
Abstract
Secretory IgA (SIgA) antibodies represent the first line of antigen-specific immune defense protecting the mucosal surfaces against environmental pathogens and antigens, and maintaining homeostasis with the commensal microbiota. The polymeric immunoglobulin receptor (pIgR) has the dual role of transporting locally produced dimeric IgA across mucosal epithelia, and serving as the precursor of secretory component, a glycoprotein that enhances the immune functions of SIgA. The complex regulation of pIgR expression and transcytosis by host and microbial factors is finely tuned to optimize the role of SIgA in mucosal immunity. Disruption of this regulatory network in disease states similar to inflammatory bowel disease can result in profound consequences for mucosal homeostasis and systemic sequelae. Future research into the function and regulation of pIgR and SIgA may offer new insights into the prevention and treatment of infectious and inflammatory diseases that originate at mucosal surfaces.
Similar content being viewed by others


Introduction
Secretory IgA (SIgA) is generated by cooperation between two distinct cell types in mucous membranes: plasma cells in the lamina propria that secrete dimeric IgA (dIgA) and epithelial cells that transport the IgA into the lumen. Transcytosis of dIgA through polarized intestinal epithelial cells (IECs) is mediated by the polymeric immunoglobulin receptor (pIgR), an epithelial glycoprotein that picks up its cargo on the basolateral surface and carries it by the process of transcytosis to the apical face. Endoproteolytic cleavage of pIgR near the plasma membrane releases the five extracellular immunoglobulin-like domains of the pIgR, also known as secretory component (SC), into the lumen together with its cargo, thus forming the SIgA complex. Because one molecule of pIgR is consumed with every round of SIgA transport, control of pIgR expression and its epithelial trafficking are critical for regulating the levels of SIgA in external secretions. Here we propose that cross talk between the commensal microbiota and IECs has a central role in this regulatory process.
Regulation of pIgR Expression by the Commensal Microbiota
SIgA antibodies in mucosal secretions contribute to immune homeostasis by limiting access of microbial and environmental antigens to the body proper, maintaining the integrity of the epithelial barrier, and shaping the composition of the commensal microbiota.1 These functions are particularly important in the intestinal tract, home to some 100 trillion bacteria, into which the average human adult secretes up to 3 g of SIgA per day. Accumulating evidence suggest that microbial-associated molecular patterns (MAMPs) stimulate expression of pIgR by IECs, as part of a homeostatic loop in which the microbiota enhances the production of SIgA, which in turn regulates the composition and function of the microbiota. It has long been appreciated that the microbiota has a critical role in development of the gut-associated immune system, including class switching to IgA and maturation of plasma cells.1 A direct role for gut bacteria in stimulating pIgR expression was first suggested by the finding that monocolonization of germ-free mice with the commensal bacterium Bacteroides thetaiotaomicron resulted in increased pIgR expression in IECs.2 More recently, using a model of reversible colonization of germ-free mice with a non-dividing mutant of Escherichia coli, Hapfelmeier et al.3 observed a long-lived SIgA response specifically recognizing the inducing bacteria. However, exposure of E. coli-colonized mice to other bacteria limited the duration of the SIgA response against the original colonizer, suggesting that the SIgA response adapts to the dominating members of the microbiota. We have compared the composition and compartmentalization of the gut microbiota in wild-type and pIgR knockout mice, which lack SIgA in gut secretions (Reikvam et al., unpublished). Interestingly, we found that the diversity of the bacterial community in the cecal lumen was reduced in pIgR knockout mice, supporting a role for SIgA in limiting overgrowth of selected species. In contrast, the community of bacteria adherent to the epithelial surface was more diverse in pIgR knockout mice than in wild-type mice, and these adherent bacteria caused enhanced expression of several epithelial antimicrobial peptides. These findings suggest that bacteria-specific SIgA anchored in the mucus layer restricts adhesion of bacteria to the epithelial surface and limits bacterial stimulation of host epithelial cells. Under homeostatic conditions, release of MAMPs by SIgA-bound bacteria in the mucus layer may stimulate epithelial cells to produce physiological levels of pIgR, thus enhancing dIgA transport and replenishing SIgA that is lost by mucosal sloughing. Under conditions of suboptimal SIgA production or penetration of the mucus layer by invasive species, direct bacterial stimulation of the epithelial surface could result in enhanced production of pro-inflammatory and antimicrobial products.
