
Abstract
Tannerella forsythia is a pathogen implicated in periodontitis, an inflammatory disease of the tooth-supporting tissues often leading to tooth loss. This key periodontal pathogen is decorated with a unique glycan core O-glycosidically linked to the bacterium's proteinaceous surface (S)-layer lattice and other glycoproteins. Herein, we show that the terminal motif of this glycan core acts to modulate dendritic cell effector functions to suppress T-helper (Th)17 responses. In contrast to the wild-type bacterial strain, infection with a mutant strain lacking the complete S-layer glycan core induced robust Th17 and reduced periodontal bone loss in mice. Our findings demonstrate that surface glycosylation of this pathogen may act to ensure its persistence in the host likely through suppression of Th17 responses. In addition, our data suggest that the bacterium then induces the Toll-like receptor 2–Th2 inflammatory axis that has previously been shown to cause bone destruction. Our study provides a biological basis for pathogenesis and opens opportunities in exploiting bacterial glycans as therapeutic targets against periodontitis and a range of other infectious diseases.
Similar content being viewed by others


Introduction
Periodontitis is a chronic inflammatory disease whose pathology is defined by interplay between bacterial factors and host immune responses. Tannerella forsythia is a Gram-negative periodontal bacterium, which together with Porphyromonas gingivalis and the spirochete Treponema denticola constitutes the so-called “red complex” consortium implicated in the etiology of periodontitis.1 These bacteria inhabit subgingival biofilms in the human oral cavity, where they modulate immune responses to instigate the periodontal tissue and bone destruction that is a hallmark of this disease. Furthermore, clinical studies also suggest a link between periodontitis and systemic diseases, such as cardiovascular disease, diabetes and obesity,2, 3 and rheumatoid arthritis.4 In some respects, the etiology of periodontitis is somewhat analogous to that of inflammatory bowel disease, where disturbances in host defense responses against the gut flora contribute to the disease progression.5
T. forsythia is a relatively understudied pathogen despite being a key member of the pathogenic consortium. Its virulence mechanisms are just beginning to be revealed.6, 7 In contrast to its red complex partners, T. forsythia lacks surface fimbriae, capsule, and is non-motile, but exclusively possesses a unique surface layer (S-layer) glycoprotein lattice. S-layers are water-insoluble proteins intrinsically capable of selfassembling into crystalline lattices on bacterial cell surfaces, and are believed to provide selective advantages to bacteria, such as resistance to predation or immune clearance.8 Interestingly, T. forsythia is the only Gram-negative bacterium that is known to possess a glycosylated S-layer.9 It is composed of two high-molecular-weight glycoproteins, TfsA and TfsB, with predicted protein molecular masses of 135 and 154 kDa, respectively.9 However, owing to glycosylation, TfsA and TfsB proteins migrate as 200- and 210-kDa molecular size bands on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), respectively.9 We previously identified a genetic operon involved in the glycosylation of these S-layer proteins and predicted a direct role for the wecC gene (TF2055; postulated to encode UDP-N-acetylmannosaminuronic acid dehydrogenase) in the glycosylation of S-layer proteins, and showed that this has a role in defining the physical properties of the cell surface affecting biofilm formation on inert surfaces.10 In line with our hypothesis, a recent study identified that the wecC operon in T. forsythia has a role in O-glycosylation of the S-layer glycoproteins with a unique oligosaccharide core.11 Specifically, wecC seems to be key to the addition of a terminal sugar motif consisting of two subterminal mannosuronic acid residues and a terminal pseudaminic acid residue (Pse5Am7Gc, 5-acetimidol-7-N-glycolyl-pseudaminic acid) on this oligosaccharide core.11
Cell surface glycosylation in bacteria can modulate immune responses during pathogenesis.12, 13 In this regard, much of our knowledge on the role of glycosylation in bacteria colonizing the mucosal surfaces has come from the studies of capsule polysaccharides, glycosylated flagella/pili, and outer-membrane glycoproteins in gut-dwelling organisms such as Bacteroides fragilis and S-layer from Gram-positive bacteria. For example, evidence obtained from Bacteroides spp. indicate that surface polysaccharides can modulate dendritic cell (DC) activation and cytokine production.14, 15 These immunomodulatory polysaccharides can regulate the differentiation of CD4(+) T cells and interleukin (IL)-10 secretion via Toll-like receptor 2 signaling.16 Likewise, the Gram-positive Lactobacillus acidophilus S-layer glycoprotein can regulate DC function by interacting with the C-type lectin receptor DC-SIGN (DC-specific intercellular adhesion molecule-3-grabbing nonintegrin) to induce specific T-cell functions.17 Similarly, P. gingivalis minor fimbriae glycoprotein targets DCs through DC-SIGN to elicit distinct effector functions.18, 19
Recent in vitro studies suggested that T. forsythia S-layer proteinaceous lattice per se could have a role in dampening the immune response during infection.20 However, the underlying mechanisms responsible for this immune suppression or the role of S-layer during periodontal inflammation in infection models have not been defined. Our hypothesis at the start of this study was that T. forsythia surface glycans would modulate DC and macrophage responses, and influence T-cell-dependent periodontal inflammation and alveolar bone homeostasis.
Collectively, our results indicate that Tannerella surface glycosylation has a role in restraining the Th17-mediated neutrophil infiltration in the gingival tissues. This probably allows the bacteria to persist and induce Toll-like receptor (TLR)2 and Th2 responses to cause tissue and bone destruction.21 Overall, these results may help to explain how this oral pathogen uses its surface glycosylation to exploit the immune response to colonize and induce disease.
Results
WecC controls surface protein glycosylation in T. forsythia
Our previous study showed that inactivation of the wecC gene in T. forsythia coding for a putative UDP-N-acetylmannosaminuronic acid dehydrogenase affects the surface glycosylation.10 This was based on the findings that in comparison to the wild-type parental strain, the wecC deletion mutant showed increased surface hydrophobicity and the S-layer proteins of the deletion mutant migrated with increased mobility on SDS-PAGE gels.10 A recent study by Posch and co-workers,11 which was published during the course of our study, showed that this mutant lacks a terminal branch of the S-layer glycan. To characterize phenotypically the mutant further, we performed ultrastructure analysis using transmission electron microscopy of ultrathin sections of the wild-type T. forsythia and its wecC deletion mutant TFM-ED1 previously constructed in our laboratory10 (hereafter called ED1). The results clearly demonstrate that while the wild-type cells show the characteristic ∼25-nm-thick regular lattice or striated “teeth-like” S-layer first observed by Anne Tanner22 (Figure 1a, left), the ED1 mutant cells possess an irregular but intact S-layer of similar thickness (25 nm) (Figure 1a, right). This indicates that removal of the terminal glycosyl branches affects the S-layer assembly but not the secretion of localization on the cell surface. To further prove this notion, we performed an outer-membrane purification and observed both by SDS-PAGE glycoprotein-specific staining (Pro-Q Emerald) and western blotting, using an anti-S-layer polyclonal antibody raised against intact purified wild-type S-layer, that the altered S-layer proteins are still present in the outer-membrane fraction of ED1, albeit at an altered molecular weight (Figure 1a and b). In addition, and in agreement with Posch et al.,11 we found that a number of other proteins appear to be glycosylated with a WecC-dependent glycosyl domain, but that the S-layer is the most abundant. In addition, we also proved the surface localization of these glycoproteins using our antibody on Tannerella cells by fluorescence microscopy and showed that these proteins are localized on the periphery of the cell in both the wild-type and ED1 mutant (Supplementary Figure S1 online).

