
Abstract
Peroxisome proliferator-activated receptor-γ (PPAR-γ) is widely expressed in macrophages and has been identified as a putative target for the development of novel therapies against inflammatory bowel disease (IBD). Computational simulations identified macrophages as key targets for therapeutic interventions against IBD. This study aimed to characterize the mechanisms underlying the beneficial effects of macrophage PPAR-γ in IBD. Macrophage-specific PPAR-γ deletion significantly exacerbated clinical activity and colonic pathology, impaired the splenic and mesenteric lymph node regulatory T-cell compartment, increased percentages of lamina propria (LP) CD8+ T cells, increased surface expression of CD40, Ly6C, and Toll-like receptor 4 (TLR-4) in LP macrophages, and upregulated expression of colonic IFN-γ, CXCL9, CXCL10, IL-22, IL1RL1, CCR1, suppressor of cytokine signaling 3, and MHC class II in mice with IBD. Moreover, macrophage PPAR-γ was required for accelerating pioglitazone-mediated recovery from dextran sodium sulfate (DSS) colitis, providing a cellular target for the anti-inflammatory effects of PPAR-γ agonists in IBD.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD), with its two clinical manifestations, ulcerative colitis and Crohn's disease, is an immune-mediated disease characterized by widespread inflammation and immune cell infiltration of the gastrointestinal tract. The etiology of IBD is multifactorial, and entails interaction among genetic predisposition, environmental factors, and the gut microbiota. Treatments targeted to downmodulate the immune and inflammatory responses, such as the corticosteroid prednisone or the anti-tumor necrosis factor-α antibody Remicade, have shown promise in reducing severity and reoccurrence of the disease. These treatments, however, are also associated with various adverse side effects, such as cushingoid appearance, weight gain, and systemic immunosuppression, thus stressing the need to develop safer alternatives for the long-term management of IBD.1
Peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists have also demonstrated efficacy in ameliorating intestinal inflammation associated with IBD.2, 3, 4 PPAR-γ is a transcription factor expressed highly in all the major cell types involved in IBD pathogenesis, including intestinal epithelial cells (IECs), macrophages, dendritic cells, and lymphocytes. Chronic administration of synthetic PPAR-γ ligands (i.e., thiazolidinedione (TZD) class of antidiabetic drugs) is also linked with adverse side effects that led to a mandatory labeling with a black box warning.5 Nonetheless, PPAR-γ can also become activated by safer compounds such as dietary lipids including conjugated linoleic acid (CLA), or endogenous lipid mediators produced during inflammation such as 15(S)-HETE, 13(S)-HODE, and other unsaturated fatty acids6, 7 or the isoprenoid abscisic acid,8, 9 thereby heightening the potential for developing safer and more efficacious therapies against gut inflammatory diseases.
We demonstrated that mice lacking PPAR-γ broadly in all immune and epithelial cells are irresponsive to the beneficial effects of the naturally occurring ligand of PPAR-γ CLA in experimental models of colitis2, 10, 11, 12 and colorectal cancer,13 although the specific immune and/or epithelial cells required for the PPAR-γ-induced anti-inflammatory effects of CLA were still unclear. By using the CD4+CD45RBhi adoptive transfer model of colitis, we demonstrated that PPAR-γ is required for the anti-inflammatory activity of regulatory T (Treg) cells against effector CD4+ T cell-induced colitis.12 Recent examination of more narrow cell-specific PPAR-γ knockout mice that underwent CD4-Cre− and Villin-Cre-mediated recombination revealed that PPAR-γ expressed in T cells and IECs, respectively, contribute to protection against experimental IBD through immunoregulatory mechanisms involving both T cells and macrophages.14, 15, 16 For instance, in T cell-specific PPAR-γ knockout (CD4-Cre+) mice, mesenteric lymph node (MLN) and blood had less Treg cells and inflammatory and cell adhesion molecules and SOCS-3 (suppressor of cytokine signaling 3) were significantly upregulated in the colonic mucosa when compared with CD4-Cre− with a wild-type phenotype.14 The IEC-specific PPAR-γ knockout (Villin-Cre+) mice had significantly worsened disease severity and upregulated lysosomal pathway and antigen presentation-related gene expression while modulating expression of genes in the p53 tumor-suppressor pathway in comparison with their littermate PPAR-γ-expressing (Villin-Cre−) controls.16
