By contrast, restimulation with TNBS induced more IL-13+ cells in WT than in Fn14 KO (Figure 5a), in agreement with the attenuated Th2 cytokine secretion shown in Figure 4d.

Interleukin (IL)-13 producing cells in mesenteric lymph node (MLN). (a) MLN cells from wild-type (WT) or Fn14 (FGF-inducible molecule 14) knockout (KO) mice with chronic colitis were stimulated with anti-CD3 (2 μg ml−1 of coating concentration) and anti-CD28 antibody (1 μg ml−1) or trinitrobenzene sulfonic acid (TNBS) and cultured for 48 h. Intracellular staining for IL-13, and surface staining for CD3 and T-cell receptor-β (TCRβ) was performed and results of CD3 gated cells are shown with percentages indicated in the quadrants, representative data of two independent culture of pooled MLN cells from two mice for each genotype. (b) MLN cells were stimulated with anti-CD3 antibody and IL-13 secretion was measured. (−) indicates without anti-CD3 antibody. Each symbol indicates an individual mouse. There was no statistically significant difference in WT, Fn14 KO, or TWEAK (tumor necrosis factor (TNF)-like weak inducer of apoptosis) KO-stimulated cells.
Fn14 expression is induced in ECs in chronic TNBS-induced colitis
In order to elucidate the mechanism whereby the TWEAK/Fn14 pathway promoted local inflammation and fibrosis in the colon as well as Th2-type immunity in the draining lymph nodes, we investigated the site of TWEAK/Fn14 pathway expression in TNBS-induced chronic colitis. For this purpose, we separated colon ECs, LPMC and MLN cells from mice with acute stage of colitis (3 day after a rectal injection of TNBS), and chronic phase of colitis (3 days after 6 weekly injections of TNBS) and compared the TWEAK and Fn14 gene expression. To avoid contamination of intraepithelial lymphocytes, ECs were purified as the EpCAM+CD45− cell fraction (Supplementary Methods and Supplementary Figure S1). Naive mice were compared with mice with acute or chronic stage colitis. In naive mice, TWEAK mRNA expression was higher in naive MLN than LPMC or ECs, whereas the level of Fn14 mRNA in naive mice was very low and comparable in the MLN, LPMC, and intestinal ECs (data not shown). By contrast, TWEAK mRNA was apparently increased in WT ECs at both the acute and chronic time points (Figure 6a), although it did not reach statistical difference at chronic time point. A similar pattern of induction of Fn14 mRNA was observed (Figure 6b). Neither TWEAK nor Fn14 changed in MLN or LPMC with colitis induction (Figure 6a,b). These data support that the TWEAK/Fn14 pathway is upregulated locally in colonic ECs after TNBS-colitis induction and that TWEAK acts through Fn14 on ECs to promote processes underlying Th2-type colitis and fibrosis. They also suggest that TWEAK/Fn14 pathway promotion of Th2-type immunity in the draining nodes is an indirect consequence of its action on ECs within the colon.

Expression of TWEAK (tumor necrosis factor (TNF)-like weak inducer of apoptosis), Fn14 (FGF-inducible molecule 14), and thymic stromal lymphopoietin (TSLP) in acute and chronic stages of chronic trinitrobenzene sulfonic acid (TNBS)-induced colitis. Gene expression was determined by quantitative reverse transcriptase–PCR using purified epithelial cells (ECs), lamina propria mononuclear cells (LPMC), or mesenteric lymph node (MLN) cells from naive, acute (after single injection of TNBS), and chronic stage (after 6 weekly injections of TNBS) colitis. (a) Expression levels of TWEAK in ECs, LPMC, and MLN. (b) Expression levels of Fn14 in ECs, LPMC, and MLN. (c) Expression levels of TSLP in ECs. In each graph, gene expression levels are expressed relative to the corresponding wild-type (WT) naive sample. Statistically significant P values (P<0.05) are shown calculated with unpaired t test. KO, knockout.