Alteration of the glycosylation pattern of surface proteins of Tannerella forsythia by deletion of the wecC gene results in an altered but intact surface (S)-layer and no changes in lipopolysaccharide (LPS). (a) Transmission electron microscopy (TEM) of negatively stained ultrathin sections of wild-type (WT) and ED1 mutant strains. Arrowheads indicate the S-layer structure covering the outer membrane. (b) Outer-membrane preparations of WT and ED1 strains were separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (4–8%) gels and stained with silver (left), Pro-Q Emerald glycostain (middle), and probed with an anti-S-layer antibody after western blotting (right). (c) LPS was extracted and separated on a 10% urea-SDS-PAGE gel and silver stained.
In addition, as lipopolysaccharide (LPS) and protein glycosylation are sometimes linked in Gram-negative bacteria, e.g., Aeromonas sp.,23 we examined whether the LPS pattern of the ED1 mutant was altered compared with the wild-type mutant. Purified LPS fractions form the wild-type strain, and the mutant strains were subjected to urea-SDS-PAGE followed by LPS silver staining.24 The results (Figure 1c) showed an identical electrophoretic pattern for the LPS of the two strains, indicating that the wecC deletion does not affect the chemical nature of LPS expression in T. forsythia. Taken together, these results imply that the ED1 mutant is defective with respect to surface glycosylation associated with the S-layer and other surface glycoproteins, but does not present any abnormality with respect to LPS.
Glycosylation-deficient mutant induces increased nuclear factor-κB activity
We postulated that the wild-type and the ED1 strains would be differentially recognized by immune cells. This hypothesis was based on the fact that: (i) pathogen-associated polysaccharides act as ligands for TLRs (TLR2 and TLR4), nucleotide oligomerization domain containing proteins, and C-type lectin receptors; (ii) surface glycosylation can modulate immune response by blocking recognition of other pathogen-associated molecules, and thus might help in immune evasion; and (iii) in a recent study, a T. forsythia mutant completely deficient in S-layer was shown to cause increased secretion of inflammatory cytokines in a monocytic cell line and human gingival fibroblasts in vitro compared with the wild-type strain.20 However, as the complete S-layer deletion strain was used in the previous study, it was not clear whether surface glycosylation per se was modulating any effects of the S-layer on the host response. Therefore, our ED1 mutant presents a unique opportunity to test the role of the novel protein glycosylation pattern of surface proteins in Tannerella on immune responses. Thus, we first investigated the inflammatory response to T. forsythia wild-type and ED1 mutant strains using a reporter cell line, THP-1-Blue. This cell line is derived from the human monocytic cell line THP-1 and is stably transfected with a reporter plasmid expressing a secreted form of embryonic alkaline phosphatase under the control of a nuclear factor-κB- and AP-1-inducible promoter. The results showed significantly elevated secreted form of embryonic alkaline phosphatase activity in THP-1-Blue cells in response to ED1 challenge over THP-1-Blue cells challenged with the wild-type bacteria at a similar dose (Supplementary Figure S2 online). These initial results provided a basis for dissecting the role of surface glycosylation in modulating the immune response to T. forsythia in detail.
Bacterial glycosylation regulates levels and nature of cytokine induction in antigen-presenting cells
As shown previously,21 T. forsythia challenge primarily induces Th2 development in vivo. We were therefore interested to investigate whether surface glycosylation in T. forsythia has a role in modulating the secretion of specific Th cell-differentiating cytokines by antigen-presenting cells. For this purpose, mouse bone marrow-derived DCs (BMDCs) and macrophages were challenged with the T. forsythia wild-type or ED1 mutant cells at a multiplicity of infection of 10. The cytokines secreted in the medium were then assayed by enzyme-linked immunosorbent assay (ELISA). BMDCs and peritoneal macrophages were purified by positive selection and confirmed to be 90–95% pure, verified by staining for CD11c, MHC-II, and CD86 (BMDCs) or CD11b (macrophages) by flow cytometry (data not shown). Pure Escherichia coli LPS was used as a positive agonist. The data showed that T. forsythia ED1 mutant induced significantly higher amounts of IL-6 and IL-1β secretion in both BMDCs and macrophages compared with the wild-type T. forsythia (Figure 2). However, no significant difference was observed with regard to IL-10 secretion in either BMDCs or macrophages (Figure 2). As expected, E. coli LPS induced potent production of these cytokines (Figure 2). Interestingly, IL-12p40 (the shared subunit for IL-12 and IL-23) and IL-23 levels were higher, but IL-12p70 (IL-12 p35 and p40 heterodimer) levels were lower in BMDCs challenged with the ED1 mutant compared with the wild-type strain. In macrophages, a similar pattern was observed for IL-12p40 and IL-23 (Figure 2). Strikingly, a heightened IL-12p40 response was observed in both DCs and macrophages. IL-12p40 can form a homodimer (IL-12p80), which can potentially block Th1 differentiation through competing with IL-12 for IL-12R.25, 26, 27 Moreover, IL-12p80 through its interaction with IL-12Rβ1 stimulates migration of DCs to chemokine CCL19,28 and thus the heightened IL-12p40 dimer secretion in response to T. forsythia challenge might have a role in DC recruitment as well. As expected,21 no significant IL-12p70 secretion was observed in macrophages in response to ED1 challenge (Figure 2b) or the wild-type bacteria (Figure 2). These results demonstrate that T. forsythia induces differential responses in antigen-presenting cells (BMDCs vs. macrophages) with respect to the Th1 cytokine IL-12. This suggests that DCs and macrophages utilize a different mechanism to handle T. forsythia. Although DCs and macrophages share many pattern-recognition receptors, differences in nucleosome remodeling for transcription and transcript stability in these cell types can impact the expression of cytokines. In support, Mycobacterium tuberculosis induces increased IL-12 secretion in DCs compared with that in macrophages through extensive nucleosome remodeling in DCs via the p40 promoter.29 In addition, DC-specific intercellular adhesion molecule-3-grabbing non-integrin lectin-like receptor can modulate the responses of other PRRs by moderating the crosstalk between intracellular signaling pathways.30, 31

Tannerella forsythia surface glycosylation differentially regulates cytokine expression in macrophages and dendritic cells (DCs). Cytokine levels (interleukin (IL)-1β, IL-6, IL-10; IL-12p40, IL-12p70-, and IL-23) were examined by enzyme-linked immunosorbent assay (ELISA) in supernatants of mouse DCs and peritoneal macrophages following challenge with either the wild-type (Tf WT) or the glycosylation-deficient mutant (ED1) of T. forsythia. The data show the means±s.d. of triplicate determinations in one of three independent sets of experiments that yielded similar results; statistically significant differences between the groups are indicated as ***P<0.001 and **P<0.01. EcLPS, Escherichia coli LPS.
Surface glycosylation protects T. forsythia from DC recognition
The increased ability of the ED1 mutant to induce IL-23 secretion in BMDCs led us to hypothesize that glycosylation affects the ability of DCs to process the bacteria. To test this hypothesis, DCs were incubated with fluorescently labeled bacteria at a multiplicity of infection of 10 for 30 min and the ability of the DCs to bind/uptake bacteria was evaluated by flow cytometry. Flow cytometry analysis (Figure 3a and b) showed that a significantly lower population of DCs stained positive when incubated with the wild-type T. forsythia compared with ED1. Correspondingly, a larger DC population remained negative when incubated with wild-type T. forsythia cells compared with ED1 and vice versa (Figure 3a and b). There was an approximately twofold increase in the mean fluorescent intensity of DCs (M2; Figure 3c) associated with ED1 compared with the wild-type T. forsythia (Figure 3c). Moreover, the flow cytometry data were confirmed by an in vitro bacterial killing assay in which bacteria were enumerated following incubation of live bacteria with DCs for different time points. The data from this assay show that a significantly greater numbers of ED1 cells than the wild-type strain associated with DCs initially (15 min; Figure 3d). Moreover, the increased recognition of ED1 translated to an increased bacterial killing as a significantly fewer numbers of ED1 cells were recovered at a later time point (60 min) compared with the wild-type strain (Figure 3d). These results indicate that loss of surface glycosylation renders T. forsythia increasingly prone to capture by DCs, which leads to enhanced bacterial processing by DCs as well.