Computational simulations using a mathematical model of the cellular interactions at the colonic mucosa and MLN during colitis identified macrophages and their mechanisms of plasticity as key targets for therapeutic interventions against IBD.17 In this regard, the effect of the macrophage-specific PPAR-γ deletion on experimental IBD was previously examined by Shah et al.,18 who found that macrophage-specific PPAR-γ knockout mice were more susceptible to dextran sodium sulfate (DSS) colitis than wild-type counterparts. In addition to showing significantly worsened disease activity, macrophage-specific PPAR-γ knockout mice showed an upregulation in colonic expression of chemokines and inflammatory cytokines.18 These findings suggest that PPAR-γ expression in macrophages may act to limit colonic recruitment of blood-derived immune cells. However, Shah et al.18 did not examine whether deletion of PPAR-γ in macrophages affected T-cell populations in effector or inductive sites of the gut-associated lymphoid tissue or the global colonic transcriptome. This is important as the balance between effector and regulatory immune cell populations can significantly modulate the inflammatory response and tissue damage in IBD. This study aimed to characterize the immunoregulatory mechanisms by which macrophage PPAR-γ ameliorates experimental IBD. We demonstrate that the deficiency of PPAR-γ in macrophages increases the expression of colonic inflammatory and fatty acid oxidation genes, impairs the peripheral Treg compartment, favors a proinflammatory macrophage phenotype in the colonic lamina propria (LP), and an influx of effector T cells at the colonic mucosa during IBD; all of which implies a possible cross-talk between macrophages and T cells coordinated by PPAR-γ.
Results
The deficiency of PPAR-γ in macrophages worsens colitis severity
To examine the effect of macrophage-specific PPAR-γ deficiency on colitis severity, Lysozyme M (LysM)-Cre+ and LysM-Cre− mice were treated with 2.5% DSS for 7 days. Figure 1a illustrates the deletion of PPAR-γ in the macrophages, spleen, MLN, and colon of LysM-Cre+ mice as well as LysM-Cre− mice. The results show that the deletion is more efficient in purified macrophages in comparison with the other tissues containing mixed cell populations. These PCR results are in line with the western blot analyses showing that the amount of PPAR-γ protein is lower in macrophages and colons from LC+ mice when compared with LC− mice (Figure 1b). In addition, the degree of deletion of PPAR-γ is more effective in macrophages than in colonic samples. This can be explained by the fact that the entire colon contains epithelial cells and other immune cells that express normal levels of PPAR-γ as the Lysozyme M promoter is expressed in myeloid cells but not in epithelial or lymphoid cells. Figure 1c illustrates the suppressive effect of macrophage-specific PPAR-γ deficiency on colonic PPAR-γ and CD36 mRNA expression in healthy mice. Similar suppression of PPAR-γ and CD36 expression was found for the other tissues (data not shown).

Genotyping of peroxisome proliferator-activated receptor-γ (PPAR-γ) flfl; Lysozyme M-Cre+ (LC+) and Lysozyme M-Cre− (LC−) control mice. Conditional deletion of the PPAR-γ gene via Lysozyme M-Cre-mediated recombination was examined in mouse tissues by PCR analysis. The flox (fl) allele at 275 bp and the null allele at 400 bp. (a, left to right) fl/fl in bone marrow-derived macrophages (BMMs) without recombination (LC−) (lane 1) or with recombination (LC+) (lane 2). Homogenized whole spleen, mesenteric lymph node (MLN), and colon without recombination (LC−) (lanes 3, 5, and 7) or with recombination (LC+) (lanes 4, 6, and 8). (b, left to right) PPAR-γ (top panel) and β-actin (bottom panel) protein expression as measured by western blot in BMM and colon of LC− (lanes 1 and 3) and LC+ (lanes 2 and 4) mice. (c) Quantification of PPAR-γ (left panel) and CD36 (right panel) colonic mRNA expression by real-time quantitative reverse transcriptase PCR (RT-PCR) in LC− and LC+ mice.