Thymic stromal lymphopoietin (TSLP) induction in ECs is reduced in chronic TNBS-induced colitis in Fn14 KO mice.
ECs are a critical innate cell type within the colon that can shape Th2-type immune responses through their production of Th2-driving cytokines, such as TSLP, IL-33, and IL-25.28, 29 Thus TWEAK may act through Fn14 upregulated on ECs to induce their production of these Th2-conditioning cytokines. The levels of TSLP, IL-33, and IL-25 mRNA in ECs from mice before and after colitis induction was measured. We found that ECs had significantly increased TSLP mRNA acutely and chronically after TNBS induction (Figure 6c), whereas IL-33 levels after induction were marginal and IL-25 levels were not detectable (data not shown). Interestingly, the increase in TSLP mRNA was greater in WT as compared with in Fn14 KO ECs at both stages. Consistent with this, at the level of whole tissue, TSLP was increased 1.7-fold in WT TNBS but not in KO TNBS as compared with their respective naive colons.
TWEAK induces TSLP expression in ECs in combination with IL-13
We further investigated whether TWEAK acts directly on ECs to induce the expression of TSLP by using naive colon tissue explants. Addition of the TWEAK to the WT colon tissue did not induce alteration of the TSLP mRNA level. As IL-13 was previously shown to upregulate TSLP,30 we investigated the potential interplay of TWEAK with IL-13 (Figure 7a). IL-13 showed a trend to upregulate TSLP transcripts but the level did not reach statistically significant difference. Combination of TWEAK and IL-13 strikingly upregulated expression of TSLP, and the difference from control reached a statistically significant level. Importantly, Fn14 KO colon was completely defective in response to TWEAK and IL-13. Therefore, the TWEAK/Fn14 pathway potentiates IL-13-induced TSLP expression. Taken together, these results support that TWEAK promotes Th2-type chronic inflammation and fibrosis by locally inducing EC expression of TSLP.

In vitro synergistic effect of TWEAK (tumor necrosis factor (TNF)-like weak inducer of apoptosis) and interleuin (IL)-13. (a) TWEAK potentiates IL-13-induced expression of thymic stromal lymphopoietin (TSLP) in epithelial cells. Whole-colon tissue from naive wild-type (WT) or Fn14 (FGF-inducible molecule 14) knockout (KO) mice was cultured for 6 h in the presence of TWEAK, IL-13, or both. Total RNA was extracted and TSLP transcripts were measured by quantitative reverse transcriptase–PCR. ‘None’ indicates control culture without cytokines. Expression levels are shown as relative to WT control culture. (b) Mouse embryonic fibroblasts obtained from WT or Fn14 KO BALB/c mice were cultured with TWEAK, IL-13, transforming growth factor-β (TGF-β), or their combination. Cell proliferation was evaluated by the incorporation of EdU (5-ethynyl-2′-deoxyuridine) detected by fluorescence. Data are shown as a mean+s.e.m.
IL-13 and TWEAK synergistically promote fibroblast proliferation
As fibrosis was decreased in Fn14 KO mice, the direct action of TWEAK on fibroblasts was investigated. Mouse embryonic fibroblasts (MEF) from WT BALB/c mice were obtained and their proliferation assessed (Figure 7b). In WT MEF, IL-13 and TGF-β both stimulated MEF proliferation modestly; however, addition of TWEAK greatly enhanced DNA synthesis, especially together with IL-13. These results suggest that TWEAK/Fn14 promotes fibrosis in the presence of IL-13. By contrast, MEF from Fn14 KO mice failed to respond to IL-13 or TWEAK, while their proliferation to TGF-β trended to increase. These results suggest that TWEAK also has a role in supporting the proliferation of fibroblasts, especially in the presence of IL-13.
Chronic colitis with fibrosis was TSLP pathway-dependent.
