
Abstract
Intranasal infection with vaccinia virus co-expressing interferon epsilon (VV-HIV-IFN-ɛ) was used to evaluate the role of IFN-ɛ in mucosal immunity. VV-HIV- IFN-ɛ infection induced a rapid VV clearance in lung that correlated with (i) an elevated lung VV-specific CD8+CD107a+IFN-γ+ population expressing activation markers CD69/CD103, (ii) enhanced lymphocyte recruitment to lung alveoli with reduced inflammation, and (iii) an heightened functional/cytotoxic CD8+CD4+ T-cell subset (CD3hiCCR7hiCD62Llo) in lung lymph nodes. These responses were different to that observed with intranasal VV-HA-IFN-α4 or VV-HA-IFN-β infections. When IFN-ɛ was used in an intranasal/intramuscular heterologous HIV prime-boost immunization, elevated HIV-specific effector, but not memory CD8+T cells responses, were observed in spleen, genito-rectal nodes, and Peyer's patch. Homing marker α4β7 and CCR9 analysis indicated that unlike other type I IFNs, IFN-ɛ could promote migration of antigen-specific CD8+T cells to the gut. Our results indicate that IFN-ɛ has a unique role in the mucosae and most likely can be used to control local lung and/or gut infections (i.e., microbicide) such as tuberculosis, HIV-1, or sexually transmitted diseases.
Similar content being viewed by others


Introduction
Interferons (IFNs) are small single-chain glycoproteins, involved in the first line of defense against pathogens such as viruses, bacteria, and parasites. They are also known to have an important role in inhibition of tumor growth and modulation of immune responses.1, 2, 3, 4 Clinically, IFN-α and IFN-β have demonstrated efficacy as treatments against cancer, chronic hepatitis B, and other infectious diseases.5, 6, 7 Categorized by the type of receptors they use for signaling, there are three types of IFN (I, II, and III) discovered to date. The type I IFN family includes IFN-α, IFN-β, IFN-ɛ, IFN-κ, IFN-τ, and IFN-ω,8, 9 and signal via the IFNAR1 and IFNAR2 receptor chains. IFN-γ is the sole member of the type II family, signaling through IFNGR1 and IFNGR2. The more recently discovered IFN-λ belongs to the type III family, which consists of three members: IFN-λ1, IFN-λ2, and IFN-λ3,9, 10, 11 which signal via a receptor complex involving IL-10R2 and IL-28Ra subunits.12
The recently discovered IFN-ɛ has been classified into the type I IFN family due to similarity of receptor type, protein structure, and gene locus. The IFN-ɛ consists of 192 amino acids, and the analysis of its protein structure has indicated that IFN-ɛ has overall similarity to IFN-β.13 Unlike other type I IFNs, IFN-ɛ has found to be expressed constitutively in the lung, brain, small intestine, and reproductive tissues.13, 14 Studies by Peng et al.,15 using aminion-derived WISH cells infected with vesicular stomatitis virus, indicated that IFN-ɛ has less antiviral, anti-proliferative, and natural killer cell enhancing activities compared with IFN-α2b. Matsumiya et al.16 showed that IFN-ɛ is the only type I IFN family member to be expressed by HeLa cells, and its expression was upregulated following TNF-α stimulation. Moreover, seminal plasma was also found to upregulate expression of IFN-ɛ in cervico-vaginal tissues.17 Therefore, IFN-ɛ is thought to have a protective role in reproductive tissue.
In our laboratory, previously Day et al.18, 19 studied the functional and biological properties of IFN-ɛ using a recombinant vaccinia virus (VV) co-expressing HIV gag/pol and murine IFN-ɛ (VV-HIV-IFN-ɛ) compared to IFN-α4 and IFN-β. The in vitro studies indicated that VV-HIV-IFN-ɛ was able to reduce growth of VV in the L929 murine cell lines, and had enhanced ability to upregulate activation markers CD69 and CD86 as well as antiviral proteins (PKR and 2′5′OAS) compared with the parental control (VV-HIV). However, its antiviral activity was lower than that induced by IFN-α4 or β. Interestingly, these studies also revealed that IFN-ɛ was constitutively expressed in the lung and in an allergic BALB/c model, and IFN-ɛ mRNA levels were upregulated compared with the control,19 suggesting that IFN-ɛ could be involved in mucosal immunity.
When developing control strategies against HIV-1 induction of strong and sustained mucosal immunity is thought to be critical as virus is first encountered at the mucosae (e.g., genito-rectal tissue and gastro-intestinal tract), and the early CD4+ depletion occurs in the gut.20, 21, 22 Heterologous prime-boost immunization using poxvirus vectors co-expressing molecular adjuvants has shown to induce robust levels of HIV-specific effector and memory T-cell responses to vaccine antigens.23, 24, 25, 26, 27 Ranasinghe et al.26, 28, 29, 30 have shown that, both the order of vector delivery, and the route of delivery could trigger variable magnitudes and avidity of the antigen-specific T cells. They have demonstrated that recombinant fowlpox (rFPV) and vaccinia virus (rVV) intranasal/intramuscular (IN/IM) prime-boost immunization can generate elevated HIV-specific cytotoxic T lymphocytes (CTL) of high avidity in systemic and mucosal compartments compared with purely systemic immunization (IM/IM).26, 28, 29, 30 Unlike IFN-α4 or IFN-β, IFN-ɛ has a unique mucosal constitutive expression profile and also possesses antiviral activity.13, 15, 16, 17, 18 Therefore, in this study we further investigated (i) the immunobiology of IFN-ɛ and its role in mucosal immunity, especially in the lung and gut, and (ii) whether co-expression of IFN-ɛ together with HIV vaccine antigens could act as an adjuvant to enhance much needed HIV-specific mucosal CTL immunity in gut following IN rFPV/IM rVV prime-boost immunization compared to systemic delivery 26, 30
Results
Intranasal VV-HIV-IFN-ɛ infection generates enhanced VV-specific CD8 CTL in the lung and rapid viral clearance
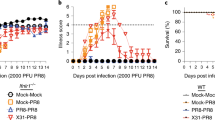
Co-workers Hardy,13 Chen,14 and also our own studies18, 19 have demonstrated that IFN-ɛ was constitutively expressed in the lung. Hence, we first evaluated its role in lung-specific immunity. Mice were infected IN with 5 × 106 plaque-forming unit (PFU) control VV-HIV or VV-HIV-IFN-ɛ, and VV-specific T-cell responses in the systemic (spleen) and mucosal (lung) compartments were evaluated by IFN-γ ELISpot. Even though at 7 days post-infection (PI), no differences in the number of spleen IFN-γ+CD8+ T cells were observed (Figure 1a), larger spot-forming units were detected following VV-HIV-IFN-ɛ infection compared with VV-HIV (Figure 1b). Therefore, IFN-γ protein levels were evaluated using enzyme-linked immunosorbent assay (Figure 1c), and data indicated that VV-HIV-IFN-ɛ-infected mice generated significantly higher spleen IFN-γ levels (60 ng ml−1) compared with the control VV-HIV infection (20 ng ml−1; *P=0.012).