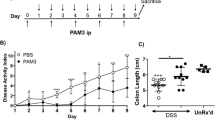
To investigate the effect of macrophage-specific PPAR-γ deficiency on the severity of experimental IBD, PPAR-γ flfl Cre+ mice and control PPAR-γ flfl Cre− littermates were treated with 2.5% DSS in the drinking water for 7 days, and disease activity was monitored daily. Macrophage-specific PPAR-γ-null mice had significantly worsened disease activity throughout the 7-day challenge when compared with PPAR-γ-expressing littermates (Figure 2a). On day 7, colons, MLNs, and spleens from mice in each group were scored on gross anatomical signs of inflammation. In line with the disease activity index scores, both the colons and spleens were significantly more inflamed in PPAR-γ flfl Cre+ mice than in PPAR-γ-expressing littermate mice (Figure 2b and c ).

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on disease severity and macroscopic lesions in spleen and colon of mice with experimental inflammatory bowel disease (IBD). PPAR-γ flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− control mice were challenged with 2.5% dextran sodium sulfate (DSS) for 7 days. The disease activity index (DAI), a composite score reflecting clinical sings of the disease, was assessed (a) daily and the macroscopic lesions in (b) colon and (c) spleen were determined on day 7. Data are represented as mean±s.e. of groups of ten mice. Points with an asterisk are significantly different (P<0.05).
To determine whether exogenous administration of a synthetic PPAR-γ agonist (pioglitazone (PIA)) affects the clinical recovery from DSS colitis, we challenged PPAR-γ flfl Cre+ mice and control PPAR-γ flfl Cre- mice with 2.5% DSS for 5 days followed by a 3-day recovery phase using a previously published protocol of colonic regeneration.19 We used selective instead of safer and equally efficacious compounds such as CLA2, 10, 13, 20 and abscisic acid,8, 9 because PIA is known to be a more selective activator of PPAR-γ than these naturally occurring compounds. During the recovery phase, mice were administered either control or PIA-supplemented diets. In the colonic mucosa by activating macrophage PPAR-γ with PIA facilitated recovery from experimental IBD (Figure 3). Moreover, the deletion of PPAR-γ in macrophages (but not in other immune or epithelial cells) abrogated the ability of PIA to induce recovery from colitis, suggesting that macrophages are important targets for the action of TZDs.

Effect of pioglitazone and macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion in the clinical recovery from experimental inflammatory bowel disease (IBD). Treatment with the PPAR-γ agonist pioglitazone (PIA) accelerates recovery from gut inflammation in Lysozyme M-Cre− mice but not in macrophage-specific PPAR-γ-null mice (Lysozyme M-Cre+). Data are represented as mean±s.e. of groups of ten mice. Means without a common letter superscript are significantly (P<0.05) different.
Colonic histopathology was significantly worsened in macrophage-specific PPAR-γ-deficient mice
To further examine the affect of macrophage-specific PPAR-γ deficiency on DSS colitis, we examined the histopathological changes in the colon resulting from the DSS challenge of LysM-Cre− and LysM-Cre+ mice for the amount of epithelial erosion, leukocyte infiltration, and mucosal thickening. Our data indicate that LysM-Cre+ mice had significantly higher scores for all three parameters tested, showing that observations of worse disease activity translate to more severe colonic immunopathology (Figure 4).

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on colon histopathology and inflammation of mice with experimental inflammatory bowel disease (IBD). PPAR-γ flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− control mice were challenged with 2.5% dextran sodium sulfate (DSS) for 7 days. Representative photomicrographs of colonic samples from (a) LC− and (b) LC+ mice with DSS colitis (original magnification × 40). Colonic specimens underwent blinded histological examination and were scored (1–4) on (c) epithelial erosion, (d) mucosal wall thickening, and (e) leukocyte infiltration on day 7 of the challenge. Data are represented as mean±s.e. of groups of ten mice. Points with an asterisk are significantly different (P<0.05).