As the attenuated inflammation and fibrosis in Fn14 KO mice was associated with less TSLP production from ECs, we wished to investigate whether the TSLP pathway has major role in this model, and chronic colitis was induced in TSLPR gene–deficient (TSLPR KO) mice. TSLPR KO mice exhibited a significantly milder colitis as compared with WT mice, very similar to the result in Fn14 KO mice. In comparison with WT mice, histologically less fibrosis was seen (Figure 8a), with more body weight gain (Figure 8b). Collagen/non-collagen protein ratio was higher in naive TSLPR KO mice than in naive WT mice, but no increase was seen after induction of chronic colitis (Figure 8c). They showed a lower total histological score, crypt loss score, and less inflammatory cell infiltration than WT mice (Figure 8d). Swelling of SLN was milder than WT mice as shown by the lower numbers of SLN cells, and they did not show evident TNBS-specific proliferative responses (Figure 8e). These results indicate that the TSLP pathway has a major role in this model. Finally, addition of exogenous TSLP to LPMC cultures from naive WT or Fn14 KO colon resulted in a similar induction of IL-13 transcripts, suggesting that the decreased IL-13 secretion in Fn14 KO mice described above was due to the reduction in TSLP production by Fn14 KO ECs but not due to a difference in other type of cells (Figure 8f).

Preserved epithelial cells and less fibrosis in thymic stromal lymphopoietin receptor (TSLPR) knockout (KO) mice in chronic colitis. (a) Formalin-fixed paraffin-embedded colon sections from naive or trinitrobenzene sulfonic acid (TNBS)-injected TSLPR KO or wild-type (WT) mice were prepared for hematoxylin and eosin (H&E) staining (left column) and detection of collagen with Sirius red and non-collagen proteins with fast green (right column). Bars=100 μm. (b) Body weight curves of WT or TSLPR KO mice with colitis. Data shown are mean with s.e.m. WT and TSLPR KO group are significantly different (P<0.001 by two-way analysis of variance analysis). (c) Ratio of collagen to non-collagen proteins extracted from formalin-fixed paraffin-embedded colon sections prepared from naive or inflamed colon of WT or TSLPR KO mice. (d) Scores for total histology, crypt loss, and leukocyte infiltration determined as CD45+ area. (e) Number of cell yield from freshly harvested sacral lymph node (SLN) and TNBS-specific proliferative responses in WT and TSLPR KO mice determined as described in Figure 4b. (f) TSLP induced interleukin (IL)-13 mRNA expression in WT and Fn14 (FGF-inducible molecule 14) KO lamina propria mononuclear cells (LPMC). Colon LPMCs obtained from naive WT or Fn14 KO mice were stimulated with TSLP (30 ng ml−1) for 16 h and mRNA of IL-13 quantified. Data are expressed relative to WT without TSLP defined as one as a mean+s.e.m. of three mice for each genotype. In c, d and e, each dot represents an individual mouse. WO, without.
Discussion
In this study, we show for the first time the role of the TWEAK/Fn14 pathway in a model of chronic fibrotic Th2-type colitis that was previously shown to depend on IL-13. In this model of chronic TNBS-induced colitis, the expression of TWEAK and Fn14 was induced locally in colonic ECs, and deficiency of Fn14 resulted in milder inflammation, tissue damage, and fibrosis than in WT mice. Fn14 KO mice with chronic colitis also exhibited attenuated Th2-type adaptive immune responses in the draining lymph nodes, including reduced IL-13 as compared with WT mice, where the major IL-13-expressing cells were identified as CD3+ TCRβ+ T cells. However, their lymph node had no intrinsic defect in IL-13 production and little, if any, expression of Fn14, implicating an indirect mechanism of TWEAK signaling through Fn14 in colonic ECs. Further, we elucidate this mechanism by demonstrating Fn14-dependent expression of the Th2-promoting cytokine TSLP by ECs in mice with acute and chronic colitis and TWEAK potentiation of IL-13-induced TSLP in WT colon explants. Importantly, we show that the TSLP pathway is also required in TNBS-induced chronic colitis, as evidenced by milder disease in TSLPR KO mice similar to that in Fn14 KO mice. Finally, we provide a second mechanism whereby the TWEAK/Fn14 pathway may locally promote fibrotic changes, showing that TWEAK and IL-13 synergize for fibroblast proliferation. Thus our results demonstrate not only the novel role of the TWEAK/Fn14 pathway but also mechanisms whereby it acts in ECs and fibroblasts to promote chronic intestinal inflammation associated with fibrosis.