Abrogation of surface glycosylation in Tannerella forsythia causes enhanced dendritic cell (DC) association. Association of fluorescein isothiocyanate (FITC)-labeled wild-type (Tf WT) and ED1 strains was assessed by flow cytometry. (a) Representative traces of bacteria bound/engulfed by bone marrow DCs (BMDCs) are demarcated by the thick gray (WT strain) and dark (ED1 strain) lines. Autofluorescence signal is demarcated by the thin line (untreated BMDCs) and corresponds to BMDCs without bound/ingested bacteria. (b) Bar graphs show percentages of BMDCs in gates M1 (representing the background autofluorescence and unbound bacteria) and M2 (representing brightly fluorescent cell population with bound and/or internalized FITC-labeled bacteria. (c) Bar graphs represent the mean fluorescent intensities (m.f.i.) (±s.d.). (d) Bacterial killing by DCs. DCs were infected with bacteria (WT or ED1) at a multiplicity of infection (MOI) of 10. The number of viable bacteria was determined (colony-forming unit (CFU)) at indicated time points postinfection. Data represent mean CFU (±s.d.) per 105 DC cells of triplicate readings from two independent experiments. ***P<0.001.
Surface glycosylation deficiency results in reduced in vivo virulence
In vitro data above show that ED1 mutant is significantly more potent than the wild-type strain in causing IL-1β and IL-6 secretion in both DCs and macrophages. As DCs are able to recognize the glycosylation-deficient ED1 mutant more efficiently and respond by secreting increased levels of IL-23, the contribution of surface glycosylation in modulating Th cell responses and periodontal inflammation in vivo was determined. For this purpose, BALB/cJ mice were infected with six doses of bacteria (108 colony-forming unit (CFU) per dose) at 48 h intervals, using either the wild-type or ED1 mutant cells by oral gavage. Control sham-infected animals were inoculated with vehicle (1% carboxymethyl cellulose) alone. At 1 week following the last infection, polymerase chain reaction (PCR) analyses of oral swabs confirmed infection via expression. T. forsythia-specific 641-bp 16s rDNA product for all mice infected with T. forsythia (wt) or ED1 mutant (n=8 per group). As expected, sham-infected mice were positive only for the 1.4-kb universal eubacteria 16s rDNA product but not for T. forsythia-specific 16s rDNA product (Supplementary Figure S3 online). Furthermore, analysis of T. forsythia-specific IgG titers 6 weeks after the first infection confirmed that the mice were successfully infected. Our results showed that the bacteria-specific serum IgG titers to the wild-type and ED1 strains increased several fold over sham-infected levels in mice infected with corresponding strains (Figure 4a). The low IgG titer to T. forsythia even in the sham-infected mice is presumably due to nonspecific crossreaction to anti-bacterial antibodies elicited to normal resident bacteria. Taken together, the results of PCR analysis and antibody responses indicated that mice orally gavaged with the T. forsythia wild-type and ED1 strains were successfully infected with each strain.

Tannerella forsythia wild-type (Tf WT) strain is more virulent than the glycosylation-deficient ED1 mutant strain in a mouse model of periodontitis. (a) Specific and crossreactive antibodies are elevated in mice following oral infection. Sera from mice after 6 weeks of first infection (sham, Tf WT, or ED1) were analyzed for T. forsythia-specific immunoglobulin (IgG) by enzyme-linked immunosorbent assay (ELISA). Antibody levels are presented as log2 titers. Data represent means and standard deviations for each group (n=8), and statistical differences between the group means were determined (*P<0.05 vs. sham-infected group; #P<0.05 vs. Tf-infected group). (b, c) ED1 mutant causes significantly less bone loss compared with the WT strain. Mice (n=8) were infected by oral gavage with six doses (109 cells per dose) of either T. forsythia wild-type strain (109 cells per dose; Tf WT) or T. forsythia ED1 mutant or sham-infected. (b) Alveolar bone destruction was assessed after 6 weeks by measuring the distance from the alveolar bone crest (ABC) to the cementoenamel junction (CEJ) at 14 maxillary buccal sites per mouse (R1–R7=right jaw; L1–L7=left jaw). (c) Net bone loss induced by WT or ED1 bacteria was calculated as mean total ABC–CEJ distance of bacterially infected group minus mean total ABC–CEJ distance of the sham-infected group. The data show that the net bone loss caused by the WT strain is significantly higher than that by the ED1 mutant. Bars indicate means and standard deviations. Data were analyzed by a Mann–Whitney unpaired t-test, and statistically significant differences are indicated as ***P<0.001 and **P<0.01. NS, not significant.
To assess periodontal bone loss induction, the distance from the alveolar bone crest (ABC) to the cementoenamel junction (CEJ) was measured at 14 buccal sites 6 weeks postinfection. As expected, significant alveolar bone loss was observed in T. forsythia wild-type infected mice at multiple sites compared with the sham-treated controls (Figure 4b and c). Strikingly, mice infected with the ED1 mutant did not show statistically significant periodontal bone loss. Moreover, the net alveolar bone loss induced by the ED1 mutant (calculated as the average total ABC-CEJ distance of the sham-treated group subtracted from the bacteria-infected group) was significantly lower than the net bone loss induced by the T. forsythia wild-type strain (Figure 4c). Thus, the T. forsythia ED1 mutant is less virulent in a periodontitis model.
Resistance to glycosylation-deficient strain correlates with Th17 responses
We compared Th cell response of mice infected with the wild-type and the ED1 strains by analyzing Th cells from draining cervical lymph nodes (cLNs) of infected mice. Mice were infected orally three times at 48 h intervals with the wild-type or the ED1 strain, and 72 h after the last infection, cLN cells were stimulated with anti-CD3 and anti-CD28 antibodies to stimulate the T-cell receptor and induce cytokine production. Cells were then treated with phorbol myristate acetate-ionomycin, stained for cell surface CD4, and intracellularly stained for IL-5, interferon (IFN)-γ, and IL-17 to detect Th2, Th1, and Th17 cells, respectively. As shown in wild-type strain-infected mice (susceptible to bone loss) (Figure 5), the number of Th2 cells increased, while the number of Th1 and Th17 cells decreased following infection (Figure 5). Conversely, the number of Th2 cells was reduced with a concomitant increase in Th17 cells in mice infected with ED1 (resistant mice) compared with sham (Figure 5). The number of Th1 cells decreased significantly in both the wild-type T. forsythia and ED1-infected groups compared with sham. This decrease was greater following wild-type T. forsythia infection compared with ED1 infection. As the ED1 mutant caused skewing to a Th17 response, we conclude that T. forsythia surface glycosylation has a role in dampening Th17 responses.

Tannerella forsythia glycosylation-deficient ED1 mutant induces T-helper (Th)17 cells following infection. Production of interferon (IFN)-γ, interleukin (IL)-5, and IL-17 in cervical lymph nodes (cLNs) of mice was seen. Mice (n=4) were infected three times at 48-h intervals. At 72 h after the final infection, cLN cells were stimulated with anti-CD3 and anti-CD28 antibodies (Abs) for 48 h. Cells were then stimulated with ionomycin and phorbol myristate acetate (PMA) for 4–6 h before intracellular staining for IL-5, IFN-γ, or IL-17. (a) Representative flow cytometry dot plots of CD4+ T cells from sham- and bacteria-infected mice intracellularly stained for IL-5, IFN-γ, or IL-17. (b) Bar graphs showing percentages (mean and standard deviations) of specific cytokine-positive T cells for each group. Statistically significant differences between the groups is indicated as *P<0.05. NS, not significant.