Effect of macrophage-specific PPAR-γ deficiency on immune cell populations in the blood, spleen, MLN, and LP
In blood, F4/80+CD11b+ monocytes from LysM-Cre+ mice expressed significantly less monocyte chemotactic protein-1 (MCP-1) but greater levels of major histocompatibility complex class II (MHC II) than control mice, as indicated by median fluorescence intensity (Figure 5a–c). Spleens from macrophage-specific PPAR-γ-deficient mice had reduced CD4+ T cells and CD4+CD25+FoxP3+ Treg cells on day 7 of DSS challenge (Figure 5d–f).

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on immune cell subsets in blood and spleen from mice with experimental inflammatory bowel disease (IBD). (a–c) Blood and (d–f) spleen from PPAR-γ flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− mice were immunophenotyped. Data were collected on day 7 of the dextran sodium sulfate (DSS) challenge and were analyzed with the FACS Diva software. Blood monocytes or spleen T-cell subsets were selected by gating first on the viable cells based on forward scatter and side scatter. Monocytes were then gated on F4/80+ CD11b+ and within this cell population (a), monocyte chemotactic protein-1 (MCP-1) (b) and major histocompatibility complex class II (MHC-II) mean fluorescence intensity (MFI) (c) were then examined. Splenocytes were gated on CD3+CD4+ (d) and CD3+CD8+ (e) or CD4+CD25+FOXP3+ (f) to characterize the T-cell subsets. Data are represented as mean±s.e. of groups of ten mice. Points with an asterisk are significantly different (P<0.05).
In MLNs, macrophage-specific PPAR-γ-deficient mice had significantly more F4/80+CD11b+ macrophages than in PPAR-γ-expressing mice (Figure 6). Additionally, macrophages from macrophage-specific PPAR-γ-deficient mice expressed greater levels of CD40, a costimulatory molecule involved in the pathogenesis of DSS-induced intestinal inflammation.21 Similar to the spleens, MLNs from macrophage-specific PPAR-γ deficient mice had significantly fewer Treg than those from LysM-Cre− mice.

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on immune cell subsets in mesenteric lymph nodes (MLNs) from mice with experimental inflammatory bowel disease (IBD). MLN-derived cells from PPAR-γ *flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− mice were immunophenotyped to identify immune cell subsets by flow cytometry. Data were collected on day 7 of the dextran sodium sulfate (DSS) challenge and were analyzed with the FACS Diva software. MLN macrophages were selected based on F4/80 and CD11b (a) expression in the cell viable gate and then CD40 expression within this population was determined by assessing mean fluorescence activity (MFI, b). For T-cells, all viable MLN cells were selected using forward scatter and side scatter and the gated on CD3+CD4+ (c) and CD4+CD25+FOXP3+ (d). Data are represented as mean ±s.e. of groups of ten mice. Points with an asterisk are significantly different (P0.05).
In the colonic LP, there were no differences in the overall percentage of F4/80+CD11b+ macrophages between LysM-Cre+ and LysM-Cre− mice, although macrophages from LysM-Cre+ mice differed phenotypically than those of LysM-Cre− mice (Figure 7). Specifically, macrophage-specific PPAR-γ-deficient macrophages expressed significantly more Ly6C, and the percentage of CD40+ and TLR-4+ macrophages were elevated. These findings suggest a more inflammatory, possibly M1, classically activated phenotype of macrophages in macrophage-specific PPAR-γ-deficient mice. We also observed a significant influx of CD8+ T cells in the colonic of LP macrophage-specific PPAR-γ-deficient mice, which is indicative of an enhanced secondary recruitment of effector T cells.

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on immune cell subsets in the colonic lamina propria (LP) from mice with experimental inflammatory bowel disease (IBD). Colonic LP cells from PPAR-γ flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− mice were immunophenotyped to identify immune cell subsets by flow cytometry. Data were collected on day 7 of the dextran sodium sulfate (DSS) challenge and were analyzed with the FACS Diva software. LP macrophages were selected based on F4/80 and CD11b expression (a) in the cell viable gate and then CD40 (b), Ly6C (c), and Toll-like receptor 4 (TLR-4) (d) expression within this population was determined by assessing mean fluorescence activity (MFI). For T cells, all viable mesenteric lymph node (MLN) cells were selected using forward scatter and side scatter and then gated on CD3+CD4+ (e) and CD3+CD8+. (f) Data are represented as mean±s.e. of groups of ten mice. Points with an asterisk are significantly different (P<0.05).