Anti-vaccinia CD8+ T-cell response in spleen and lung at 7 days post IN infection of type I IFNs. Mice (n=4–5) per group were infected with 5 × 106 PFU of VV-HIV, VV-HIV-IFN-ɛ, VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4. (a) Spleen and (d) lung cells were stimulated with vaccinia-specific KdA52R75−83 (KYGRLFNEI) peptide described as in Methods, and IFN-γ-producing T-cell responses were measured by IFN-γ ELISpot and/or ICS. (b) Picture represents four wells of an ELISpot plate, dots represent IFN-γ spot-forming unit (SFU) in spleen, and (c) bar chart represents IFN-γ protein levels measured by enzyme-linked immunosorbent assay (ELISA) following VV-HIV-IFN-ɛ or VV-HIV infection, upon re-stimulation with KdA52R75−83. (e) Histogram analysis of CD69 and CD103 surface markers on lung CD8+ T cells (gated on total CD8+ T cells). (f) Lung VV titers at days 3 and 6 following IN VV-HIV or VV-HIV-IFN-ɛ infection. The dotted line indicates the limit of detection of the assay, which is <102 PFU. (g) Lung H&E staining of uninfected naive control and VV-HIV and VV-HIV-IFN-ɛ IN-infected mice, (h) Lung H&E staining of VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4 IN-infected mice 7 days PI. Images were analyzed by Olympus Microscope at × 4 (top) and × 60 (bottom) magnification, respectively. The × 60 magnification represents the boxed area at the top. Error bars represent ±s.d. APC, allophycocyanin; HA, hemagglutinin; H&E, hematoxylin and eosin; ICS, intracellular cytokine staining; IFN, interferon; IN, intranasal; PI, post infection; intranasal; VV, vaccinia virus.
Strikingly at 7 days PI, unlike the other two type I IFNs (α4 or β), IN VV-HIV-IFN-ɛ infection induced elevated lung VV-specific CD8+ T cell responses measured by IFN-γ ELISpot, following KdA52R75−83 peptide stimulation (Figure 1d, left). Intracellular cytokine staining indicated that 100% of these IFN-γ-producing CD8+ T cells were also positive for lysosomal-associated membrane protein-1 or CD107a,31 suggesting they were cytotoxic (Figure 1d, right). Moreover, CD8+ T cells obtained from the VV-HIV-IFN-ɛ-infected lung expressed enhanced activation markers CD69 and CD103 compared with its control or VV-HA-IFN-β or VV-HA-IFN-α4 groups (Figure 1e). Even though at 3 days PI, the VV-HIV-IFN-ɛ-infected group showed only a slight reduction in lung virus titers compared with the control, a rapid clearance of VV was observed by 6 days PI (**P=0.036) and no detectable viral titers were observed in the lung (note that in these assays 102 PFU is the limit of detection; Figure 1f). Similarly, with VV-HA-IFN-β no virus was detected. In contrast, VV-HA-IFN-α4 group showed very few virus particles just above 102 PFU detection level at 6 days PI (data not shown). This is consistent with our previous observations in ovaries 3 days PI.18
Hematoxylin and eosin (H&E) staining of VV-HIV-IFN-ɛ-infected lungs at 7 days PI (Figure 1g, right panels) showed reduced lung inflammation ( × 4 magnification) and high lymphocyte infiltration ( × 60 magnification) to lung alveoli compared with VV-HIV-infected lungs (Figure 1g, middle panels). The VV-HIV-IFN-ɛ-infected lung morphology at × 4 magnification was very similar to that of the naïve controls (Figure 1g, left panels), which further substantiated the rapid viral clearance. Interestingly, when we compared the lung morphology of VV-HA-IFN-β (Figure 1h, middle panels), VV-HA-IFN-α4 (Figure 1h, right panels), and control VV-HA ((Figure 1h, left panels)-infected groups to that of VV-HIV-IFN-ɛ group, at 7 days PI VV-HA-IFN-β- and VV-HA-IFN-α4-infected lung showed greater inflammation and lower lymphocyte recruitment compared with VV-HIV-IFN-ɛ group. Collectively, the efficiency of lymphocyte recruitment to lung alveoli was found to be VV-HIV-IFN-ɛ > VV-HA-IFN-β > VV-HA-IFN-α4 (Figure 1g and h).
Intranasal VV-HIV-IFN-ɛ infection induced elevated highly functional CD8+CD4+ double-positive T-cell population in lung lymph nodes
Seven days post VV-HIV-IFN-ɛ infection, twofold higher CD8+CD4+ double-positive (DP) T-cell population (5.7%) was observed in lung lymph nodes (LLNs) compared with the control VV-HIV infection (1.71%, *P=0.039). In naive control group, this population totaled to 0.74% of the CD8+ T cells (Figure 2a). To further understand the functionality of this DP population, we first evaluated whether these CD8+CD4+ T cells induced by VV-HIV-IFN-ɛ were able to produce VV-specific IFN-γ at effector (7 days) and/or memory (28 days) stage of infection. By 7 days PI, a greatly enhanced IFN-γ+CD8+CD4+ T cell subset was observed in VV-HIV-IFN-ɛ-infected mice compared with the control (Figure 2b, fluorescence-activated cell sorting (FACS) plots). However, no such differences were observed at 28 days PI compared with 7 days PI (Figure 2b, bar chart). Similar CD8+CD4+ T-cell subsets that express memory phenotype and cytokines/chemokines and serine proteases have been reported in previous studies.32, 33, 34, 35, 36 Hence, we further characterized this CD8+CD4+ positive LLN subset and found that nearly all the DP T cells were CD3+ and CD107a+, but no granzyme B expression was detected 7 days PI. Also 80% of the DP T cells expressed memory markers CCR7 and 20% were found to be CD62L positive (Figure 2c).

Intranasal VV-HIV-IFN-ɛ infection can induce functional and cytotoxic CD8+CD4+ double-positive CTL in LLN. BALB/c mice (n=5–6) per group were IN infected with 5 × 106 PFU of VV-HIV, VV-HIV-IFN-ɛ, VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4, and unimmunized mice were used as naive controls. (a) FACS plots represent one pooled experiment (left to right: naive, VV-HIV, and VV-HIV-IFN-ɛ), and bar chart on the right indicates average values from three individual pooled experiments. Upper right quadrants (R3) indicate the percentage of CD8+CD4+ DP T cells out of total CD8+ T-cell population (R2) in LLN. (b) FACS plot represent percentage of IFN-γ+CD8+CD4+ T cells (out of total DP population–R3) at 7 days PI (28 days is not shown), following VV-HIV (left) and VV-HIV-IFN-ɛ (right) infections. The cells were stimulated for 16 hrs with KdA52R75−83-specific peptide (bottom FACS plots) and unstimulated cells were used as background controls (top FACS plots). The bar chart below represents the percentage of IFN-γ+CD8+CD4+ T cells at 7 and also 28 days PI, and the plotted data indicate A52R75−83 stimulated minus the unstimulated values. (c) CD107a, CD3, CCR7 and CD62L expression on CD8+CD4+ DP T cells (R3) following VV-HIV-IFN-ɛ infection. (d) The left graph indicates the percentage of CD8+CD4+ T cells in LLN following IN, IM, and IP VV infection. The right graph represents the percentage of CD8+CD4+ T cells in the LLN at 3, 7, and 28 days post IN infection. (e) Percentage of CD8+CD4+ T cells (left), and IFN-γ+CD8+CD4+ T cells (right) in lung at 7 and 28 days post IN infection. (f) FACS plots from left to right represent percentage of LLN CD8+CD4+ population obtained with VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4-infected groups, respectively. (g and h) Surface markers CD107a, CD3, CCR7, and CD62L expression on CD8+CD4+ DP T cells (R3) following VV-HA-IFN-β and VV-HA-IFN-α4 IN infection. Except where stated, data represent pooled values, error bars represent ±s.d., and experiments were repeated for 2–3 times. DP, double positive; FACS, fluorescence-activated cell sorting; HA, hemagglutinin; IFN, interferon; IN, intranasal; IM, intramuscular; IP, intraperitoneal; LLN, lung lymph node; PFU, plaque-forming unit; PI, post infection; VV, vaccinia virus.