Resistance to glycosylation-deficient strain correlates with decreased osteoclastic activity and increased neutrophil infiltration
To evaluate osteoclastic activity, maxillary bones were stained for TRAP- (tartrate resistance alkaline phosphatase, a marker of osteoclasts) positive (TRAP+) cells. Approximately a twofold increase in the numbers of TRAP+ cells was observed in the wild-type T. forsythia-infected maxillae compared with those of ED1 mutant or sham-infected mice (Figure 6a and b). Thus, induction of alveolar bone loss induced by wild-type T. forsythia correlated with increased osteoclastic activity in maxillary bones. Strikingly, the ED1 infection did not enhance osteoclastic activity, even though increased Th17 response was observed in ED1-infected mice (Figure 5). Interestingly, contrary to the role of Th17 in bone destruction as seen in rheumatoid arthritis,32 Th17 cells have a protective role in infection-induced bone loss in the context of the oral niche.

Tannerella forsythia wild-type (Tf WT) strain induces increased osteoclastic activity in alveolar bone. (a) Representative histological sections showing tartrate resistance alkaline phosphatase (TRAP)+ cells from sham- and bacteria-infected mice. (b) Average number of TRAP+ cells in 10 high power magnification fields per slide (4 mice per group). Statistically significant differences between groups is indicated as **P<0.01.
To evaluate inflammatory cells in the gingival space, maxillae were stained with anti-CD45 and anti-neutrophil-specific marker antibody. The results indicated that T. forsythia wild-type and ED1 infection caused an increase in the CD45+ lymphocytic population in gingival tissues compared with the sham-infected tissues (Figure 7). In line with the increased ability of the ED1 to induce inflammatory cytokines (Supplementary Figure S1 online and Figure 2a), this mutant also caused increased infiltration of CD45+ lymphocytes in gingival tissues (Figure 7). There was approximately a threefold increase in neutrophil numbers in ED1-infected mice over wild-type strain-infected mice (Figure 7b). Not surprisingly, the increase in neutrophil numbers following ED1 infection parallels the increase in Th17 response in ED1-infected mice.

Tannerella forsythia glycosylation-deficient ED1 mutant induces enhanced lymphocytes/neutrophil infiltration following infection. (a) Immunohistochemical staining for total inflammatory cells (CD45+) and neutrophils in gingival tissue 3 weeks following infection. All images are representative of original magnification × 400. (b) Slide images were viewed with Aperio Image Scope viewing software and the interdental areas from the first to third molar were used to quantify inflammatory cells. Bar graphs show the number of CD45+ and neutrophil antibody-positive cells. Statistically significant differences between the respective T. forsythia- and sham-infected groups are indicated as ***P<0.001 and **P<0.01. NS, not significant; Tf WT, Tannerella forsythia wild-type.
Glycosylation-deficient strain is cleared faster than the wild-type strain from gingival tissues
Reduced alveolar bone loss paralleling increased neutrophil infiltration in gingival tissues in glycosylation-deficient strain-infected mice suggested that neutrophils might have a role in bacterial clearance. To evaluate this possibility, bacterial burdens were estimated in gingival tissues of mice following infection. Our results show that in comparison to the T. forsythia wild-type strain, the survival of glycosylation-deficient strain was significantly reduced in mice (Figure 8), suggesting that neutrophils might have a protective role in the current infection setting. However, the contribution of surface glycosylation in providing protection against killing by antigen-presenting cells might also be responsible for the increased survival of this pathogen in the gingival tissues.

Glycosylation deficiency impairs bacterial survival. Bacterial loads in gingival tissues from mice infected with Tannerella forsythia wild-type (Tf WT) or ED1 strains on day 10 postinfection as determined by quantitative real-time polymerase chain reaction (PCR). The bars and error bars indicate the means and standard deviations for four mice in each group. *P<0.05. CFU, colony-forming unit; gapdh, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
In this study, we investigated the immunomodulatory effect of WecC-dependent O-linked protein glycosylation in T. forsythia. We observed increased secretion of Th17-differentiating cytokines IL-23 and IL-6 secretion in BMDCs and macrophages in response to the T. forsythia strain ED1 lacking the terminal sugar motif of a O-glycan core compared with the T. forsythia ATCC 43037 strain having the complete glycosylation repertoire. Moreover, as would be predicted based on these cytokine expression profiles, an increase in Th17 cells was detected in draining lymph nodes of mice orally infected with the glycosylation-deficient mutant ED1 compared with the wild-type strain. Further, the glycosylation deficiency impaired the bacterium's ability to induce alveolar bone loss, which our evidence suggests is due to an increase in the Th17 response following infection and a resultant increased neutrophil infiltration in the gingival tissues. Moreover, in line with an increase in neutrophil infiltration, reduced bacterial loads were found associated with mouse gingival tissues. Therefore, the major finding of our study is that Tannerella surface glycosylation associated with S-layer and other surface glycoproteins helps in inhibiting bacterial association with DCs and in restraining Th17 response to bacterial challenge in a murine model.
The T. forsythia ED1 mutant used in this study was previously generated by our group by a specific deletion of the wecC gene (TF2055) postulated to encode UDP-N-acetylmannosaminuronic acid dehydrogenase.10 This mutation results in altered glycosylation of S-layer glycoproteins,10 specifically it causes deletion of a terminal trisaccharide motif that consists of a terminally located mannosaminuronic acid (ManpNAcA) and pseudaminic acid (Pse5Am7Gc) residue linked to another mannosaminuronic acid residue.11 Importantly, both our transmission electron microscopy and outer-membrane purification experiments illustrate that the ED1 strain contains an intact, if altered, S-layer that still forms a protein coat around the cell surface but lacks full glycosylation resulting in altered immune system interactions. Moreover, there are no obvious changes in LPS presentation as judged by electrophoretic analysis of LPS preparations, thus indicating that the observed phenotype of the ED1 mutant with respect to its ability to modulate the immune response is due to the loss of surface glycosylation and not due to LPS abnormality.
In addition, it is thought that the S-layer of T. forsythia is a member of a set of proteins that are secreted by a novel protein secretion mechanism.33 These proteins are thought to be secreted via a largely uncharacterized protein secretion machinery that relies on a conserved C-terminal secretion domain (CTD) and has been most well characterized in the partner periodontal pathogen P. gingivalis.34, 35 In P. gingivalis, it is becoming clear that the CTD acts as a recognition site for a sortase-like protease that cleaves the CTD sequence linking C-terminal carbonyl to a sugar amine of A-LPS.35 Our findings that the altered S-layer (and other) glycoproteins of the ED1 strain can still be exported to the surface suggest that the glycosylation pattern of CTD proteins in T. forsythia is not likely to be part of the recognition signal for CTD system in T. forsythia.