Fn14 is normally expressed at relatively low levels in healthy tissues9 but induced by IL-13, TNF-α, or a Toll-like receptor ligand, namely an oligodeoxynucleotide containing CpG motifs in colon mucosa.10, 11 Therefore, with breach of the mucosal barrier with EC damage in colitis, stimuli from bacterial components derived from commensal flora are likely to promote and maintain high expression of Fn14 in the ECs. Indeed, we found that TWEAK and Fn14 were both upregulated in the ECs in TNBS-induced colitis. As TWEAK signaling through Fn14 promotes EC death,11 it likely further contributes to breakdown of the EC barrier as evidence by the more severe histological scores in WT as compared with Fn14 KO mice. TWEAK/Fn14 signaling in ECs also induces secretion of MMP, cytokines, and chemokines,10 as shown in our gene profiling, thereby recruiting immune cells, which can also produce TWEAK and signal through the Fn14 upregulated in ECs. Indeed, in this chronic colitis model, chemokines that selectively recruit eosinophils, those for macrophages, and lymphocytes were all significantly higher in WT than Fn14 KO colon after induction of chronic colitis. Thus, the TWEAK/Fn14 pathway exacerbates and perpetuates the colon damage likely by promoting continuous breakdown of the EC barrier and infiltration of immune effector cells.
Although the loss of the intestinal barrier and wide range of exposure to microbial components in the colon are associated with TNBS injection, additionally, as TNBS is a hapten, it induces development of TNP-specific Th2-type adaptive immune responses in the draining lymph nodes and innate and Th2-type adaptive immune cell infiltrates in the colon in WT mice. As this is dependent on IL-13,19 we identified the major IL-13-expressing cells, which are CD3+ TCRβ+ T cells with little apparent contribution of innate IL-13-producing cells. In contrast to WT mice, the Fn14 KO exhibited a reduced TNP-specific Th2-type response in the draining lymph nodes, including IL-13 expression, and reduced Th2-type profile shown by histological and gene expression analysis in the colon. This is a novel finding, as previously, little, if any, contribution of TWEAK/Fn14 signaling to systemic acquired immunity was shown in models of collagen-induced arthritis,14, 15 hapten-induced acute colitis,10 and autoantibody-mediated nephritis induced by chronic graft-vs.-host disease.16 As the immune response to haptenized lumenal antigen, such as with repeated intrarectal TNBS, may be more enhanced than that in response to antigen administered subcutaneously or directed at target tissues that are relatively sterile, we speculate that this may explain why the TWEAK/Fn14 pathway impacted the adaptive immune response in the draining lymph nodes in chronic colitis but did not have such impact in acute colitis10 or the other disease models as noted above. Thus, this would constitute an indirect mechanism to explain the reduced TNP-specific response in our current study. Consistent with this, we found little, if any, Fn14 expression in the MLN cells, precluding a direct effect of TWEAK signaling at that site, but rather there is increased expression of Fn14 in the colonic ECs. Thus TWEAK/Fn14 signaling locally in colonic ECs appears to promote a downstream Th2-type response in TNBS-induced chronic colitis in mice.