Effect of macrophage-specific PPAR-γ deficiency on colonic inflammation
To assess the effect of macrophage-specific PPAR-γ deletion on colonic inflammation, real-time quantitative reverse transcriptase PCR (RT-PCR) was performed on colonic samples of LysM-Cre+ and LysM-Cre− mice with DSS colitis. As anticipated, the expression of PPAR-γ was greater in LysM-Cre− mice, although the data were not statistically significant (Figure 8), because of the fact that PPAR-γ was deleted in macrophages but not in lymphocytes, IECs, endothelial cells, dendritic cells, fibroblasts, and other colonic cell types that express normal PPAR-γ levels. In contrast to the effects of T cell-specific PPAR-γ deletion,14 interleukin (IL)-6 and IL-1β did not differ significantly between groups. However, interferon-γ (IFN-γ) levels were significantly greater in LysM-Cre+ mice, a finding in line with the greater abundance of CD8+ T cells in the LP and increased levels of macrophage inflammatory markers. The expressions of cellular adhesion molecule MAdCAM-1 (mucosal addressin cellular adhesion molecule 1) and ICAM-1 (intracellular adhesion molecule 1) also showed a strong numerical elevation in LysM-Cre+ mice, although the data did not reach statistical significance. In their study using macrophage-specific PPAR-γ-deficient mice, Shah et al.18 reported a significant increase in colonic expression of MCP-1 and CCR2 following treatment with DSS. Here, we found that MCP-1 expression was greater in colons of LysM-Cre+ mice, although differences were not statistically significant.

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on target gene expression in the colonic mucosa of mice with experimental inflammatory bowel disease (IBD). Expression of (a) PPAR-γ, (b) interleukin (IL)-1β, (c) mucosal addressin cellular adhesion molecule 1 (MAdCAM-1), (d) monocyte chemotactic protein-1 (MCP-1), (e) IL-6, (f) interferon-γ (IFN-γ), (g) intracellular adhesion molecule 1 (ICAM-1), and (h) macrophage inflammatory protein 1α (MIP-1α) was assessed in colonic mucosal specimens recovered from PPAR-γ flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− mice. Data are represented as mean±s.e. of groups of ten mice. Points with an asterisk are significantly different (P<0.05).
Global gene expression analyses in the colonic mucosa of macrophage-specific PPAR-γ-deficient mice and floxed littermates
A total of 124 genes had statistically significant differences in colonic expression (P<0.01 and twofold change) between groups as described in the heat map (Supplementary Figure S1 online). Gene ontology analyses revealed that the deletion of PPAR-γ in macrophages during IBD is significantly enriched for genes involved in response to stimulus, immune response, immune system process, cellular metabolic process, and oxidation/reduction (Supplementary Table S1 online). These differentially expressed genes were submitted to ingenuity pathway for network analyses and the top two networks retrieved were carbohydrate metabolism (Supplementary Figure S2 online) and immunological disease (Supplementary Figure S3 online).
The carbohydrate metabolism network reveals that the deletion of PPAR-γ in macrophages caused a downregulation of oxidation genes stearoyl-coenzyme A desaturase (SCD), PPAR-α, CYP4B1, a member of the cytochrome P450 monooxygenase family, casein kinase 1 (CK1), plasma membrane calcium-transporting ATPase 1 (ATP2B1), and immunoglobulin mu chain C region (IGHM). Additionally, the deletion of PPAR-γ in macrophages caused an upregulation of DNA primase large subunit 2 (PRIM2), macrophage scavenger receptor 1 (MSR1), nuclear factor IL-3 regulated (NFIL3), the zinc-finger transcription factor EGR2, the atypical member of the E2F transcription factor family E2F8, the antiapoptotic molecule BCL2A1, and the GTP cyclohydrolase I (GCH1). The microarray results of two key metabolically active genes (i.e., PPAR-α and CYP4B1) were validated by real-time RT-PCR (Figure 9), thereby confirming the microarray results.