A mechanism through which MAMPs may stimulate pIgR expression was suggested by our finding that expression of pIgR was enhanced by stimulation of a human IEC line with double-stranded RNA or lipopolysaccharide, ligands for Toll-like receptors (TLRs) 3 and 4.4, 5 Similar upregulation of pIgR expression was observed when cells were stimulated with intact reovirus, an intestinal virus with a double-stranded RNA genome,6 or bacteria of the family Enterobacteriaceae, which express a form of lipopolysaccharide that stimulates TLR4.7 Although caution must be observed in the interpretation of experiments using IEC lines that may not have normal patterns of TLR expression, our recent in vivo studies in mice support the significance of these results. We found that expression of pIgR and mucosal secretion of SIgA were significantly reduced in mice genetically deficient in MyD88,7 an intracellular adaptor protein that transmits signals from TLR4 and other TLRs that respond to bacterial ligands, as well as the receptors for interleukin (IL)-1 and IL-18. We further demonstrated that pIgR expression is reduced in mice with a targeted deletion of MyD88 only in IECs (Frantz et al., unpublished), suggesting that MyD88-dependent signaling within epithelial cells is crucial for maintaining optimal pIgR expression. We did not observe marked abnormalities in the architecture of the intestinal epithelium in mice with IEC-targeted depletion of MyD88, as has been reported for mice genetically deficient in MyD88 in all cell types.8 However, the marked increase in numbers of bacteria in the mesenteric lymph nodes of mice with IEC-targeted depletion of MyD88 suggested that MyD88-dependent signaling in epithelial cells is crucial for maintenance of normal intestinal immunity and barrier function.
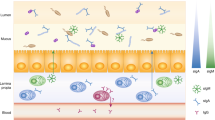
A proposed model of bacterial–epithelial cross talk in the regulation of pIgR expression and dIgA transport is illustrated in Figure 1. A dense layer of mucus, which increases in thickness from the proximal to the distal colon, serves to separate bacteria from direct contact with the epithelial surface.9, 10 Secretion of MAMPs by commensal bacteria stimulates TLR signaling and enhances pIgR expression, resulting in enhanced transcytosis of SIgA as well as free SC, which contributes to innate immune functions (see article by Mantis et al.11 in this issue). SIgA and SC have mucus-binding properties, which serve to trap bacteria in the mucus layer, stimulate biofilm formation, and limit access of bacteria to the epithelial surface.

Regulation of production of secretory IgA (SIgA) and free secretory component (SC) in mucosal epithelia. The single-layered epithelium is covered with a thick mucus layer that physically excludes members of the resident microbiota. (1) Stimulation of epithelial Toll-like receptors (TLRs) with microbial-associated molecular patterns activates MyD88-dependent signaling pathways that trigger upregulation of PIGR gene transcription. Activation of TLRs may also stimulate polymeric immunoglobulin receptor (pIgR) transcytosis. (2) Newly synthesized pIgR molecules are sorted to the basolateral surface of epithelial cells. (3) Dimeric IgA secreted by lamina propria plasma cells binds to pIgR on the epithelial membrane and stimulates transcytosis. (4) IgA-bound and unoccupied pIgR are transcytosed through epithelial cells. (5) Proteolytic cleavage of pIgR at the apical surface releases SIgA and free SC. (6) Binding of SIgA and SC to luminal bacteria promotes association with the mucin layer and biofilm formation, and prevents direct access of bacteria to the epithelial surface.