As CD8+CD4+ T cells are normally found in the thymus, we then compared thymic DP T cells following infection with VV-HIV and VV-HIV-IFN-ɛ to assess whether VV-HIV-IFN-ɛ could also modulate thymic CD8+CD4+ T-cell numbers. However, no notable differences were observed between the two groups (results not shown). Then we further investigated whether the presence of CD8+CD4+ T cells in the LLN was infection route- and/or time-dependent. When different routes of infections were performed, no elevated DP T-cell subset was observed in LLN following IM or intraperitoneal (IP) VV-HIV-IFN-ɛ infection compared with IN infection (Figure 2d, left). Next we assessed whether the CD8+CD4+ T cells induced by IN VV-HIV-IFN-ɛ infection was transient or long-lasting by evaluating the responses at 3, 7, and 28 days PI. At 3 days, no difference in CD8+CD4+ population was observed between the two groups; however, highly enhanced number of CD8+CD4+ T cells were observed at 7 days PI (∼6%) in VV-HIV-IFN-ɛ group, and this was reduced to around 2% by 28 days (Figure 2d, right). Interestingly, this correlated well with increased numbers of IFN-γ-producing lung T cells and the rapid viral clearance in lung at 7 days PI (Figure 1d, f and g). Given the above findings, we subsequently tested whether the CD8+CD4+ T-cell population could also be detected in the lung itself. At 7 days PI, the number of lung CD8+CD4+ T cells in each infected group was similar (Figure 2e, left), but mice infected with VV-HIV-IFN-ɛ generated twice the number of IFN-γ+CD8+CD4+ T cells (∼5%) compared with the control (∼2%) at day 7, but not at 28 days PI (Figure 2e, right).
Then we evaluated whether this elevated LLN CD8+CD4+ T-cell population could also be found following VV-HA-IFN-β and VV-HA-IFN-α4 infections. For this study we used the same VV constructs that were used in our previous comparative studies.18 Data indicated that even though IFN-β did not induce an elevated CD8+CD4+ DP T-cell subset, VV-HA-IFN-α4 infection induced much greater CD8+CD4+ T-cell subsets compared with the control VV-HA (Figure 2f). Interestingly, unlike VV-HIV-IFN-ɛ the DP cells induced by both VV-HA-IFN-β and VV-HA-IFN-α4 found to be CD3lo (20%), CD62Lhi (80%), and CCR7 negative (Figure 2g and h).
Intranasal VV-HIV-IFN-ɛ infection induces gut-specific KdA52R75−83 tetramer reactive CD8+ T cells in Peyer's patch
As IFN-ɛ was originally found in the small intestine and reproductive tissues,13, 14 7 days post IN infection with VV-HIV-IFN-ɛ, VV-HIV-IFN-β, VV-HIV-IFN-α4, and the respective controls the VV-specific CD8+ T cell response in Peyer's patch (PP) were evaluated using KdA52R tetramer staining together with gut-specific homing marker analysis. Data clearly indicated that IN VV-HIV-IFN-ɛ infection induced greatly elevated numbers of VV-specific CD8+ T cells compared with the control VV-HIV (Figure 3a and b). These T cells also showed twofold higher expression frequency of α4β7 and CCR9 in PP (Figure 3c). In contrast, both IFN-β and IFN-α4 infections did not induce any such enhancement in gut-specific T cells (Figure 3d, e, f and g).

Co-expression of IFN-ɛ induced a higher level of gut-specific homing markers on KdA52R75−83-specific CD8+ T cell in PP following IN infection. BALB/c mice (n=5 per group) were infected IN with 5 × 106 PFU of (a) VV-HIV, (b) VV-HIV-IFN-ɛ, (d) VV-HA, (e) VV-HA-IFN-β, and (f) VV-HA-IFN-α4. Seven days post infection, the percentage of KdA52R75−83-specific CD8+ T cells that expressed gut-specific homing markers (α4β7 and CCR9) were analyzed. (c and g) The expression frequencies of homing markers on KdA52R75−83-specific CD8+ T cells following IN IFN-ɛ, IFN-β, and IFN-α4 infections were calculated as indicated in Methods. All FACS plots, the upper right quadrants (R3) represent gates R1+R2. Data represent pooled values, and experiments were repeated two times. FACS, fluorescence-activated cell sorting; FITC, fluorescein isothiocyanate; HA, hemagglutinin; IFN, interferon; IN, intranasal; PFU, plaque-forming unit; VV, vaccinia virus.
Intranasal delivery of IFN-ɛ can generate elevated effector KdGag197−205-specific T-cell responses in the mucosae
The above results indicate that when IFN-ɛ is delivered IN it can affect the cell milieu at the mucosae, and potentiate the generation of VV-specific CTL. In previous studies we have shown that rFPV is an excellent mucosal (IN or intrarectal) delivery vector.26, 30, 37 Therefore, we next evaluated the adjuvant activity of IFN-ɛ on the encoded vaccine antigen HIV gag/pol, using a recombinant FPV and VV prime-boost strategy (FPV-HIV-IFN-ɛ/VV-HIV-IFN-ɛ). In this study, we mainly investigated whether co-expression of IFN-ɛ could act as a trigger to generate enhanced mucosal T-cell immunity specifically in gut. Mice were primed IN or IM with FPV-HIV or FPV-HIV-IFN-ɛ and boosted IM with VV-HIV or VV-HIV-IFN-ɛ, respectively, and KdGag197−205-specific CD8+ T-cell immune response evaluated in the spleen, lung, PP, and genito-rectal nodes (GNs) 2 weeks following the booster immunization. Results indicate that IN/IM prime-boost vaccination with IFN-ɛ co-expression generated significantly enhanced KdGag197−205-specific T cells in the spleen (*P=0.033), PP, and GN, but not in the lung, compared with the control vaccination (Figure 4a). Conversely, when IFN-ɛ was used as a systemic adjuvant (IM/IM), a decrease in KdGag197−205-specific T-cell numbers was observed in all mucosal compartments compared with the control vaccination (Figure 4b). However, independent of IFN-ɛ co-expression overall, the IM/IM immunization strategy induced greater T-cell responses in PP compared with the IN/IM strategy (Figure 4a and b). Therefore, we assessed the expression frequency of gut-specific homing markers α4β7 and CCR9 on these KdGag197−205-specific T cells to evaluate whether following IM/IM delivery the T cells could transiently migrated from systemic compartment to the mucosal compartment. Homing marker analysis clearly indicated that the control vaccine delivered IN/IM generated elevated gut HIV-specific T cells compared with IM/IM strategy (Figure 5c). However, when IFN-ɛ was used as an adjuvant, elevated numbers of HIV-specific CD8+ T cells expressing homing markers α4β7 (60–66%) and CCR9 (68–73%) were induced in gut independent of the route of vaccine delivery (Figure 5a–c). Collectively, our comparative studies indicate that the gut-specific homing was associated with IFN-ɛ, but not IFN-α4 or IFN-β (Figure 3a–g).

HIV-specific CD8+ T effector cell function in systemic and mucosal compartments following rFPV/rVV prime-boost immunization. BALB/c mice (n=4–5 per group) were immunized with 1 × 107 PFU of FPV-HIV/VV-HIV or FPV-HIV-IFN-ɛ/VV-HIV-IFN-ɛ using (a) combined mucosal systemic (IN/IM) or (b) pure systemic (IM/IM) immunization. Two weeks post boost, the percentage of KdGag197−205-specific CD8+ T cells were evaluated in the spleen, lung, genito-rectal nodes, and Peyer's patch using tetramer staining. Spleen data represent average and ±s.d. of five mice (IN/IM, *P=0.033) and due to small sample size mucosal data represent pooled values, spleen error bars represent ±s.d. All experiments were repeated at least three times. HA, hemagglutinin; IFN, interferon; IM, intramuscular; IN, intranasal; PFU, plaque-forming unit; rFPV, recombinant fowlpox; rVV, recombinant vaccinia virus; VV, vaccinia virus.