Periodontitis is an inflammatory disease in which both innate and adaptive immune responses have critical roles. For instance, impairment in neutrophil and macrophage recruitment due to deficiencies in adhesion molecules increases P. gingivalis-induced alveolar bone destruction,36 and destructive roles for T and B cells in periodontal pathogenesis is well documented.37, 38 Similar to other inflammatory diseases, a disruption of the proper balance between individual Th subtypes (Th1, Th2, and Th17) contributes to the progression of periodontitis. The role of these different Th cell types and how bacterial factors promote or downregulate specific Th cell types and disrupt this balance is a major focus of many investigations.39, 40, 41, 42 With respect to T. forsythia, we have previously shown that T. forsythia infection causes a Th2 bias because of TLR2 activation, and that TLR2–Th2 inflammatory axis is primarily responsible for T. forsythia-induced alveolar bone loss.21 In this study, we show that surface glycosylation of Tannerella specifically acts to prevent Th17 generation, allowing generation of TLR2-dependent Th2 bias beneficial to bacteria by causing inflammatory tissue destruction and releasing components needed for bacterial growth. The protective role of Th17 against T. forsythia-induced alveolar bone loss in the periodontitis mouse model is in agreement with the findings of a previous study showing increased susceptibility of IL-17 receptor-deficient mice to the fellow periodontal pathogen P. gingivalis.43 However, Th17 responses have also been implicated in the progression of periodontitis.39, 44 It is likely that Th17 (IL-17) responses have protective or destructive roles depending on the disease phase and the pathogens involved. Th17 during early stages may provide anti-microbial immunity and have a role in controlling periodontal disease. Moreover, the presence of Th17 cells may not always translate to excessive IL-17 production deleterious to the periodontal tissue. This has been recently demonstrated in a study investigating the P. gingivalis-induced inflammatory bone loss in older mice in which neutrophils and γδT cells, but not Th17 cells, were found to be the major source of IL-17 in the gingival tissue.45 Another recent study found that IFN-γ, but not IL-17, drives P. gingivalis-induced inflammatory alveolar bone destruction.46
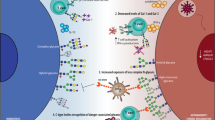
This study highlights the fundamental role of T. forsythia surface glycosylation in restraining the Th17 response, which otherwise might be detrimental to the pathogen by facilitating its clearance through neutrophils. Our model of events, which is outlined in Figure 9, envisages that T. forsythia engagement of TLR2 by surface ligands such as BspA and lipoproteins then skew the responses toward Th2, inducing alveolar bone loss. The exact mechanism by which surface layer glycans help to suppress Th17 response is unknown. However, it is likely that glycosylation, by blocking APC uptake of bacteria as shown in this study, might minimize the interactions of bacterial ligands with intracellular pattern-recognition receptors (NODs, TLR9). Another possibility exists that surface glycans by interacting with C-type lectin receptors skew the response towards Th2. In this regard, DC-SIGN as well as cytoplasmic NOD-like receptors have been shown to control Th2 responses.47 While TLR2 signaling has a dominant role in DCs against T. forsythia as shown previously, O-glycan-mediated signaling and its crosstalk with TLR2 impacts the net outcome of the DC–Tannerella interactions. This is because the differential expression of IL-12p40, IL-12p70, and IL-23 cytokines was lost when both TLR2- and O-glycan-mediated signaling were abolished (i.e., tlr2−/− DCs stimulated with ED1; Supplementary Figure S4 online). Here, it is also tempting to speculate that the terminal trisaccharide lacking in the S-layer glycoproteins of the ED1 mutant, which contains pseudaminic acid (a modified form of siaic acid in bacteria) can putatively interact with Siglecs (sialic acid binding Ig-like lectins) receptors on immune cells. Siglecs can act both as positive and negative regulators of immune response.48 The presence of pseudaminic acid is not unique to the T. forsythia S-layer, with the flagellin molecule of a range of species containing versions of this sugar.23 Although its role in flagellar glycosylation is thought to be largely structural, the nonglycosylated Pseudomonas aeruginosa flagellin is less immunostimulatory in inducing an IL-8 response than the wild-type glycosylated form of flagellin.49 As outlined above, our data suggest control of Th17 responses as key to periodontal disease progression, and it is of note that several other types of infection by other pathogens and human-dwelling bacteria also influence this arm of the immune system. For example, another oral organism Candida albicans dampens Th17 responses in vivo by secreting yet to be identified factors.50 In addition, the polysaccharide antigens of the gut organism Bacteroides fragilis account for the suppression of Th17 responses critical for its persistence in the gut.51 Taken together, these, and our, data suggest that therapeutic modulation of Th17 immunity might be an attractive strategy for clinical intervention of a range of pathologies including periodontitis.

A model depicting the role of Tannerella forsythia surface glycosylation in pathogenesis. The surface (S)-layer glycoproteins modulate dendritic cell (DC) effector functions to suppress T-helper (Th)17 responses, promoting bacterial persistence in the oral cavity. Bacteria then exploit Toll-like receptor (TLR)2 signaling to favor dominance of Th2 responses. The TLR2–Th2 inflammatory axis causes tissue destruction and drives osteoclastogenesis. Concurrently, inflammation promotes bacterial growth by making available peptides, heme, and other factors as nutrients.
In summary, S-layer glycosylation in T. forsythia may act by suppressing Th17 and innate neutrophil responses to increase pathogen survival in the host. In addition, it may also contribute in pathogen survival by increasing pathogen resistance to killing by antigen-presenting cells. Thus, the evolution of this trait by T. forsythia presumably increases its persistence in the human cavity by acting as an immune evasion tactic. At the same time, the organism exploits TLR2-mediated Th2 responses to its advantage in colonization of its primary environmental niche within periodontal pockets, which ultimately results in alveolar bone loss and expansion of this habitat. Overall, these results highlight how glycosylation of its surface layer proteins is part of a range of novel strategies that this persistent colonizer of the oral cavity utilizes to both cause periodontal disease but also survive in the body for many years and cause recurring bouts of this condition. These data have wide-ranging implications for the mechanisms used by persistent bacterial pathogens in immune manipulation, evasion, and exploitation, and reveal yet more of the intricacies of bacterial–host interactions that have evolved over time.
Methods
Bacterial strains and culture conditions. T. forsythia ATCC 43037 was grown in TF broth (brain heart infusion media containing 5 μg ml−1 hemin, 0.5 μg ml−1 menadione, 0.001% N-acetylmuramic acid, 0.1% L-cysteine, and 5% fetal bovine serum) as liquid cultures or on plates containing 1.5% agar in broth under anaerobic conditions as described previously.52 An isogenic T. forsythia mutant TFM-ED1 (hereafter called ED1)10 inactivated of wecC gene encoding UDP-N-acetyl-mannosaminuronic dehydrogenase was grown in broth or agar plates containing 5 μg ml−1 erythromycin.
Transmission electron microscopy method. For electron microscopy, 1 ml of plate-grown bacterial cell suspension of OD600 1.0 was centrifuged and the supernatant was discarded. The cell pellet was washed twice with cacodylate buffer (0.1 m cacodylate buffer (pH 7.4)) for 5 min before fixing with 3% glutaraldehyde for 2–4 h at 4°C. This was then rinsed in cacodylate buffer (pH 7.4) 3 × for 20 min at 4°C. The pellet was then soaked with 1% aqueous osmium tetraoxide for 1 h room temperature and washed three times (2 min) with dH2O at room temperature, followed by two washes (3 min) with 70% ethanol at room temperature, before two washes with 95% ethanol and two with 100%. After drying, the pellet was then soaked twice for 15 min in propylene oxide. This sample was then incubated overnight in propylene oxide:avaldite solution (1:1, v v−1) for 16 h (lids on) before removal and addition of 100% avaldite for 8 h (lids off). The pellet was then embedded in fresh avaldite and left to polymerize at 60°C for 24–48 h. Avaldite was prepared using 20 ml of Agar 100 epoxy resin, 16 ml of hardener DDSA, 8 ml of hardener MNA, and 1.3 ml of accelerator BDMA (C.3%). The embedded cell pellet was sectioned at 70–90 nm and placed on to a formvar-coated grid.
The grids were stained using uranyl acetate and lead citrate. Uranyl acetate (7%) was saturated in 50% methanol and used for staining. Lead citrate was prepared by adding 1.33 g lead nitrate and 1.76 g sodium citrate in 50 ml H2O/NaOH. Stained crosssections were viewed by transmission electron microscopy at the University of Sheffield Electron Microscopy service in the Department of Biological Sciences.