Effect of macrophage-specific peroxisome proliferator-activated receptor-γ (PPAR-γ) deletion on metabolically active gene expression in the colonic mucosa of mice with experimental inflammatory bowel disease (IBD). Expression of (a) cytochrome P450 (CYP4B1) and (b) PPAR-α was assessed in colonic mucosal specimens recovered from PPAR-γ flfl; Lysozyme M-Cre+ and Lysozyme M-Cre− mice. Data are represented as mean±s.e. of groups of ten mice. Points with an asterisk are significantly different (P<0.05).
The immunological disease network demonstrates for the first time that the deletion of PPAR-γ in macrophages caused a downregulation of retinoid acid orphan receptor γt (ROR γt, also described as RORC), Bmp, and activin membrane-bound inhibitor (BAMBI, initially described as a pseudoreceptor antagonizing TGF-β receptor activation) and Staphylococcal complement inhibitor (SCIN) in the colon of mice with IBD. Additionally, the deletion of PPAR-γ in macrophages caused an up-regulation of chemokine (C-X-C motif) ligand 9 (CXCL9), CXCL10, IL-22, IL1RL1, CCR1, SOCS3 (a gene responsive to IFN-γ), MHC class II, ADM, IFN-induced tetratricopeptide repeat protein 2 (IFIT-2), which belongs to the type I IFN response genes, receptor interacting protein kinase 3 (RIPK3), C3a receptor gene (C3AR1) an important receptor of the complement system, IIGP is a member of the p47 GTPase family induced upon exposure to Escherichia coli LPS,22 cytochrome b-245, βpolypeptide (CYBB), receptor 3A for the Fc region of immunoglobulin G (FCGR3A).
Discussion
The appeal of PPAR-γ as a putative therapeutic target for human IBD lies in its high expression levels in the majority of cells found in mucosal barriers, including macrophages, lymphocytes, and IECs, although PPAR-γ expression in the colonic epithelium is decreased in ulcerative colitis patients.23 In previous experiments we have shown that PPAR-γ expressed in IECs and T cells protects mice from experimental IBD.2, 9, 12, 14, 16 In IECs, PPAR-γ expression was shown to be important in the lysosomal and p53 tumor-suppressor pathways and antigen presentation complex genes, whereas deletion of PPAR-γ in T cells resulted in an increase in proinflammatory genes and cellular adhesion molecules.14, 16 Macrophage PPAR-γ has also been reported to protect mice from experimental IBD,18 a finding consistent with its noted anti-inflammatory effects in this cell type24, 25, 26, 27 and the purported role of colonic macrophages as possible sites for therapeutic intervention against gut inflammation.17
Rosiglitazone, a TZD also known as Avandia, has been proven to be efficacious in treating human ulcerative colitis,4, 28 which provides clinical validation in humans for the use of PPAR-γ as target for new IBD treatments. Additionally, PPAR-γ can become activated by naturally occurring, non-toxic compounds such as dietary lipids and phytochemicals (e.g., abscisic acid and CLA) that are safer than Avandia (rosiglitazone) and Actos (pioglitazone). The long-term objective of this program was to investigate the immunoregulatory mechanisms by which macrophage PPAR-γ prevents or ameliorates mucosal inflammation, thereby adding to the development of safer, more effective therapies against gut immunoinflammatory disorders.
Macrophage-specific PPAR-γ deficiency significantly worsened disease activity, colonic histopathology, and abrogated pioglitazone's ability to accelerate recovery from DSS colitis. These clinical and pathological differences were consistent with the predictions of our computational and mathematical model of IBD, which suggested that macrophages and their mechanisms of plasticity are key targets for therapeutic intervention against IBD.17 Additionally, these clinical and pathological findings are associated with significant differences in peripheral and colonic immune cell populations. More specifically, macrophage-specific PPAR-γ-null mice overall had significantly fewer Treg cells in spleen and MLN than their littermates expressing PPAR-γ in macrophages, suggesting a possible role of macrophage populations in defining T-cell ontogeny and development; a process that may be regulated by macrophage PPAR-γ. We have previously shown that Treg accumulation in mucosal inductive sites is associated with the prevention of chronic colitis caused by the adoptive transfer of CD4+CD45RBhi T cells in SCID mice.12 Further studies examining the role of PPAR-γ in modulating the cross-talk between macrophages and T cells at the mucosal surfaces and the effects of this cross-talk on gastroenteric inflammation are warranted.