Gut-specific homing marker expression on KdGag197−205-specific CD8+ T cell in Peyer's patch. BALB/c mice (n=6 per group) were immunized with 1 × 107 PFU of FPV-HIV/VV-HIV or FPV-HIV-IFN-ɛ/VV-HIV-IFN-ɛ using (a) combined mucosal-systemic (IN/IM) and/or (b) pure systemic (IM/IM) immunization. Two weeks post boost, the percentage of KdGag197−205-specific CD8+ T cells that expressed gut-specific homing markers (α4β7 and CCR9) were analyzed. (c) Comparison of the frequency of homing marker expression on KdGag197−205-specific CD8+ T cells following IN/IM and IM/IM strategies, and percentage was calculated as indicated in Methods. All fluorescence-activated cell sorting plots, the upper right quadrants (R3) represent gates R1+R2 (similar to Figure 3). Data represent pooled values, and experiments were repeated two times. HA, hemagglutinin; IFN, interferon; IM, intramuscular; IN, intranasal; PFU, plaque-forming unit; FPV, fowlpox; VV, vaccinia virus.
IFN-ɛ used as an adjuvant in IN/IM immunization can enhance IFN-γ+CD8+ T cells but not avidity
Studies were then performed to evaluate whether IFN-ɛ used as an IN adjuvant could also enhance mucosal CTL cytokine expression or alter CTL avidity. Mice were primed IN or IM with FPV-HIV or FPV-HIV-IFN-ɛ and boosted IM with VV-HIV or VV-HIV-IFN-ɛ, and IFN-γ+CD8+ T-cell responses were evaluated by intracellular cytokine staining in spleen, lung, PP, and GN 2 weeks post booster immunization. Both vaccination strategies showed no difference in IFN-γ-producing T-cell numbers in spleen in the presence or absence of IFN-ɛ (Figure 6a and b). In contrast, IN FPV-HIV-IFN-ɛ/IM VV-HIV-IFN-ɛ-immunized group showed elevated IFN-γ+CD8+ T cells in PP and GN (Figure 6a), also three times higher interleukin (IL)-2+CD8+ T cells were observed in GN (Figure 6c). However, following IM/IM immunization, a reduction in the numbers of IFN-γ-producing CTL were observed in all mucosal compartments tested (Figure 6b). In previous studies we have found that IN/IM prime-boost immunization can generate high-avidity CTL.29 Hence, to evaluate whether IFN-ɛ had any effect on T-cell avidity both in systemic and/or mucosal compartments, CTL avidity was measured in the spleen (Figure 6d) and lung (data not shown) using tetramer dissociation assay, as described in methods. Results showed that, expression of IFN-ɛ did not impact on the KdGag-specific T-cell avidity.

Magnitude and avidity of HIV-specific effector T cells in systemic and mucosal compartments following rFPV/rVV (IN/IM and IM/IM) prime-boost immunization. BALB/c mice (n=4–5) were (a) primed IN with 1 × 107 PFU FPV-HIV or FPV-HIV-IFN-ɛ and boosted IM with 1 × 107 PFU VV-HIV or VV-HIV- IFN-ɛ; or (b) primed IM with 1 × 107 PFU FPV-HIV or FPV-HIV-IFN-ɛ and boosted IM with 1 × 107 PFU VV-HIV or VV-HIV-IFN-ɛ. IFN-γ production was assessed by intracellular cytokine staining following H-2Kd AMQMLKETI Gag peptide stimulation. (c) Following IN/IM immunization, the number of IL-2-producing CD8+ T cells were evaluated by ELISpot. (d) Avidity of KdGag197−205-specific CD8+ T splenocytes measured by tetramer dissociation assay. In all graphs (a, b, and c), unstimulated cells were used as background controls and were subtracted from each sample before plotting the data. Spleen data represent average and ±s.d. of five mice, and due to small sample size mucosal data represent pooled values. All experiments were repeated at least three times. HA, hemagglutinin; IFN, interferon; IL, interleukin; IM, intramuscular; IN, intranasal; PFU, plaque-forming unit; rFPV, recombinant fowlpox; rVV, recombinant vaccinia virus; SFU, spot-forming unit; VV, vaccinia virus.
Co-expression of IFN-ɛ does not generate enhanced memory CTL response
It is well established that when designing effective vaccines, long-lasting memory T-cell responses are critically important. As, in the current studies, the co-expression of IFN-ɛ induced elevated mucosal effector T-cell responses following IN/IM prime-boost immunization, the memory HIV-specific T-cell responses were also evaluated at 8 weeks following the booster immunization. However, results show that IFN-ɛ does not potentiate a sustained mucosal or systemic KdGag197−205-specific CTL response (Figure 7a and b). This is also consistent with our immunobiology studies, in which no VV-specific responses were detected at 28 days (Figure 2b, d right, and Figure 2e right).

HIV-specific memory T-cell responses in systemic and mucosal compartments following IN/IM prime-boost immunization. BALB/c mice (n=5) were primed IN with 1 × 107 PFU FPV-HIV or FPV-HIV-IFN-ɛ and boosted IM with 1 × 107 PFU VV-HIV or VV-HIV-IFN-ɛ. Mice were killed 8 weeks post booster immunization, and (a) the percentage of KdGag197−205-specific memory CD8+ T cell was measured by tetramer staining in the spleen, lung, Peyer's patch, and genito-rectal nodes following 16 h stimulation with H-2Kd-binding AMQMLKETI Gag peptide, (b) the HIV-specific IFN-γ-producing CD8+ T-cell responses were measured by intracellular cytokine staining. All above mucosal data represent pooled values, with the exception of spleen data. Experiments were repeated at least three times. HA, hemagglutinin; IFN, interferon; IM, intramuscular; IN, intranasal; PFU, plaque-forming unit; FPV, fowlpox; VV, vaccinia virus.
Discussion
IFN-ɛ is found in the brain, lung, intestine, and reproductive tissues, and molecular and protein structure studies have found IFN-ɛ to be present both in mice and humans.13, 15, 16, 17 Peng et al.15 were the first to evaluate antiviral activity of IFN-ɛ in vitro and they found that IFN-ɛ elicited lower antiviral activity to that of IFN-α. In our previous studies when VV expressing IFN-α4, IFN-β, and IFN-ɛ were injected systemically (intravenous or IP) and vaccinia viral titers were evaluated 3 days PI in ovaries, magnitude of the antiviral activity across the type I IFN family were in the order of IFN-β > IFN-α4 > IFN-ɛ.18 The unique constitutive expression of IFN-ɛ in the lung,13, 14, 18, 19 reproductive tract,17 and intestinal14, 15 mucosae made us postulate that unlike other type I IFNs, IFN-ɛ may have a role in mucosal immunity.19 Therefore, by delivering IFN-ɛ intranasally we then investigated whether IFN-ɛ could induce any protective mucosal immunity. Following IN VV-HIV-IFN-ɛ infection, no difference in VV-specific IFN-γ+CD8+ T-cell numbers were observed in the spleen; however, larger spot-forming units were detected in the VV-HIV-IFN-ɛ group compared with the VV-HIV control. IFN-γ enzyme-linked immunosorbent assay confirmed that although there were no differences in the total numbers of the IFN-γ-expressing T cells, VV-HIV-IFN-ɛ induced increased IFN-γ levels in the spleen.
In our previous studies we also found elevated IFN-ɛ mRNA levels in an allergic BALB/c lung model compared with control BALB/c lung, and the levels were found to be IFN-α4>IFN-β>IFN-ɛ.19 Interestingly, in the current immunobiology studies IN VV-HIV-IFN-ɛ delivery induced greatly elevated CD8+CD107a+IFN-γ+ T-cell responses in the lung compared with VV-HA-IFN-α4 and VV-HA-IFN-β infections. Following IN delivery when the lung CD8 responses were further evaluated, expression of the early activation marker CD69 and slow onset activation marker CD103 on the CD8+ T cells were found to be upregulated only in the VV-HIV-IFN-ɛ group, but not in VV-HA-IFN-α4 or VV-HA-IFN-β groups. Several other studies have shown that CD69+CD103+CD8+ T cells can trigger lung-specific CTL recruitment and promote better surveillance of lung,38, 39 which is consistent with our 6 days PI H&E lung staining data, where the pathology and inflammation observed were found to be naive ≤VV-HIV-IFN-ɛ<VV-HA-IFN-β<VV-HA-IFN-α4<control infection. This correlates with rapid VV clearance in lung at 6 days PI, following IN VV-HIV-IFN-ɛ infection compared with the VV-HIV control and also the very low viral titers (just above detection level) detected in the VV-HA-IFN-α4-infected group.