Outer-membrane purification and analysis. Cells were incubated at 37°C in an anaerobic atmosphere (10% CO2, 10% H2, and 80% N2) for 3 days, harvested using sterile loops, and resuspended by pipetting in 1 ml prechilled 50 mm HEPES and 50 mm NaCl (pH 7.4). Cell suspensions were placed in ethanol/ice water bath and lysed by sonication. Undisrupted cells and cell debris were pelleted by centrifugation at 15,000 r.p.m. for 30 min at 4°C and discarded. The supernatants were ultracentrifuged at 60,000 r.p.m. in a Beckman Ti70.1 fixed-angle rotor for 2 h at 4°C to pellet the inner and outer membranes. The supernatants were carefully decanted and discarded, while the membrane pellets were gently washed twice in 50 mm HEPES and 50 mm NaCl (pH 7.4). The pellets were then resuspended in 1 ml 50 mm HEPES and 50 mm NaCl (pH 7.4). To solubilize the inner membrane, an equal volume of 2% (v v−1) sodium N-lauroyl sarcosinate in 50 mm HEPES and 50 mm NaCl (pH 7.4) was added and mixed thoroughly by pipetting, followed by an incubation at 37°C for 30 min. The suspensions were centrifuged at 20,000 r.p.m. for 30 min at 4°C, and the supernatants containing the inner-membrane fraction were carefully decanted. The outer-membrane pellets were gently washed twice in 50 mm HEPES and 50 mm NaCl (pH 7.4), after which these pellets were resuspended in 500 μl 50 mm HEPES and 50 mm NaCl (pH 7.4), and stored at −20°C.
Samples were then analyzed by SDS-PAGE on Invitrogen NuPAGE 4–8% gradient gels and silver stained (SilverStainPlus; Bio-Rad, Hercules, CA). In addition, parallel gels were oxidized with periodic acid before staining with a Pro-Q Emerald glycoprotein staining kit from Molecular Probes (Eugene, OR) according to the manufacturer's instructions (visualized on a UV transilluminator). Samples were also transferred to nitrocellulose membrane before probing with a rabbit antibody raised against purified intact S-layer and detected with anti-rabbit horseradish peroxidase and Pierce Supersignal WestPico chemiluminescent substrate before exposure to Kodak X-ray film and development in a Kodak X-Omat developer.
Production of anti-S-layer antibody. Intact native S-layer from the wild-type T. forsythia 43037 was purified by cesium chloride density gradient ultracentrifugation according to a previously described protocol,9 and 100 μg of this mixture was used to prepare antibodies in rabbits within the Sheffield Antibody Unit (Bioserv, Sheffield, Yorkshire, UK).
Lipopolysaccharide. LPS was extracted from agar-grown T. forsythia wild-type and ED1 strains using the equivalent of 1 ml of an OD600 of 105 cells resuspended in phosphate-buffered saline (PBS) and using an LPS extraction kit from ChemBio (Bricket Wood, Hertfordshire, UK) with modifications. Briefly, after extraction the LPS was treated with proteinase K and DNAse before re-extraction using the same kit according to the manufacturer's instructions. LPS was visualized after separation of samples on a 10% urea-SDS-PAGE gel and oxidation with periodic acid (0.7%) before silver staining as described previously.24
Inoculation of mice. Specific pathogen-free BALB/cJ mice (Jackson Laboratory, Bar Harbor, ME), 5–6 weeks old (8–10 female mice per group), at the start of the experiment were maintained in the Laboratory Animal Facility of the University at Buffalo. The Institutional Animal Care and Use Committee approved all the experimental protocols used in the study. After 1-week quarantine, mice were infected with T. forsythia as per the previously described protocol.21 After 1 week of kanamycin (1 mg ml−1) in drinking water ad libitum, followed by a 3-day antibiotic-free period to suppress the resident bacteria, mice were infected by oral gavage with 109 CFU ml−1 of live bacteria (T. forsythia wild-type ATCC 43037, or the mutant strain ED1) in 100 μl of PBS with 100 μl of 2% carboxymethyl cellulose three times per week with 48-h intervals and the schedule was repeated the following week. Control (sham-infected) mice received antibiotic pretreatment and the 200 μl of 1% carboxymethyl cellulose without the bacteria.
Assessment of infection by PCR. Subgingival plaque samples were obtained from the molars of each mouse by placing sterile paper points (Johnson & Johnson, Piscataway, NJ) subgingivally for 5 s and then transferring and vortexing into 1 ml T. forsythia growth medium. Following 1 week of incubation, the medium was spun at 12,000 × g for 10 min, and the total bacterial genomic DNA was isolated with use of the Pure Gene genomic DNA isolation kit (Gentra, Minneapolis, MN). PCR was performed on 250 ng of genomic DNA using a primer pair specific for T. forsythia 16S RNA gene (amplicon length 641 bp) or a universal primer pair that matches almost all bacterial 16S RNA genes (amplicon length 1.4 kb) as described previously.53 PCR products were analyzed by agarose (1%) gel electrophoresis and ethidium bromide staining.
Assessment of alveolar bone loss. Horizontal bone loss around the maxillary molars was assessed by a morphometric method. After 6 weeks of first infection, the mice were killed and the serum samples were collected for IgG antibody response against the T. forsythia wild-type and ED1 strains by ELISA as described earlier.21 Skulls were autoclaved, de-fleshed, and jaws were immersed in 3% hydrogen peroxide overnight and stained with 1% methylene blue. The distances between the ABC and CEJ, considered as alveolar bone loss, were measured by two independent evaluators in a blinded manner at seven buccal sites on each side with the help of a dissecting microscope attached to an imaging system with a software (Aquinto Imaging System; a4i America, Brook-Anco, Rochester, NY).
Real-time PCR quantification of bacterial burden. Maxillary periodontal tissues (soft gingival and hard tissue) were excised on day 10 postinfection from the mice. DNA was extracted using a DNeasy tissue kit (Qiagen, Valencia, CA), and T. forsythia loads were determined by real-time PCR with primers targeting 16S rRNA gene described above using an iCycler IQ and SYBR green master mix from Bio-Rad. In addition, mouse glyceraldehyde-3-phosphate dehydrogenase (gapdh) gene amplified (primers 5′-GCACAGTCAAGGCCGAGAAT-3′ and 5′-GCCTTCTCCATGGTGGTGAA-3′) from the same sample was used to normalize bacterial loads in extracted tissues as described;54 bacterial loads were expressed as the number of log10 CFUs per 109 copies of gapdh. The real-time Ct values were converted to weights of DNA by interpolation from DNA weight vs. Ct standard curve. For calculating CFUs from the weights of DNA, the genome size of 3.4 Mb for T. forsythia (http://www.homd.org) was used.
T. forsythia -specific IgG antibody response by ELISA. T. forsythia wild-type and ED1 strain-specific IgG antibody responses were measured by ELISA as described previously.21 Briefly, 96-well Immuno-Maxisorp plates (Nalgene Nunc International, Rochester, NY) coated with formalin-fixed bacteria (109 cells per ml and 100 μl per well) were incubated with serial dilutions of mouse sera, followed by horseradish peroxidase-conjugated goat anti-mouse IgG (Bethyl Laboratories, Montgomery, TX). ELISA wells were color developed with TMB Microwell enzyme substrate (Kirkgaards and Perry, Gaithersburg, MD) and plates were read at 495 nm. Titers were defined as the log2 of the highest dilution with a signal that was 0.1 optical density unit above the background level.