We further delineated the mechanism whereby Fn14 deficiency reduced the Th2-type response by signaling through Fn14 upregulated in ECs in TNBS-induced chronic colitis. Emerging roles of ECs in bridging the innate immune response and Th2-type immunity have been reported.28, 29 TSLP as well as IL-25 and IL-33 are primary Th2-driving cytokines derived from ECs that act to condition DC, which then leave colon and go to draining nodes and induce/promote Th2-cell responses. Furthermore, recent reports showed that TSLP can induce Th2 cells directly in the absence of antigen-presenting cells.31, 32, 33 In addition, accumulation of mast cells, eosinophils, and basophils in the mucosa of chronic colitis, the cell types known to produce IL-4, IL-5, IL-13, and Th2-cell-recruiting chemokines in response to TSLP,34, 35, 36 contributes to the local amplification of the Th2-type response. IL-13 induces TSLP production from various types of ECs involved in diseases such as airway hypersensitivity, allergic rhinitis and dermatitis, and intestinal parasite infection.28, 29 Notably, we found that the IL-13-induced expression of TSLP in colonic explant cultures is dependent on Fn14 and potentiated by TWEAK. In vivo, TSLP, but not IL-25 or IL-33, was significantly increased in WT ECs in acute and chronic TNBS colitis and reduced at both stages in Fn14 KO mice, consistent with their blunted IL-13 response. We also demonstrate that the TSLP pathway is key in this model, as evidenced by milder disease and TNBS-specific responses in TSLPR KO mice, similar to those in Fn14 KO mice. Thus, we propose a mechanistic model in which Fn14 is acutely induced in ECs after TNBS-induced damage, enabling TWEAK to promote EC expression of TSLP acutely in WT but not in Fn14-deficient mice, thereby driving the development of Th2-type immunity, including the expression of IL-13.
Consistent with our model, we found no intrinsic defect in IL-13 expression as a result of Fn14 deficiency, in that WT and Fn14 KO LPMC respond similarly to TSLP, and WT and Fn14 KO draining lymph node cells and LPMC respond similarly to non-specific stimuli. Thus our proposed mechanism, an IL-13-TWEAK/Fn14-TSLP axis in ECs, constitutes a feedforward loop exacerbating and sustaining the chronic Th2-driven TNBS-induced colitis. The detailed mechanism as to how the TWEAK/Fn14 signaling promotes IL-13-induced TSLP expression remains to be investigated. We suggest several possibilities that are not mutually exclusive. As TSLP promoter activation is mediated by an upstream NF-κB site,37 it is possible that TSLP expression was promoted through NF-κB activation by TWEAK/Fn14 pathway.38, 39 Secondly, as IL-13 and TWEAK in concert activate endogenous TNF-α,11 the induction of TSLP expression may be mediated by TNF-α, which is also known to promote TSLP expression in ECs.37
Another important point in our study is the link between the TWEAK/Fn14 pathway and fibrosis. Although not yet reported in the context of colitis, studies from multiple other systems have indicated a role for TWEAK/Fn14 in driving fibrogenic responses. For example, TWEAK/Fn14 dependence of collagen deposition has been reported in the liver injury,40 kidney after ischemia reperfusion injury,41 skeletal muscle in a model of denervation-induced skeletal muscle atrophy,42 and heart in the context of dilated cardiomyopathy.43 Although the TWEAK/Fn14 pathway may drive fibrosis through its promotion of chronic inflammatory activity, particularly Th2-type activity, there are also accumulating evidence supporting that the TWEAK/Fn14 pathway contribute to fibrosis through its direct action on fibroblasts/myofibroblasts and their progenitors. As Fn14 is upregulated by TGF-β on fibroblasts,44, 45 and is likely also expressed on myofibroblasts, the TWEAK/Fn14 pathway may directly contribute to a fibrogenic response by regulating the proliferation of these fibroblasts, their differentiation to myofibroblasts, and extracellular matrix production. Indeed, our current study shows that TWEAK synergizes with IL-13 or TGF-β to promote the expansion of fibroblasts, and this is dependent on Fn14.