Inducible Transcription of the PIGR Gene
Studies in our laboratories and others over the past 20 years have demonstrated that pIgR expression in mucosal epithelial cells is upregulated by a number of host cytokines, including interferon (IFN)-γ, tumor necrosis factor (TNF), IL-4, and IL-1 (reviewed in refs. 12,13). An insight into possible regulatory mechanisms was gained by the observation that pIgR mRNA levels rise slowly in response to cytokine stimulation, peaking around 24 h. The cytokine-induced increase in pIgR mRNA could be blocked by inhibiting protein synthesis with cycloheximide, suggesting that de novo synthesis of one or more intermediary proteins is required. Molecular cloning of the human PIGR gene revealed the presence of several regulatory elements that bind cytokine-inducible transcription factors; the complex pathways of PIGR gene regulation are summarized in Figure 2. Signaling through the IFN-γ receptor causes activation and nuclear translocation of STAT1 dimers, resulting in de novo transcription of IFN regulatory factor 1 and association of IFN regulatory factor 1 with a cognate element in exon 1 of the PIGR gene. Stimulation with IL-4 causes nuclear translocation of activated STAT6 dimers, which associate with a complex element in intron 1 of the PIGR composed of a STAT6-binding site, a putative binding site for an as yet-unidentified IL-4-induced co-activator, and a TNF-inducible nuclear factor κB (NF-κB)-binding site. Interactions among these cytokine-inducible elements in exon 1 and intron 1 of the PIGR gene may be responsible for the observed synergy between IFN-γ, IL-4, and TNF in upregulation of pIgR expression. The sequences of the exon 1 IFN regulatory factor site and the intron 1 STAT6 and NF-κB sites are identical in the human PIGR and mouse Pigr genes, suggesting evolutionary conservation of these regulatory mechanisms. We subsequently demonstrated that induction of PIGR gene transcription via TLR3 and 4 signaling involves the same NF-κB element in intron 1 that is induced by TNF,4 suggesting cooperativity in PIGR gene regulation by signaling received from microbial products and host cytokines. We recently reported that both the classical and alternative pathways of NF-κB activation are involved in PIGR gene regulation, including the novel discovery that signaling through the lymphotoxin-β receptor upregulates pIgR.5 Experiments with small interfering RNA-mediated knockdown of individual NF-κB subunits suggested that both RelA/p50 and RelB/p52 dimers can activate the NF-κB element in intron 1. It is likely that this NF-κB element is also involved in regulation of pIgR by IL-1, which activates a MyD88-dependent signaling pathway that shares many elements with TLR pathways. Another pro-inflammatory cytokine, IL-17, has recently been shown to enhance pIgR expression in mucosal epithelial cells;14 it will be interesting to determine whether IL-17 acts through the intron 1 NF-κB element and/or additional regulatory elements in the PIGR gene. It is significant that expression of pIgR is upregulated by cytokines characteristic of multiple T-cell lineages, including Th1, Th2, and Th17, as well as pro-inflammatory cytokines such as TNF and IL-1. The responsiveness of the PIGR gene to such diverse cytokines suggests that the ability to enhance transport of IgA antibodies in response to a wide range of immunological stimuli may have conferred an evolutionary advantage.

Regulation of PIGR gene transcription by microbial products and host cytokines. Ligation of Toll-like receptors (TLRs) and cytokine receptors results in translocation of activated transcription factors to the nucleus and de novo synthesis of additional transcription factors. The PIGR gene contains binding sites for members of the interferon regulatory factor (IRF), signal transducer and activator of transcription (STAT), and nuclear factor-κB (NF-κB) families of transcription factors. The complex signaling pathways that regulate PIGR gene transcription are described in the text. IL, interleukin; TNF, tumor necrosis factor.
Downregulation of pIgR Expression in Inflammatory Bowel Disease
Given the well-documented upregulation of pIgR expression by pro-inflammatory cytokines, it is paradoxical that expression of pIgR in IECs is downregulated during intestinal inflammation associated with inflammatory bowel disease (IBD). In a study comparing Crohn's disease patients with healthy controls, we found that downregulation of pIgR mRNA in the colonic mucosa was correlated with disease severity.15 We have subsequently observed that downregulation of pIgR in colonic epithelial cells is also a feature of ulcerative colitis in humans, and in experimental murine colitis induced by oral administration of dextran sulfate sodium (Bruno et al., unpublished). We further noted aberrant cellular localization of pIgR in colonic epithelial cells, dysregulated epithelial transcytosis of dIgA, accumulation of IgA in the intestinal lamina propria, and increased levels of circulating IgA in humans and mice with IBD. We hypothesize that inflammation-induced damage to the intestinal epithelium may disrupt intracellular signaling pathways and alter protein trafficking, resulting in profound defects in the pIgR–IgA system. These changes could result in impaired mucosal immunity and increased exposure of the systemic immune system to intestine-derived antigens, further exacerbating inflammation. We and others have reported that expression of pIgR is downregulated in sporadic colorectal tumors in humans.16, 17, 18 A common feature of these conditions is rapid proliferation of relatively undifferentiated epithelial cells, which may not respond properly to microbial and host factors that maintain pIgR expression under homeostatic conditions and enhance pIgR expression during infections.
Regulation of Epithelial Transcytosis of pIgR
The pIgR has five extracellular transmembrane domains, a single membrane-spanning helix and an intracellular tail of 103 amino acids (reviewed in refs. 13,19). After its translation and insertion into the endoplasmic reticulum, the pIgR is sorted through the Golgi and trans-Golgi network and targeted to the basolateral membrane of polarized epithelial cells. Transcytosis of dIgA occurs in three biochemically and morphologically distinct steps: (i) endocytosis from clathrin-coated pits and delivery to basolateral early endosomes; (ii) microtubule-dependent translocation to apical recycling endosomes; and (iii) delivery from the apical endosomes to the plasma membrane.20 Proteolytic cleavage of pIgR at the apical plasma membrane ensures that transport of dIgA is unidirectional, although there can be a certain level of recycling at each step. During transcytosis, dIgA becomes covalently bound to the pIgR via a disulfide exchange reaction and the extracellular SC domain of the pIgR remains bound to dIgA in external secretions. The presence of bound SC confers protection against proteases and mucosal anchoring properties to the SIgA molecule (see article by Mantis et al.11 in this issue).