In the MLN, macrophage infiltration was significantly increased in macrophage-specific PPAR-γ-null mice in comparison with PPAR γ-expressing littermates. MLN macrophages also expressed significantly greater amounts of surface costimulatory molecule CD40 than PPAR-γ-expressing littermate mice. PPAR-γ activation has been shown to reduce CD40 expression in the hippocampus,29 atherosclerotic plaques,30 and renal tubular epithelial cells,31 although this is the first study to demonstrate a similar effect on colonic macrophages. Combined with our results showing a significantly decreased Treg compartment, these results indicate that macrophage PPAR-γ deficiency may increase effector responses in mucosal inductive sites.
Similar to our findings in the MLN, macrophage CD40 expression was also enhanced in the colonic LP. Our findings are consistent with the elevated levels of CD40+ macrophages in colons of Crohn's disease and ulcerative colitis patients32, 33, 34, 35 where they have been linked to activation of IECs,36 proinflammatory cytokine production,32 and costimulation of T cells;35 all of which can lead to effector and inflammatory responses at the gut mucosa. Combined with increases in Ly6C and Toll-like receptor 4 (TLR-4), our findings suggest that a greater percentage of colonic LP macrophages in macrophage-specific PPAR-γ-null mice were in a classically activated M1, proinflammatory state. These findings are consistent with studies showing that PPAR-γ has an important role in alternative M2 macrophage differentiation.24 Although we did not find significant increases in proinflammatory cytokines derived from macrophages, such as IL-6 and IL-1β, or in chemokines MCP-1 or macrophage inflammatory protein-1α, there were consistent numerical increases in chemokines and cytokines in the colons of macrophage-specific PPAR-γ-null mice.
Colonic IFN-γ expression and the percentages of LP CD8+ T cells were significantly increased in macrophage-specific PPAR-γ-null mice, indicating the predominance of a Th1 phenotype, and that macrophage PPAR-γ may be an important component in cross-talk between macrophages and lymphocytes in the colon. Our results on colonic gene expression differ somewhat from those previously reported by Shah et al.,18 who found no significant difference in IFN-γ and significant increases in IL-1β and MCP-1 in macrophage-specific PPAR-γ knockout mice. The differences between our results and those reported may be because of the different genetic backgrounds of the mice. Whereas mice from our experiments are from a C57BL/6 background, Shah et al.18 used mice from a mixed Sv129/C57BL/6/CB.20 background. Our group and others have demonstrated that the mouse genetic background has an important role in delineating disease activity in and colonic inflammatory lesions during DSS colitis.16, 37 As gut microbial populations vary across mouse vivaria, possible differences in the gut microbiota between our mice and those used by Shah et al.18 may also have contributed to differential clinical and pathological findings reported.
In conclusion, our results show that macrophage PPAR-γ deficiency worsens experimental IBD by modulating the expression of inflammatory/effector (CXCL9, CXCL10, IL-22, IFN-γ, CCR1, SOCS3, and IL1RL1) and metabolic (SCD1, PPAR-α, and CYP4B1) genes, impairing the peripheral Treg compartment and increasing both the influx and activation of LP macrophages and T cells at the colonic mucosa. We also provide in vivo evidence demonstrating that colonic macrophages are important targets for TZDs in the gut mucosa during experimental IBD.