Furthermore, an elevated route-dependent CD8+CD4+ DP T-cell subset was observed in LLN, following IN VV-HIV-IFN-ɛ infection compared with the control VV-HIV. This LLN DP T-cell population peaked at 7 days PI and then declined to similar levels to that observed in the control infection at day 28. Interestingly, this enhanced LLN-specific CD8+CD4+ DP T-cell population was also detected in VV-HIV-IFN-α4 group, but the DP T-cell numbers were found to be lower than that of IFN-ɛ group. In contrast, no enhanced CD8+CD4+ DP T cells were found following VV-HIV-IFN-β infection. Induction of similar antigen-specific, functional/cytotoxic CD8+CD4+ T-cell subset has been observed and reported following some virus and parasitic infections.35, 40 DP T cells that secrete cytokines have also been reported in peripheral blood and intestinal sites, especially lamina propria at early stage of viral infection.32, 41, 42 In viral infections, such as Simian Immunodeficiency Virus and Epstein–Barr virus, circulating CD8+CD4+ effector T cells composing up to 20% of total T-cell number have been reported in macaques.34, 43 In autoimmunity and chronic inflammatory disorders, similar functional CD8+CD4+ T cells have also been detected.44 In our studies, only the mice that received VV-HIV-IFN-ɛ elicited higher vaccinia-specific IFN-γ+CD8+CD4+ T cells in LLN at 7 days PI, and they were found to be CD3hi and also cytotoxic (CD107ahi).31 Similarly a IFN-γ+CD8+CD4+ T-cell population was also observed in lung. These observations clearly suggested that IFN-ɛ-induced functional and cytotoxic DP T cells are very similar to that reported by other groups.33, 35 Interestingly, VV-HA-IFN-α4 and VV-HA-IFN-β induced CD3lo DP T cells, which indicated that these DP T cells were most likely distinctly different to that observed following VV-HIV-IFN-ɛ infection. The above IFN-α4- and IFN-β-induced DP T-cell population was very similar to the unusual T-cell-like lymphocyte population (CD8+CD4+CD3−), reported by Penttila et al.45 in the lung during the early stage of IN Chlamydia pneumniae and influenza A infections, that did not produce IFN-γ. They also suggested that lung-derived CD8+CD4+CD3− cells have a distinct function to that of lamina propria–derived CD8+CD4+CD3+ T cells32, 41, 42 and that they may secrete different inflammatory cytokines.45
Interestingly, the IFN-ɛ-induced DP T-cell population was CD62Llo and CCR7hi, whereas VV-HA-IFN-α4 and VV-HA-IFN-β infection induced a DP T-cell population that was CD62Lhi, but did not express CCR7. In general, CCR7 is known to be an important chemokine receptor that mediates T-cell migration to secondary lymphoid organs.46 However, whether CCR7 has any role in DP T-cell migration to specific tissues or lymphoid organs is not yet known. In our studies we have clearly observed a difference in the CD8+CD4+ T-cell populations induced by IFN-ɛ compared with IFN-α4 or IFN-β. Our data indicate that following IFN-ɛ infection the heightened CCR7 expression together with increased IFN-γ production by CD8+CD4+ T cells may contribute to the enhanced lymphocyte recruitment and the reduced inflammation in the lung. We believe this warrents further investigation.
In summary, above data indicate that IN delivery of IFN-ɛ is able to induce an antiviral state and a functional/cytotoxic CD8+CD4+ T-cell population in the lung mucosae. The H&E staining clearly indicates that these factors most likely contributed to the enhanced lung-specific effector T-cell response and viral clearance. Hence, our findings substantiate previous reports that IFN-ɛ has a unique role in lung mucosae, compared with the other type I IFNs.13, 14, 18 This potentially opens the doors to the development of new, IFN-ɛ-based therapies for the control of infections in the lung such as pulmonary chlamydia, tuberculosis, or chronic influenza.
We have also found that IN VV-HIV-IFN-ɛ infection could induce not only elevated VV-specific (KdA52R-specific) CD8+ T cells in PP (1.29%), but these were also gut-specific, as the expression frequency of α4β7 and CCR9 were much greater compared with the control. This gut-specific homing was unique to IFN-ɛ, as the other two type I IFNs tested did not show any such responses. As in our immunobiology studies, IFN-ɛ induced effective VV clearance in the lung mucosae and also excellent VV-specific CD8+ T cells in gut, we then assessed whether IFN-ɛ could be used as an adjuvant to induce mucosal HIV-specific CD8+ T-cell immunity, especially in the gut mucosae.
In our previous studies, Day et al.18 have shown that, when IFN-ɛ was delivered in the prime but not in the booster vaccination (IM VV-HIV-IFN-ɛ/IM FPV-HIV), no enhanced HIV Gag-specific T-cell responses or IFN-γ-producing CD8+ T cells were observed in the spleen. Our consequent studies have now clearly established that only IN rFPV/IM rVV delivery can generate excellent systemic and mucosal CTL immunity, but rVV prime followed by rFPV booster vaccination does not induce effective antigen-specific T-cell immunity26, 30 (C Ranasinghe et al., in preparation). Therefore, in this study we have delivered IN FPV-HIV-IFN-ɛ followed by IM VV-HIV-IFN-ɛ, where IFN-ɛ was also given both in the prime and the booster vaccination and have demonstrated that it can induce elevated KdGag-specific effector CD8+ T-cell responses in the spleen, PP, and GN. No significant responses were observed in the lung 14 days post booster immunization, which is consistent with the immunobiology studies, where elevated lung responses were only observed at 7 days not 28 days PI. In contrast, the IM/IM delivery of IFN-ɛ elicited a reduction of KdGag-specific T cells in all the mucosal compartments compared with the control vaccination.
Interestingly, we also found that following IM/IM delivery in PP both control and IFN-ɛ vaccine strategies were able to activate a greater number of HIV-specific T cells compared with IN/IM strategy. One point of debate in the literature is whether systemic vaccine delivery has the capacity to generate long-lasting mucosal immunity.47, 48 Kaufman et al.47 have indicated that antigen-specific CD8+ T cells could traffic to mucosal surface following IM delivery. Our results clearly indicated that following IM delivery, even though elevated KdGag-specific T cells were observed in the mucosae following control vaccination, these CD8+ T cells most likely were temporarily migrating from the blood compartment, as these did not express α4β7 and CCR9 (gut-specific homing markers). However, animals that received IFN-ɛ following IN/IM or IM/IM vaccination were able to induce KdGag-specific T cells with higher levels of α4β7 and CCR9, compared with the respective controls. Thus, our data clearly indicate that the presence of IFN-ɛ in the cell milieu can induce elevated numbers of antigen-specific T cells that home to gut mucosae. We believe the mechanism governing this induction warrants further investigation. Our observation also indicates that when evaluating vaccine-specific mucosal immunity analysis of mucosal homing markers together with tetramer staining could be of great importance.