Intracellular targeting of the pIgR is similar to other integral membrane proteins destined for the apical plasma membrane in polarized epithelial cells, which generally follow an indirect route involving initial targeting the basolateral surface and subsequent transcytosis to the apical surface.21 In the absence of dIgA there is constitutive transcytosis of pIgR, which is the source of free SC in mucosal secretions. However, binding of dIgA to pIgR at the basolateral surface increases the transcytotic rate of the pIgR–dIgA complex by initiating a signaling cascade that activates effector proteins acting on the third step in the transcytosis pathway. The cytoplasmic domain of the pIgR does not contain known signaling motifs, but dimerization of the receptor activates the tyrosine kinase p62YES, which phosphorylates several substrates including phospholipase Cγ1. Downstream effectors of the phospholipase Cγ1 signaling pathway mediate an increased rate of exocytosis of pIgR–dIgA complexes from apical recycling endosomes.22 More recently, it was demonstrated that p62YES could also phosphorylate the epidermal growth factor receptor in an IgA-dependent manner, suggesting that other extracellular signals might also affect transcytosis.23 Indeed, stimulation of cells with epidermal growth factor itself increased the rate of pIgR-mediated dIgA transport, but the effect was shorter lived than the effect of dIgA. Enhanced transcytosis of pIgR depended on phosphorylation and activation of extracellular regulated kinase, which in turn was found to phosphorylate the Rab11 effector Rab11-FIP5. However, overstimulation of this pathway, achieved by co-transfection of constitutively active epidermal growth factor receptor variants, disrupted epithelial cell polarity and transcytosis rather than enhancing constitutive transcytosis.
The ability of dIgA to stimulate its own transcytosis makes teleological sense, but transcytosis might also be stimulated by other extracellular signals. As described above, members of the resident microbiota enhance the expression of pIgR in a MyD88-dependent manner; recent data suggest that MAMPs can also stimulate transcytosis of pIgR.24 Stimulation of a human IEC line with live or dead E. coli or with lipopolysaccharide stimulated both receptor expression and the rate of dIgA transport across polarized epithelial monolayers. Because transcytosis was significantly enhanced before increased pIgR expression, the authors concluded that lipopolysaccharide could also directly stimulate transcytosis. Although the signal transduction pathway was not investigated in this study, it is possible that TLR stimulation and dIgA stimulation converge at the point of extracellular regulated kinase phosphorylation. Additional stimulation may have been mediated by other components of whole bacteria, such as formyl peptides, which have recently been shown to activate extracellular regulated kinase in IECs without stimulating pro-inflammatory pathways.25
References
Macpherson, A.J., McCoy, K.D., Johansen, F.-E. & Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 1, 11–22 (2008).
Hooper, L.V. et al. Molecular analysis of commensal host-microbial relationships in the intestine. Science 291, 881–884 (2001).
Hapfelmeier, S. et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328, 1705–1709 (2010).
Schneeman, T.A. et al. Regulation of the polymeric Ig receptor by signaling through Toll-like receptors 3 and 4: linking innate and adaptive immune responses. J. Immunol. 175, 376–384 (2005).
Bruno, M.E., Frantz, A.L., Rogier, E.W., Johansen, F.-E. & Kaetzel, C.S. Regulation of the polymeric immunoglobulin receptor by the classical and alternative NF-κB pathways in intestinal epithelial cells. Mucosal Immunol. 4, 468–478 (2011).
Pal, K., Kaetzel, C.S., Brundage, K., Cunningham, C.A. & Cuff, C.F. Regulation of polymeric immunoglobulin receptor expression by reovirus. J. Gen. Virol. 86, 2347–2357 (2005).
Bruno, M.E.C. et al. Regulation of the polymeric immunoglobulin receptor in intestinal epithelial cells by Enterobacteriaceae: implications for mucosal homeostasis. Immunol. Invest. 39, 356–382 (2010).