Thus, the TWEAK/Fn14 pathway is involved together with IL-13 in mediating an IL-13-TWEAK/Fn14-TSLP axis in ECs, as well as through its interplay with IL-13 and TGF-β in fibroblasts, to aggravate and perpetuate Th2-type chronic inflammation with fibrosis. As fibrosis is associated with fistula and stricture formation in Crohn’s disease and there is widespread damage of mucosa in ulcerative colitis, we propose that targeting TWEAK/Fn14 as possible treatment of inflammatory bowel diseases. Our findings also have interesting implications, warranting investigation of a role for the TWEAK/Fn14 pathway in other diseases involving ECs and prominent roles for IL-13, TSLP, and fibrotic changes, such as chronic respiratory and skin diseases.
methods
Mice. Female 7-week-old BALB/c mice or Fn14 KO mice46 with BALB/c background were used. All experimental protocols were approved by the local institutional animal care and use committees of the National Center for Global Health and Medicine, Japan. TSLP receptor–deficient (TSLPR KO) mice were generated as described previously47 and backcrossed to BALB/c for 12 generations.
Induction of TNBS colitis. To induce chronic colitis, under anesthesia with ketamine and xylazine, 0.2% TNBS (Research Organics, Cleveland, OH) in phosphate-buffered saline: ethanol=1:1 was rectally injected with a stainless gavage tube weekly for 6 consecutive weeks, with modification of a previously reported method.21 The dose of TNBS solution was sequentially increased: 1.0, 1.5, 1.5, 1.8, 2.0, and 2.0 μl per g body weight. Tissues and cells were harvested 3 days after the last injection. To induce acute colitis, mice were injected with 1.0 μl per g body weight on day 0, and tissues were obtained on day 3. The colon was opened longitudinally, rolled, and snap frozen for preparation of frozen sections or RNA extraction. In some experiments, formalin-fixed paraffin-embedded sections were prepared for hematoxylin and eosin staining or detection of collagen (Supplementary Methods). Collagen and non-collagen proteins of tissue sections were measured using a Quantitative Micro-Assay Kit for Collagen and Non-Collagen Proteins (Chondrex, Redmond, WA) following the vendor’s protocol.
Total RNA purification, gene profiling and quantitative reverse transcriptase–PCR. Total RNA was extracted and purified with DNAse. After reverse transcription, expression level of each gene was quantified using TaqMan gene expression kit (ABI, Carlsbad, CA) or a SYBR Green PCR Master Mix. For gene profiling, RNA was isolated from snap-frozen whole-colon tissues, including five naive WT mice, five naive Fn14 KO mice, six WT mice, and seven Fn14 KO with repeated TNBS-induced colitis and subjected to gene profiling performed with automated probe labeling, hybridization, and scanning as previously described.10 Details for the reverse transcriptase–PCR and analysis of Affymetrix data are as described in Supplementary Methods.
Primary tissue culture. The mouse colon tissues were washed with phosphate-buffered saline and cut into 1-cm lengths, opened, placed in 24-well plates, and kept in RPMI 1640 with 5% fetal calf serum, Tissues were then cultured with 50 ng ml−1 of recombinant IL-13 (PeproTech, Rocky Hill, NJ) or 100 ng ml−1 of recombinant soluble TWEAK (prepared by Biogen Idec, Cambridge, MA)3 at 37 °C, 5% CO2 for 6 h. The tissues were frozen in liquid nitrogen and stored at −80°C until use.
Statistical analysis. Data were statistically analyzed by the Mann–Whitney test using a software Prism 4 (GraphPad Software, La Jolla, CA), unless indicated in the legend.
Methods for histological analysis, cell separation, cell culture, and cytokine quantification are described in the Supplementary information.