In conclusion, our studies clearly demonstrate that unlike IFN-α or IFN-β, novel IFN-ɛ has a unique antiviral role in the mucosae. In this study we have shown that irrespective of the route of vaccine delivery IFN-ɛ can induce good gut-specific CD8+ T-cell immunity. As the gut is the primary site of HIV virus replication and CD4+ T-cell depletion,22 we believe a compound that can prevent HIV viral replication or clearance in the gut and/or genito-rectal mucosae could be of great importance in controlling HIV-1 infection. Both our immunobiology and adjuvant studies indicate that the IFN-ɛ induces a localized mucosal immune response that is short-lived. Therefore, we believe that, more than a vaccine adjuvant its utility as a topical microbicide to prevent or reduce mucosally related infections, such as tuberculosis, HIV-1 or other sexually transmitted diseases at the first line of defense, would be an area of future investigation.
Methods
Animal source. Female pathogen-free 7- to 9-week-old BALB/c (H-2d) mice were obtained from the Animal Breeding Facility, The Australian National University (ANU). All mice were maintained and used in accordance with animal ethics and experimentation guidelines of ANU.
Structure of recombinant poxvirus vaccine. Recombinant VV and rFPV virus used in the study were VV-HIV, VV-HIV-IFN-ɛ, VV-HA, VV-HA-IFN-α4, and VV-HA-IFN-β constructed, as described by Day et al.,18 FPV-HIV (FPV-086) as described by Coupar et al.50, and FPV-HIV-IFN-ɛ was constructed in our laboratory using pAF09, as described by Heine and Boyle.51
Infection and immunizations. Mice (n=4–5) were infected intranasally (IN), intramuscularly (IM), and intraperitoneally (IP) with 5 × 106 PFU VV-HIV or VV-HIV-IFN-ɛ for 7 days. Ten to twenty microliter virus per mouse IN, 50 μl per quadriceps IM, or 100 μl per mouse IP were delivered. Similarly for comparative immunobiology studies VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4 were used, note that these were the same constructs that were used in Day et al.18 For prime-boost immunization studies, mice were given 1 × 107 PFU of FPV-HIV or FPV-HIV-IFN-ɛ followed by 1 × 107 PFU of VV-HIV or VV-HIV-IFN-ɛ 2 weeks later, using either combined mucosal systemic IN/IM or pure systemic IM/IM immunization routes, and the effector and memory T-cell responses were measured 2 or 8 weeks post booster immunization, respectively. Mice were anesthetized either under mild methoxyfluorane for IN or avertin for IM delivery. All viruses were diluted in sterile phosphate-buffered saline (PBS) and then sonicated, to obtain homogeneous viral suspensions before delivery.
Lung virus titration. Mice were IN infected with 5 × 106 PFU of VV-HIV, VV-HIV-IFN-ɛ for 3 and 6 days, similarly VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4 for 6 days. Whole lungs were collected and cells were disrupted by dissociation through cell strainers. Recovered cells were then homogenized and titrated as follows. Cells (143B) were seeded onto six-well plates (Linbro Chemical, New Haven, CT) and incubated overnight until 90% confluency was reached. Homogenized lung cells were digested with an equal volume of trypsin at 37 °C for 30 min. Tenfold titrations of the virus were done in sterile PBS, and media was removed from wells, the dilutions were plated onto 143B cells and incubated at 37 °C with 5% CO2 for up to 1 h. Following incubation, 2 ml of modified Eagle's medium (supplemented with 5% fetal calf serum, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 30 μg ml−1 penicillin) was added back to each well and cells were incubated for further 48 h. Cells were then stained with 0.1% crystal violet in 20% ethanol to visualize plaques and enumerate virus titers.
H&E staining. Mice were IN infected with 5 × 106 PFU of VV-HIV, VV-HIV-IFN-ɛ, VV-HA, VV-HA-IFN-β, and VV-HA-IFN-α4 at 7 days PI, whole lungs were collected in 10% Neutrac-buffered formalin. H&E staining was performed at The John Curtin School of Medical Research (JCSMR)-MCRF (Microscopy and Cytometry Research Facility). Uninfected naive mice were used as controls. Images were analyzed by Olympus Microscope at × 4 and × 60 magnification, respectively.
Mucosal and systemic lymphocytes preparation. Immunized mice were killed by CO2 asphyxiation, and spleen, GN (or iliac lymph nodes), LLN (or mediastinal lymph nodes), lung, and PP were harvested and single-cell suspensions prepared as described previously.26 Briefly, spleen cells were dissociated through a nylon cell strainer and then resuspended in red blood cell lysis buffer (0.15 mM NH4Cl, 10.0 mM KHCO3, 0.1 mM Na2EDTA) at room temperature for 10 min to lyse the red blood cells. The cells were washed, and suspended in complete RPMI (RPMI supplemented with 7% fetal calf serum, 1 mM L-glutamine, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 30 μg ml−1 penicillin, 50 μg ml−1 streptomycin, 2 mM sodium pyruvate (Gibco-Invitrogen, Auckland, NZ), and 50 μM 2-mercaptoethanol (Sigma, St Louis, MO), and kept at 4 °C until use. Similarly, mucosal lymphocytes (GN, LLN and PP) were prepared as for spleen without the red cell lysis step.
Lung samples were first cut into small pieces and digested in 1 ml of complete RPMI buffer containing 2 mg ml−1 collagenase (Sigma), 2.4 mg ml−1 dispase (Gibco), and 5 Units ml−1 DNAse (Calbiochem, Long Beach, CA) at 37 °C for 1 h with gentle vortexing every 10–15 min. The digestion solution was then filtered through a cell strainer, and the pieces were further mashed and rinsed with complete RPMI. Following centrifugation, erythrocytes were lysed as above. The sample was further filtered through sterile gauze to remove connective tissue and debris, centrifuged and final single-cell suspensions were prepared. Lung cell suspensions were kept at 37 °C for minimum of 12 h following digestion to allow for recovery prior to performing assays, as we have observed that digestion can downregulate expression of some surface markers such as CD4 and CD8.
IFN-γ and IL-2 ELISpot assay. The IFN-γ or IL-2 ELISpot assay was used to measure VV or HIV-specific CD8+ T-cell responses as described in Ranasinghe et al.26 Briefly, 96-MultiScreen HTS filter plates (Millipore, Cork, Ireland) were precoated with 5 μg ml−1 purified rat anti-mouse IFN-γ or IL-2 capture antibody (BioLegend, San Diego, CA) in PBS. Splenocytes and lung lymphocytes (2 × 105 cells per well) were stimulated with 5 μg ml−1 immunodominant H-2Kd CD8+ T-cell epitope HIV-Gag197−205 (AMQMLKETI) or VV-A52R75−83 (KYGRLFNEI), respectively (synthesized at the Bio-Molecular Resource Facility, JCSMR, ANU), 5 μg ml−1 ConA (Sigma-Aldrich, Rockville, MD), or were left unstimulated. After 24 h or 12 h incubation for IFN-γ or IL-2, cells were washed and incubated for 2 h at room temperature with 1:1,000 biotinylated rat anti-mouse IFN-γ antibody (BD Pharmingen, San Diego, CA) diluted in 1% bovine serum albumin/PBS, and further incubated for 1.5 h with 1:1,000 streptavidin alkaline phosphatase (Amersham Life Science, Buckinghamshire, UK). Spot-forming units were developed using BCIP/NBT alkaline phosphatase substrate (Moss Inc., Pasadena, MD) and counted using ELISpot Bioreader-4000 PRO-X (BIOSYS, GmbH, Germany). Results are representative of spot-forming units per 106 CD8+ T cells minus the background count. Data are representative of at least three experiments.