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Swidsinski, A., Loening-Baucke, V., Lochs, H. & Hale, L.P. Spatial organization of bacterial flora in normal and inflamed intestine: a fluorescence in situ hybridization study in mice. World J. Gastroenterol. 11, 1131–1140 (2005).
Johansson, M.E., Larsson, J.M. & Hansson, G.C. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4659–4665 (2011).
Mantis, N.J., Rol, N. & Corthésy, B. Secretory IgA's complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 4, 603–611 (2011) (this issue).
Johansen, F.-E. & Brandtzaeg, P. Transcriptional regulation of the mucosal IgA system. Trends Immunol. 25, 150–157 (2004).
Kaetzel, C.S. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev. 206, 83–99 (2005).
Jaffar, Z., Ferrini, M.E., Herritt, L.A. & Roberts, K. Cutting edge: lung mucosal Th17-mediated responses induce polymeric Ig receptor expression by the airway epithelium and elevate secretory IgA levels. J. Immunol. 182, 4507–4511 (2009).
Arsenescu, R. et al. Signature biomarkers in Crohns disease: toward a molecular classification. Mucosal Immunol. 1, 399–411 (2008).
Koretz, K., Schlag, P., Quentmeier, A. & Moller, P. Evaluation of the secretory component as a prognostic variable in colorectal carcinoma. Int. J. Cancer 57, 365–370 (1994).
Krajci, P. et al. Secretory component mRNA and protein expression in colorectal adenomas and carcinomas. Br. J. Cancer 73, 1503–1510 (1996).
Traicoff, J.L. et al. Characterization of the human polymeric immunoglobulin receptor (PIGR) 3′UTR and differential expression of PIGR mRNA during colon tumorigenesis. J. Biomed. Sci. 10, 792–804 (2003).
Kaetzel, C.S. & Bruno, M.E.C. Epithelial transport of IgA by the polymeric immunoglobulin receptor. In Mucosal Immune Defense: Immunoglobulin A (Kaetzel, C.S., ed) 43–89 ( Springer, New York, 2007 ).
Luton, F., Cardone, M.H., Zhang, M. & Mostov, K.E. Role of tyrosine phosphorylation in ligand-induced regulation of transcytosis of the polymeric immunoglobulin receptor. Mol. Biol. Cell 9, 1787–1802 (1998).
Mostov, K.E., Verges, M. & Altschuler, Y. Membrane traffic in polarized epithelial cells. Curr. Opin. Cell Biol. 12, 483–490 (2000).
Rojas, R. & Apodaca, G. Immunoglobulin transport across polarized epithelial cells. Nat. Rev. Mol. Cell Biol. 3, 944–955 (2002).
Su, T. et al. A kinase cascade leading to Rab11-FIP5 controls transcytosis of the polymeric immunoglobulin receptor. Nat. Cell Biol. 12, 1143–1153 (2010).
Diebel, L.N. & Liberati, D.M. Disparate effects of bacteria and toll-like receptor-dependant bacterial ligand stimulation on immunoglobulin a transcytosis. J. Trauma 70, 691–700 (2011).
Wentworth, C.C., Jones, R.M., Kwon, Y.M., Nusrat, A. & Neish, A.S. Commensal-epithelial signaling mediated via formyl peptide receptors. Am. J. Pathol. 177, 2782–2790 (2010).
Acknowledgements
This work was supported by NIH grant R21AI069027 (CSK), a Senior Research Award from the Crohn's and Colitis Foundation of America (C.S.K.), and Research Council of Norway and University of Oslo (F.E.J.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Johansen, FE., Kaetzel, C. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol 4, 598–602 (2011). https://doi.org/10.1038/mi.2011.37
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2011.37
This article is cited by
-
Polymeric immunoglobulin receptor deficiency exacerbates autoimmune hepatitis by inducing intestinal dysbiosis and barrier dysfunction
Cell Death & Disease (2023)
-
Polymeric immunoglobulin receptor (pIgR) in cancer
Journal of Cancer Research and Clinical Oncology (2023)
-
Pretreatment with probiotics Enterococcus faecium NCIMB 11181 attenuated Salmonella Typhimurium-induced gut injury through modulating intestinal microbiome and immune responses with barrier function in broiler chickens
Journal of Animal Science and Biotechnology (2022)
-
IκBζ controls IL-17-triggered gene expression program in intestinal epithelial cells that restricts colonization of SFB and prevents Th17-associated pathologies
Mucosal Immunology (2022)
-
M1 macrophages evoke an increase in polymeric immunoglobulin receptor (PIGR) expression in MDA-MB468 breast cancer cells through secretion of interleukin-1β
Scientific Reports (2022)