Accession codes
References
Fichtner-Feigl, S., Strober, W., Geissler, E.K. & Schlitt, H.J. Cytokines mediating the induction of chronic colitis and colitis-associated fibrosis. Mucosal immunol. 1 (Suppl 1), S24–S27 (2008).
Wynn, T.A. & Ramalingam, T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 18, 1028–1040 (2012).
Chicheportiche, Y. et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 272, 32401–32410 (1997).
Kaplan, M.J. et al. The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. J. Immunol. 169, 6020–6029 (2002).
Maecker, H. et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell 123, 931–944 (2005).
Nakayama, M. et al. Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK-induced cell death. J. Immunol. 170, 341–348 (2003).
Justo, P. et al. Cytokine cooperation in renal tubular cell injury: the role of TWEAK. Kidney Int. 70, 1750–1758 (2006).
Burkly, L.C., Michaelson, J.S. & Zheng, T.S. TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol. Rev. 244, 99–114 (2011).
Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 7, 411–425 (2008).
Dohi, T. et al. TWEAK/Fn14 pathway: a nonredundant role in intestinal damage in mice through a TWEAK/intestinal epithelial cell axis. Gastroenterology 136, 912–923 (2009).
Kawashima, R. et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice—a pathway associated with ulcerative colitis. Gastroenterology 141, 2119–2129 (2011).
Burkly, L.C. & Dohi, T. The TWEAK/Fn14 pathway in tissue remodeling: for better or for worse. Adv. Exp. Med. Biol. 691, 305–322 (2011).
Zheng, T.S. & Burkly, L.C. No end in site: TWEAK/Fn14 activation and autoimmunity associated- end-organ pathologies. J. Leukoc. Biol. 84, 338–347 (2008).
Perper, S.J. et al. TWEAK is a novel arthritogenic mediator. J. Immunol. 177, 2610–2620 (2006).
Kamata, K. et al. Involvement of TNF-like weak inducer of apoptosis in the pathogenesis of collagen-induced arthritis. J. Immunol. 177, 6433–6439 (2006).
Zhao, Z. et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J. Immunol. 179, 7949–7958 (2007).
Wilson, M.S. & Wynn, T.A. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2, 103–121 (2009).
Ingram, J.L. et al. Opposing actions of Stat1 and Stat6 on IL-13-induced up-regulation of early growth response-1 and platelet-derived growth factor ligands in pulmonary fibroblasts. J. Immunol. 177, 4141–4148 (2006).
Fichtner-Feigl, S. et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J. Immunol. 178, 5859–5870 (2007).
Wu, F. & Chakravarti, S. Differential expression of inflammatory and fibrogenic genes and their regulation by NF-kappaB inhibition in a mouse model of chronic colitis. J. Immunol. 179, 6988–7000 (2007).
Lawrance, I.C. et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology 125, 1750–1761 (2003).
Gordon, S. & Martinez, F.O. Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604 (2010).
Pemberton, A.D. et al. Innate BALB/c enteric epithelial responses to Trichinella spiralis: inducible expression of a novel goblet cell lectin, intelectin-2, and its natural deletion in C57BL/10 mice. J. Immunol. 173, 1894–1901 (2004).
Gu, N. et al. Intelectin is required for IL-13-induced monocyte chemotactic protein-1 and -3 expression in lung epithelial cells and promotes allergic airway inflammation. Am. J. Physiol. Lung. Cell. Mol. Physiol. 298, L290–L296 (2010).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Neill, D.R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Saenz, S.A. et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464, 1362–1366 (2010).
Bulek, K., Swaidani, S., Aronica, M. & Li, X. Epithelium: the interplay between innate and Th2 immunity. Immunol. Cell. Biol. 88, 257–268 (2010).
Saenz, S.A., Taylor, B.C. & Artis, D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 226, 172–190 (2008).
Biton, M. et al. Epithelial microRNAs regulate gut mucosal immunity via epithelium-T cell crosstalk. Nat. Immunol. 12, 239–246 (2011).