Surface marker detection. To detect surface marker expression, 2 × 106 lymphocytes were incubated with directly labeled antibodies diluted in FACS buffer (PBS+2% fetal calf serum), for 30 min on ice in the dark. The antibodies used in the study were CD8-APC, CD4-PerCP (BD Pharmingen), CD69-PE (BD Pharmingen), and CD103-FITC (eBioscience, San Diego, CA), as well as CD8-APC, CD4-PerCP together with CD3-PE, or CD8-APC, CD4-FITC together CCR7-PE, CD62L-PerCPcy5.5 (BioLegend or BD Pharmingen). The stained cells were washed and fixed with 0.5% paraformaldehyde buffer prior to analysis. A total of 100,000 gated events of each sample were acquired by four-colour FACS Calibur flow cytometer (Becton-Dickinson, San Diego, CA), and analyzed using Cell Quest Pro software (Becton-Dickinson).
Intracellular cytokine analysis. Intracellular cytokine staining was used to assess IFN-γ or granzyme-producing VV or HIV-specific CD8T cells, as described in Ranasinghe et al.26, 29 Lymphocytes (2 × 106) per well were seeded in a 96-U-bottom plate and stimulated with the VV-KdA5275−83 (KYGRLFNEI) or HIV-KdGag197−205 (AMQMLKETI) peptide at 37 °C for 16 h, and further incubated with Brefeldin A (eBioscience) for 4 h. Cells were surface stained with CD8-APC (BD Pharmingen) and/or CD4-PerCP for 30 min on ice, then fixed and permeabilized prior to IFN-γ-FITC (eBioscience), and/or CD107a-PE (BD Pharmingen)/or granzyme B-APC (eBioscience) intracellular staining for 30 min at 4 °C. A total of 100,000 gated events per sample were collected using FACS Calibur flow cytometer (Becton-Dickinson), and results were analyzed using Cell Quest Pro software. Unstimulated background was subtracted from the data.
Tetramer staining. Lymphocytes (2 × 106) were incubated with CD8-FITC or PerCPcy5.5 (BD Pharmingen, San Diego, CA) and allophycocyanin (APC)-conjugated HIV-specific KdGag197−205 or VV-specific KdA52R75−83 tetramer (synthesized at the Bio-Molecular Resource Facility, JCSMR) at room temperature for 45 min in the dark. When homing marker analysis was performed, the cells were also staining with α4β7-PE and/or CCR9-FITC. Cells were washed, fixed with 0.5% paraformaldehyde and 200,000 total gated events were acquired for each of the sample by FACsCalibur flow cytometer (Becton-Dickinson), and results analyzed using Cell Quest Pro software. Note that Figures 3g and 5c the bar graphs, homing marker expression was calculated out of the total CD8+KdGag+ or CD8+KdA52R+ T-cell population, respectively, as follows: (example, IN FPV-HIV/IM VV-HIV immunization group the KdGag197−205 tetramer-specific T-cell population=0.28% (Figure 5a). If this value was considered as 100%, the frequency of α4β7 or CCR9 in this population was calculated as 100/0.28 × 0.16=57% or 100/0.28 × 0.14=50%, respectively (Figure 5c).
Tetramer dissociation assay. Tetramer dissociation was used to measure the avidity of T cells, as described in Ranasinghe et al.29 Briefly, 2 × 106 cells were stained with FITC-anti CD8α and APC-KdGag197−205 tetramer as described above, and an anti-H-2Kd competitive binding antibody (BD Pharmingen) was added. At 10, 30, and 60 min, cells were transferred into cold FACs buffer (PBS+2% FCS) to stop the reaction, and then cells were washed, and resuspended in 100 μl of 0.5% paraformaldehyde buffer for analysis as above.
IFN-γ enzyme-linked immunosorbent assay. Splenocytes (2 × 106) were cultured in 200 μl of complete RPMI in the presence of KdA52R75−83-specific peptide at 37 °C for 16 h, or were left unstimulated. Supernatants were collected and tested for IFN-γ production using the mouse IFN-γ ELISA Kit II (BD Bioscience) as per manufacturer's instructions. Absorbance was read using a Thermo Max Microplate Reader at 450 nm, and standard curves used to calculate the IFN-γ levels.
Statistical analysis. Standard deviation (s.d.) was calculated and P-values determined using two-tailed, two sample equal variance or unequal variance Student's t-test. All the experiments have been repeated at least three times with 4–6 mice per group. The P-value of <0.05 was considered significant. When GNs, PP, LLN, or lung were used, the data represent a pooled values of 4–6 mice. Owing to the small mucosal sample size, it was often not possible to process the tissues and obtain enough cells to perform assays in individual mice. Where possible we have calculated statistics using average values of three separate experiments.
References
Huber, GGAC. The biology of interferon actions. Blut. 56, 7 (1988).
Isaacs, A. & Lindenmann, J. Virus interference. I. The interferon. Proc. R Soc. Lond. B Biol. Sci. 147, 258–267 (1957).
Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809 (2001)Table of contents.
Hertzog, P.J., Hwang, S.Y. & Kola, I. Role of interferons in the regulation of cell proliferation, differentiation, and development. Mol. Reprod. Dev. 39, 226–232 (1994).
Mazzella, G. et al. Long-term results with interferon therapy in chronic type B hepatitis: a prospective randomized trial. Am. J. Gastroenterol. 94, 2246–2250 (1999).
Roffi, L. et al. Breakthrough during recombinant interferon alfa therapy in patients with chronic hepatitis C virus infection: prevalence, etiology, and management. Hepatology 21, 645–649 (1995).
Borden, E.C. et al. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990 (2007).
Pestka, S., Krause, C.D. & Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32 (2004).
Pestka, S., Langer, J.A., Zoon, K.C. & Samuel, C.E. Interferons and their actions. Annu. Rev. Biochem. 56, 727–777 (1987).
Kotenko, S.V. et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77 (2003).
Dornhoff, H., Safrit, J.T. & Neurath, M.F. Potential role of interferon-lambda in the treatment of inflammation and cancer: an update International Journal of Interferon. Cytokine Mediator Res. 3, 51–57 (2011).
Uze, G., Schreiber, G., Piehler, J. & Pellegrini, S. The receptor of the type I interferon family. Curr. Top Microbiol. Immunol. 316, 71–95 (2007).
Hardy, M.P., Owczarek, C.M., Jermiin, L.S., Ejdeback, M. & Hertzog, P.J. Characterization of the type I interferon locus and identification of novel genes. Genomics 84, 331–345 (2004).
Chen, J., Godowski, P., Wood, W.I. & Zhang, D. Vol. 6569420. (U.S. patent, ed.) 13 Genentech Inc. ( South San Francisco, CA, USA, 2003 ).
Peng, F.W. et al. Purification of recombinant human interferon-epsilon and oligonucleotide microarray analysis of interferon-epsilon-regulated genes. Protein Expr. Purif. 53, 356–362 (2007).
Matsumiya, T., Prescott, S.M. & Stafforini, D.M. IFN-epsilon mediates TNF-alpha-induced STAT1 phosphorylation and induction of retinoic acid-inducible gene-I in human cervical cancer cells. J. Immunol. 179, 4542–4549 (2007).
Sharkey, D.J., Macpherson, A.M., Tremellen, K.P. & Robertson, S.A. Seminal plasma differentially regulates inflammatory cytokine gene expression in human cervical and vaginal epithelial cells. Mol. Hum. Reprod. 13, 491–501 (2007).
Day, S.L., Ramshaw, I.A., Ramsay, A.J. & Ranasinghe, C. Differential effects of the type I interferons alpha4, beta, and epsilon on antiviral activity and vaccine efficacy. J. Immunol. 180, 7158–7166 (2008).
Day, S.L. In: John Curtin School of Medical Research Vol. PhD 123 ( Australia National University, Canberra, 2008 ).
Ogra, P.L., Faden, H. & Welliver, R.C. Vaccination strategies for mucosal immune responses. Clin. Microbiol. Rev. 14, 430–445 (2001).
Wijesundara, D.K., Jackson, R.J., Ramshaw, I.A. & Ranasinghe, C. Human immunodeficiency virus-1 vaccine design: where do we go now? Immunol. Cell Biol. 89, 367–374.