Generation of BMDCs and in vitro stimulation. Immature DCs were generated by using EasySep mouse CD11c-positive selection kit (Stem Cell Technologies, Vancouver, BC, Canada) following the manufacturer's instructions. Briefly, bone marrow progenitor cells were flushed into the recommended medium with a 23-gauge needle. The clumps that were formed were dispersed by gently passing the cell suspension through the syringe and removing the debris by passing cell suspension through 70-μm nylon cell strainer. After centrifugation, cells were resuspended in RPMI 1640, which contains 10% heat-inactivated fetal calf serum, 2 mm glutamine, penicillin, and streptomycin, 5 μm 2-mercaptoethanol, and 10 ng ml−1 granulocyte-macrophage colony-stimulating factor . Cells were incubated in 20 ml plate/150 mm Petri dish at 37°C for 5 days. Non-adherent cells were collected and wash once, and the CD11c+ cells were purified using Purple EasySep Magnet (Stem Cell Technologies). This protocol yielded BMDCs with >93% purity. Purified BMDCs were stimulated with bacteria for 12 h and the cytokine levels in the medium were quantified by ELISA.
For DC-mediated bacterial processing, BMDCs were infected with live bacteria at a multiplicity of infection of 1:10 in antibiotic-free medium, and at time points 15 and 60 min postinfection, BMDCs were extensively washed, lysed with sterile water, serially diluted, and bacterial CFU determined by plating.
Isolation of peritoneal macrophages and challenge with stimulants. Mouse peritoneal macrophages were prepared as described previously.55 Purified macrophages were seeded in 48-well culture plates at a density of 105 cells per well in 250 μl growth medium. T. forsythia cells were enumerated with Petroff–Hausser chamber immediately before each experiment. For challenge, multiplicity of infection (number of bacteria per mammalian cell) of 10 was used. E. coli LPS (100 ng ml−1) was used as a positive control. After 12 h of stimulation, the cytokine levels were quantified by ELISA.
Determination of cytokine levels by ELISA. Culture supernatants were collected from BMDCs pulsed with TF strains (TF 43037, TFM-ED1-Wec C mutant) to assess the levels of IL-1β, IL-6, IL-12p40, IL-12p70, IL-23, and IL-10 by ELISA with Ready-SET-Go kits (eBioscience, San Diego, CA) as per the manufacturer's instructions. Supernatants were used as undiluted and 1:10-fold dilutions. The detection limit for all cytokines was 5 pg ml−1.
Intracellular staining and fluorescence-activated cell sorting analysis. The following antibodies and isotype controls were purchased from eBiosciences: phycoerythrin-conjugated anti-IFN-γ, IL-5, and rat IgG2a; phycoerythrin-Cy5-labeled anti-CD4; anti-CD80 (16-10A1) and anti-CD83 (Michel-17); phycoerythrin-conjugated anti-CD11c (N418); hamster IgG; and phycoerythrin-conjugated anti-IL-17A (eBio17B7). For intracellular staining, cell suspensions from cLNs were stimulated with anti-CD3 and anti-CD28 antibodies (eBioscience) for 48 h, followed by phorbol myristate acetate (50 ng ml−1), ionomycin (1 μg ml−1), and brefeldin A (10 μg ml−1) for 6 h. Cells were washed, incubated with FcR block (1 μg ml−1; eBiosciences), and stained for CD4. Cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% saponin (Sigma, St Louis, MO), and stained for IL-5, IL-17, or IFN-γ. Cells were analyzed on a FACS Calibur (BD Biosciences, Franklin Lakes, NJ) with FCS Express software (DeNovo Software, Los Angeles, CA).
Quantification of CD45+ cells and infiltrated neutrophils. The right and left halves of the maxillary and mandibular bones were removed, fixed, and embedded in paraffin, and 4-μm sections were cut and mounted. They were deparaffinized in xylene and hydrated in graded ethanol. After antigen retrieval by incubating at 90°C for 10 min with BD Retrievagen A (BD PharMingen, Franklin Lakes, NJ), specimens were sequentially incubated in: (i) blocking solution containing 0.1% Triton X-100 in 0.1 m PBS for 1 h at room temperature; and (ii) monoclonal rat anti-mouse CD45 (BD PharMingen) antibody diluted 1:30 in PBS containing 0.1% Triton X-100 or antineutrophil marker antibody (NIMP-R14; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature. The slides were incubated with a biotinylated secondary antibody (goat anti-rat), followed by an avidin–biotin complex developed with 3, 3-diaminobenzidine (Vector Labs, Burlingame, CA); the counterstain used was hematoxylin. After each step, slides were rinsed in PBS with Tween 20 (3 × 10 min). Slides were scanned at an absolute magnification of × 400 using the Aperio Scan Scope CS system (Aperio Technologies, Vista, CA). Slides (six slides per mouse) were viewed and analyzed remotely using desktop personal computers employing the virtual image viewer software (Aperio). Antibody-positive cells (stained brown) were enumerated manually in the interdental areas from the first to third molar at randomly selected locations in each slide (six per slide). The areas associated with these locations were then obtained from the software to calculate the average cell densities per square millimeter.
TRAP staining. Mice maxillary and mandibular bones (n=4) were fixed in 10% phosphate-buffered formalin and decalcified in 10% ethylenediaminetetraacetic acid. The samples were then embedded in paraffin, and sections at 4 μm were prepared and stained for TRAP (Sigma). The TRAP-stained whole slides were digitally scanned immediately with a Scan Scope CS system (Aperio) to minimize color fading. The scanned slides were viewed with Image Scope viewing software (Aperio). The right maxillary and mandibular interdental areas (average of 10 higher power fields per slide) of the crestal alveolar bone from the first molar to third molar were used to quantify osteoclasts.
Data analysis. Data were analyzed on the Graph Pad Prism software (Graph Pad, San Diego, CA). Comparisons between groups were made using a Student's t-test (between two groups) or analysis of variance (multiple group comparisons), as appropriate. Statistical significance was defined as P<0.05.
References
Socransky, S.S., Haffajee, A.D., Cugini, M.A., Smith, C. & Kent, R.L. Jr Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144 (1998).
Seymour, G.J., Ford, P.J., Cullinan, M.P., Leishman, S. & Yamazaki, K. Relationship between periodontal infections and systemic disease. Clin. Microbiol. Infect. 13 (Suppl 4), 3–10 (2007).
Pischon, N. et al. Obesity, inflammation, and periodontal disease. J. Dent. Res. 86, 400–409 (2007).
Moen, K. et al. Synovial inflammation in active rheumatoid arthritis and psoriatic arthritis facilitates trapping of a variety of oral bacterial DNAs. Clin. Exp. Rheumatol. 24, 656–663 (2006).
Xavier, R.J. & Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007).
Sharma, A. Virulence mechanisms of Tannerella forsythia. Periodontology 2000 54, 106–116 (2010).
Stafford, G., Roy, S., Honma, K. & Sharma, A. Sialic acid, periodontal pathogens and Tannerella forsythia: stick around and enjoy the feast!. Mol. Oral Microbiol. 27, 11–22 (2012).
Sara, M. & Sleytr, U.B. S-layer proteins. J. Bacteriol. 182, 859–868 (2000).
Lee, S.W. et al. Identification and characterization of the genes encoding a unique surface (S-) layer of Tannerella forsythia. Gene 371, 102–111 (2006).
Honma, K., Inagaki, S., Okuda, K., Kuramitsu, H.K. & Sharma, A. Role of a Tannerella forsythia exopolysaccharide synthesis operon in biofilm development. Microb. Pathog. 42, 156–166 (2007).
Posch, G. et al. Characterization and scope of S-layer protein O-glycosylation in Tannerella forsythia. J. Biol. Chem. (2011).
Lebeer, S., Vanderleyden, J. & De Keersmaecker, S.C. Host interactions of probiotic bacterial surface molecules: comparison with commensals and pathogens. Nat. Rev. Microbiol. 8, 171–184 (2010).
Szymanski, C.M. & Wren, B.W. Protein glycosylation in bacterial mucosal pathogens. Nat. Rev. Microbiol. 3, 225–237 (2005).
Eynon, E.E., Zenewicz, L.A. & Flavell, R.A. Sugar-coated regulation of T cells. Cell 122, 2–4 (2005).