Blazquez, A.B., Mayer, L. & Berin, M.C. Thymic stromal lymphopoietin is required for gastrointestinal allergy but not oral tolerance. Gastroenterology 139, 1301–1309 (2010).
He, R. et al. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc. Natl. Acad. Sci. USA 105, 11875–11880 (2008).
Omori, M. & Ziegler, S. Induction of IL-4 expression in CD4(+) T cells by thymic stromal lymphopoietin. J. Immunol. 178, 1396–1404 (2007).
Allakhverdi, Z. et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 204, 253–258 (2007).
Allakhverdi, Z., Smith, D.E., Comeau, M.R. & Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 179, 2051–2054 (2007).
Siracusa, M.C. et al. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature 477, 229–233 (2011).
Lee, H.C. & Ziegler, S.F. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc. Natl. Acad. Sci. USA 104, 914–919 (2007).
Brown, S.A., Ghosh, A. & Winkles, J.A. Full-length, membrane-anchored TWEAK can function as a juxtacrine signaling molecule and activate the NF-kappaB pathway. J. Biol. Chem. 285, 17432–17441 (2010).
Roos, C. et al. Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-kappa B pathway. J. Immunol. 185, 1593–1605 (2010).
Tirnitz-Parker, J.E. et al. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology 52, 291–302 (2010).
Hotta, K. et al. Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int. 79, 179–188 (2011).
Mittal, A. et al. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell. Biol. 188, 833–849 (2010).
Jain, M. et al. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation 119, 2058–2068 (2009).
Hosokawa, Y., Hosokawa, I., Ozaki, K., Nakae, H. & Matsuo, T. Proinflammatory effects of tumour necrosis factor-like weak inducer of apoptosis (TWEAK) on human gingival fibroblasts. Clin. Exp. Immunol. 146, 540–549 (2006).
Meighan-Mantha, R.L. et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J. Biol. Chem. 274, 33166–33176 (1999).
Jakubowski, A. et al. TWEAK induces liver progenitor cell proliferation. J. Clin. Invest. 115, 2330–2340 (2005).
Carpino, N. et al. Absence of an essential role for thymic stromal lymphopoietin receptor in murine B-cell development. Mol. Cell. Biol. 24, 2584–2592 (2004).
Acknowledgements
We are grateful to Dr Suzanne Szak (Biogen Idec) for valuable review and suggestions on this manuscript, Mr Yasuhiko Nagasaka (Beckman Coulter) for his help with flow cytometric analysis and Dr M Tamura-Nakano (NCGM EM Support Unit) for her technical support for histological analysis. Grant support: This work was supported partly by grants and contracts from the program Grants-in-Aid for Scientific Research (B and C) and Grants-in Aid for Young Scientists (B) from the Ministry of Education, Cultures, Sports, Science, and Technology; Japan Science and Technology Agency; The Grant of National Center for Global Health and Medicine (21-110, 22-205); and Grants for Research on New Drugs Development and Research on intractable diseases from Ministry of Health, Labor and Welfare of Japan. Transcripts profiling: The microarray files and information have been uploaded to the NCBI GEO public repository (accession number GSE36806).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Ping Wu, and Linda C. Burkly are employees and stockholders in Biogen Idec. All the other authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article
Cite this article
Son, A., Oshio, T., Kawamura, Y. et al. TWEAK/Fn14 pathway promotes a T helper 2-type chronic colitis with fibrosis in mice. Mucosal Immunol 6, 1131–1142 (2013). https://doi.org/10.1038/mi.2013.10
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2013.10
This article is cited by
-
Mechanisms of norcantharidin against renal tubulointerstitial fibrosis
Pharmacological Reports (2024)
-
Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis
Mucosal Immunology (2019)
-
Role of the TWEAK/Fn14 pathway in autoimmune diseases
Immunologic Research (2016)