Veazey, R.S. et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280, 427–431 (1998).
Dale, C.J. et al. Prime-boost strategies in DNA vaccines. Methods Mol. Med. 127, 171–197 (2006).
Harrison, J.M. et al. 4-1BBL coexpression enhances HIV-specific CD8T cell memory in a poxvirus prime-boost vaccine. Vaccine 24, 6867–6874 (2006).
Hodge, J.W., McLaughlin, J.P., Kantor, J.A. & Schlom, J. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T-cell immunity and antitumor responses. Vaccine 15, 759–768 (1997).
Ranasinghe, C. et al. Evaluation of fowlpox-vaccinia virus prime-boost vaccine strategies for high-level mucosal and systemic immunity against HIV-1. Vaccine 24, 5881–5895 (2006).
Ranasinghe, C. & Ramshaw, I.A. Genetic heterologous prime-boost vaccination strategies for improved systemic and mucosal immunity. Expert Rev. Vaccines 8, 1171–1181 (2009).
Ranasinghe, C. & Ramshaw, I.A. Immunisation route-dependent expression of IL-4/IL-13 can modulate HIV-specific CD8(+) CTL avidity. Eur. J. Immunol. 39, 1819–1830 (2009).
Ranasinghe, C. et al. Mucosal HIV-1 pox virus prime-boost immunization induces high-avidity CD8+ T cells with regime-dependent cytokine/granzyme B profiles. J. Immunol. 178, 2370–2379 (2007).
Ranasinghe, C. et al. A comparative analysis of HIV-specific mucosal/systemic T cell immunity and avidity following rDNA/rFPV and poxvirus-poxvirus prime boost immunisations. Vaccine 29, 3008–3020 (2011).
Aktas, E., Kucuksezer, U., Bilgic, S., Erten, G. & Deniz, G. Relationship between CD107a expression and cytotoxic activity. Cell Immunol. 254, 149–154 (2009).
Nascimbeni, M., Shin, E.C., Chiriboga, L., Kleiner, D.E. & Rehermann, B. Peripheral CD4(+)CD8(+) T cells are differentiated effector memory cells with antiviral functions. Blood 104, 478–486 (2004).
Pahar, B., Lackner, A.A. & Veazey, R.S. Intestinal double-positive CD4+CD8+ T cells are highly activated memory cells with an increased capacity to produce cytokines. Eur. J. Immunol. 36, 583–592 (2006).
Weiss, L. et al. Persistent expansion, in a human immunodeficiency virus-infected person, of V beta-restricted CD4+CD8+ T lymphocytes that express cytotoxicity-associated molecules and are committed to produce interferon-gamma and tumor necrosis factor-alpha. J. Infect. Dis. 178, 1158–1162 (1998).
Giraldo, N.A. et al. Increased CD4+/CD8+ double-positive T cells in chronic Chagasic patients. PLoS Negl. Trop. Dis. 5, e1294 (2011).
Xie, D. et al. Peripheral CD4+CD8+cells are the activated T cells expressed granzyme B (GrB), Foxp3, interleukin 17 (IL-17), at higher levels in Th1/Th2 cytokines. Cell Immunol. 259, 157–164 (2009).
Kent, S.J. et al. Mucosally-administered human-simian immunodeficiency virus DNA and fowlpoxvirus-based recombinant vaccines reduce acute phase viral replication in macaques following vaginal challenge with CCR5-tropic SHIVSF162P3. Vaccine 23, 5009–5021 (2005).
Morgan, A.J. et al. CXCR6 identifies a putative population of retained human lung T cells characterised by co-expression of activation markers. Immunobiology 213, 599–608 (2008).
Lee, Y.T. et al. Environmental and antigen-receptor derived signals support sustained surveillance of the lungs by pathogen-specific CTL. J. Virol..
Howe, R. et al. Phenotypic and functional characterization of HIV-1-specific CD4+CD8+ double-positive T cells in early and chronic HIV-1 infection. J. Acquir. Immune Defic. Syndr. 50, 444–456 (2009).
Periwal, S.B. & Cebra, J.J. Respiratory mucosal immunization with reovirus serotype 1/L stimulates virus-specific humoral and cellular immune responses, including double-positive (CD4(+)/CD8(+)) T cells. J. Virol. 73, 7633–7640 (1999).
Zuckermann, F.A. & Husmann, R.J. Functional and phenotypic analysis of porcine peripheral blood CD4/CD8 double-positive T cells. Immunology 87, 500–512 (1996).
Ortolani, C., Forti, E., Radin, E., Cibin, R. & Cossarizza, A. Cytofluorimetric identification of two populations of double positive (CD4+,CD8+) T lymphocytes in human peripheral blood. Biochem. Biophys. Res. Commun. 191, 601–609 (1993).
Parel, Y. & Chizzolini, C. CD4+ CD8+ double positive (DP) T cells in health and disease. Autoimmun. Rev. 3, 215–220 (2004).
Penttila, J.M., Pyhala, R., Sarvas, M. & Rautonen, N. Expansion of a novel pulmonary CD3(−) CD4(+) CD8(+) cell population in mice during Chlamydia pneumoniae infection. Infect. Immun. 66, 3290–3294 (1998).
Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).
Kaufman, D.R. et al. Trafficking of antigen-specific CD8+ T lymphocytes to mucosal surfaces following intramuscular vaccination. J. Immunol. 181, 4188–4198 (2008).
Belyakov, IM & Ahlers, J.D. Comment on ‘trafficking of antigen-specific CD8+ T lymphocytes to mucosal surfaces following intramuscular vaccination’. J. Immunol. 182, 1779 (2009)author reply 1779-1780.
Fung, K. et al. Interferon epsilon regulates reproductive tract immunity to Chlamydia infection. J. Reprod. Immunol. (Poster Abstracts) 86, 1 (2010).
Coupar, B.E. et al. Fowlpox virus vaccines for HIV and SHIV clinical and pre-clinical trials. Vaccine 24, 1378–1388 (2006).
Heine, H.G. & Boyle, D.B. Infectious bursal disease virus structural protein VP2 expressed by a fowlpox virus recombinant confers protection against disease in chickens. Arch. Virol. 131, 277–292 (1993).
Acknowledgements
We thank the Bio-Molecular Resource Facility, at The John Curtin School of Medical Research (JCSMR), The Australian National University for synthesizing the HIV-specific KdGag197−205 and VV-specific KdA5275−83 peptides and tetramers. We also thank JCSMR MCRF, Anne Prins for assistance with H&E staining, and ABRF (animal facility) staff for their assistance, as well as Professor Ian A Ramshaw for some early discussions. We also acknowledge Lisa Pavlinovic, Jill Medveczky, and Annette Buchanan for their technical assistance with various aspects of the project. This work was supported by the Australian National Health and Medical Research Council Project Grant award 525431 (CR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Xi, Y., Day, S., Jackson, R. et al. Role of novel type I interferon epsilon in viral infection and mucosal immunity. Mucosal Immunol 5, 610–622 (2012). https://doi.org/10.1038/mi.2012.35
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2012.35
This article is cited by
-
Mucosal and systemic SIV-specific cytotoxic CD4+ T cell hierarchy in protection following intranasal/intramuscular recombinant pox-viral vaccination of pigtail macaques
Scientific Reports (2019)
-
Methyltransferase of a cell culture-adapted hepatitis E inhibits the MDA5 receptor signaling pathway
Journal of Microbiology (2019)
-
Vaccination route can significantly alter the innate lymphoid cell subsets: a feedback between IL-13 and IFN-γ
npj Vaccines (2018)
-
Differential regulation of IFNα, IFNβ and IFNε gene expression in human cervical epithelial cells
Cell & Bioscience (2017)
-
Blocking HIV-1 transmission in the female reproductive tract: from microbicide development to exploring local antiviral responses
Clinical & Translational Immunology (2015)