Mazmanian, S.K., Liu, C.H., Tzianabos, A.O. & Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118 (2005).
Round, J.L. & Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 107, 12204–12209 (2010).
Konstantinov, S.R. et al. S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc. Natl. Acad. Sci. USA 105, 19474–19479 (2008).
Zeituni, A.E., Jotwani, R., Carrion, J. & Cutler, C.W. Targeting of DC-SIGN on human dendritic cells by minor fimbriated Porphyromonas gingivalis strains elicits a distinct effector T cell response. J. Immunol. 183, 5694–5704 (2009).
Zeituni, A.E., McCaig, W., Scisci, E., Thanassi, D.G. & Cutler, C.W. The native 67-kilodalton minor fimbria of Porphyromonas gingivalis is a novel glycoprotein with DC-SIGN-targeting motifs. J. Bacteriol. 192, 4103–4110 (2010).
Sekot, G. et al. Potential of the Tannerella forsythia S-layer to delay the immune response. J. Dent. Res. 90, 109–114 (2011).
Myneni, S.R. et al. TLR2 signaling and Th2 responses drive Tannerella forsythia-induced periodontal bone loss. J. Immunol. 187, 501–509 (2011).
Tanner, A.C.R., Listgarten, M.A., Ebersole, J.L. & Strzempko, M.N. Bacteroides forsythus sp. nov., a slow growing, fusiform Bacteroides sp. from the human oral cavity. Int. J. Syst. Bacteriol. 36, 213–221 (1986).
Tabei, S.M. et al. An Aeromonas caviae genomic island is required for both O-antigen lipopolysaccharide biosynthesis and flagellin glycosylation. J. Bacteriol. 191, 2851–2863 (2009).
Stafford, G.P. & Hughes, C. Salmonella typhimurium flhE, a conserved flagellar regulon gene required for swarming. Microbiology 153, 541–547 (2007).
Holscher, C. The power of combinatorial immunology: IL-12 and IL-12-related dimeric cytokines in infectious diseases. Med. Microbiol. Immunol. 193, 1–17 (2004).
Nigg, A.P. et al. Dendritic cell-derived IL-12p40 homodimer contributes to susceptibility in cutaneous leishmaniasis in BALB/c mice. J. Immunol. 178, 7251–7258 (2007).
Piccotti, J.R., Chan, S.Y., Li, K., Eichwald, E.J. & Bishop, D.K. Differential effects of IL-12 receptor blockade with IL-12 p40 homodimer on the induction of CD4+ and CD8+ IFN-gamma-producing cells. J. Immunol. 158, 643–648 (1997).
Slight, S.R., Lin, Y., Messmer, M. & Khader, S.A. Francisella tularensis LVS-induced interleukin-12 p40 cytokine production mediates dendritic cell migration through IL-12 receptor beta1. Cytokine 55, 372–379 (2011).
Pompei, L. et al. Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J. Immunol. 178, 5192–5199 (2007).
Geijtenbeek, T.B. & Gringhuis, S.I. Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol. 9, 465–479 (2009).
Hovius, J.W. et al. Salp15 binding to DC-SIGN inhibits cytokine expression by impairing both nucleosome remodeling and mRNA stabilization. PLoS Pathog. 4, e31 (2008).
Onishi, R.M. & Gaffen, S.L. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129, 311–321 (2010).
Veith, P.D. et al. Outer membrane proteome and antigens of Tannerella forsythia. J. Proteome Res. 8, 4279–4292 (2009).
Seers, C.A. et al. The RgpB C-terminal domain has a role in attachment of RgpB to the outer membrane and belongs to a novel C-terminal-domain family found in Porphyromonas gingivalis. J. Bacteriol. 188, 6376–6686 (2006).
Slakeski, N. et al. C-terminal domain residues important for secretion and attachment of RgpB in Porphyromonas gingivalis. J. Bacteriol. 193, 132–142 (2011).
Baker, P.J., DuFour, L., Dixon, M. & Roopenian, D.C. Adhesion molecule deficiencies increase Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect. Immun. 68, 3103–3107 (2000).
Baker, P.J. The role of immune responses in bone loss during periodontal disease. Microbes Infect. 2, 1181–1192 (2000).
Baker, P.J., Evans, R.T. & Roopenian, D.C. Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch. Oral Biol. 39, 1035–1040 (1994).
Gaffen, S.L. & Hajishengallis, G. A new inflammatory cytokine on the block: re-thinking periodontal disease and the Th1/Th2 paradigm in the context of Th17 cells and IL-17. J. Dent. Res. 87, 817–828 (2008).
Gemmell, E., Yamazaki, K. & Seymour, G.J. Destructive periodontitis lesions are determined by the nature of the lymphocytic response. Crit. Rev. Oral Biol. Med. 13, 17–34 (2002).
Teng, Y.T. Protective and destructive immunity in the periodontium: part 1—innate and humoral immunity and the periodontium. J. Dent. Res. 85, 198–208 (2006).
Teng, Y.T. Protective and destructive immunity in the periodontium: part 2—T-cell-mediated immunity in the periodontium. J. Dent. Res. 85, 209–219 (2006).
Yu, J.J. et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109, 3794–3802 (2007).
Ohyama, H. et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J. Dent. Res. 88, 633–638 (2009).
Eskan, M.A. et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 13, 465–473 (2012).
Arizon, M. et al. Langerhans cells down-regulate inflammation-driven alveolar bone loss. Proc. Natl. Acad. Sci. USA 109, 7043–7048 (2012).
Pulendran, B., Tang, H. & Manicassamy, S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat. Immunol. 11, 647–655 (2010).
Crocker, P.R. & Redelinghuys, P. Siglecs as positive and negative regulators of the immune system. Biochem. Soc. Trans. 36, 1467–1471 (2008).
Verma, A., Arora, S.K., Kuravi, S.K. & Ramphal, R. Roles of specific amino acids in the N terminus of Pseudomonas aeruginosa flagellin and of flagellin glycosylation in the innate immune response. Infect. Immun. 73, 8237–8246 (2005).
Cheng, S.C. et al. Candida albicans dampens host defense by downregulating IL-17 production. J. Immunol. 185, 2450–2457 (2010).
Round, J.L. et al. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332, 974–977 (2011).
Sharma, A. et al. Cloning, expression, and sequencing of a cell surface antigen containing a leucine-rich repeat motif from Bacteroides forsythus ATCC 43037. Infect. Immun. 66, 5703–5710 (1998).
Sharma, A. et al. Tannerella forsythia-induced alveolar bone loss in mice involves leucine-rich-repeat BspA protein. J. Dent. Res. 84, 462–467 (2005).
Fang, R., Ismail, N., Shelite, T. & Walker, D.H. CD4+ CD25+ Foxp3-T-regulatory cells produce both gamma interferon and interleukin-10 during acute severe murine spotted fever rickettsiosis. Infect. Immun. 77, 3838–3849 (2009).
Zheng, X., Goncalves, R. & Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 83, 14.1.1–14.1.14 (2008).
Acknowledgements
This work was supported by US Public Health R01 grants DE14749 and DE19424. Work in the Stafford lab was supported by a Dunhill Medical Trust grant (R185/0211) to G.S. and a Wellcome VIP Fellowship to S.R. We thank Dr Sarah Gaffen for her helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article
Cite this article
Settem, R., Honma, K., Nakajima, T. et al. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol 6, 415–426 (2013). https://doi.org/10.1038/mi.2012.85
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2012.85
This article is cited by
-
Insights into bacterial protein glycosylation in human microbiota
Science China Life Sciences (2016)
-
Role of S-layer proteins in bacteria
World Journal of Microbiology and Biotechnology (2015)
-
Lactobacillus surface layer proteins: structure, function and applications
Applied Microbiology and Biotechnology (2013)