Methods
Animal procedures. Eight-week-old PPAR-γ flfl Cre+ mice, with a Cre recombinase targeted to the LysM-Cre promoter (LysM-Cre+, n=20), and control LysM-Cre− (LysM-Cre−, n=20) littermates in a C57BL/6 background were housed at the animal facilities at Virginia Polytechnic Institute and State University in a room maintained at 23.9 °C, with a 12:12 h light–dark cycle starting from 0600 hours. All experimental procedures were approved by the institutional animal care and use committee of Virginia Polytechnic Institute and State University and met or exceeded requirements of the Public Health Service/National Institutes of Health and the Animal Welfare Act. Colitis was induced with 2.5% DSS, 36,000–44,000 mol wt (ICN Biomedicals, Aurora, OH), in the drinking water. After the DSS challenge, mice were weighed on a daily basis and examined for clinical signs of disease associated with colitis (i.e., perianal soiling, rectal bleeding, diarrhea, and piloerection). For the DSS challenge, the disease activity indices and rectal bleeding scores were calculated using a modification of a previously published compounded clinical score.2 Briefly, disease activity index consisted of a scoring for diarrhea and lethargy (0–3), whereas rectal bleeding consisted of a visual observation of blood in feces and the perianal area (0–4). Mice in the DSS study were killed on day 7 of the DSS challenge by CO2 narcosis and blood was drawn from the heart. Colons and spleens were scored based on size and macroscopic inflammatory lesions (0–3).
Pioglitazone treatment during recovery from DSS colitis. To determine whether the activation of PPAR-γ in macrophages using pioglitazone (Actos) would accelerate the recovery from DSS colitis, we challenged PPAR-γ flfl LysM-Cre+ mice (n=20) and LysM-Cre− (n=20) mice with 2.5% DSS for 5 days. On day 6 of the study, the DSS was withdrawn from the drinking water and half of the mice within group were given either control or pioglitazone (70 mg kg–1)-supplemented diets during the recovery phase for 3 additional days.
Histology. Colonic sections were fixed in 10% buffered neutral formalin, later embedded in paraffin, and then sectioned (5 μm) and stained with hematoxylin and eosin stain for histological examination. Colons were graded with a compounded histological score including the extent of (i) epithelial cell erosion, (ii) leukocyte infiltration, and (iii) mucosal thickening. The sections were graded with a score of 0–4 for each of the previous categories and data were analyzed as a normalized compounded score.
Protein extraction and western blot. Macrophages were washed three times with ice-cold phosphate-buffered saline, harvested and lysed with RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630 (Sigma-Aldrich, St Louis, MO), 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) containing protease and phosphatase inhibitors and incubated for 30 min on ice. Colons were collected, cleaned, and weighed. The tissue was mixed with RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) containing protease and phosphatase inhibitors. Cells and tissues were then homogenized and incubated on ice for 30 min. Whole lysates were cleared by centrifugation (10,000 g for 10 min) and protein concentration was measured using a DC protein assay kit (Bio-Rad Laboratories, Hercules, CA). Diluted samples containing equal amounts of protein were mixed with 2 × Laemmli Sample Buffer (Bio-Rad Laboratories). Proteins were separated on a 10% SDS-PAGE gel and transferred to polyvinylidene difluoride membrane. The membrane was blocked with 1 × Tris-buffered saline with Tween-20 (TBS-T; 20 mM Tris-HCl pH 7.6, 8.5% NaCl, and 0.1% Tween-20) containing 5% nonfat dry milk for 30 min at room temperature. Anti-PPAR-γ (Santa Cruz Biotechnology, Santa Cruz, CA) primary antibody at 1:250 dilution in 1 × TBS-T 5% nonfat dry milk was incubated overnight at 4 °C. After three washes with 1 × TBS-T, membrane was incubated for 45 min at room temperature with anti-mouse IgG conjugated to horseradish peroxidase at 1:2,000 dilution. The antigen detection was performed with the enhanced chemiluminescence (Bio-Rad) detection system.
Isolation of colonic LP immune cells. Colons from mice were excised, placed in calcium/magnesium free (CMF) (Hank's buffered salt solution/10% fetal bovine serum/0.251 EDTA)/HEPES (Mediatech, Manassas, VA), cleaned of contents, and sectioned into 5-mm pieces. To remove intraepithelial lymphocytes, tissues were incubated for 45 min in 15 ml of Calcium/magnesium fire CMF/EDTA. Medium was changed every 15 min. To release LP cells, tissues were digested for 3 h with type VIII collagenase (Sigma-Aldrich) in Hank's buffered salt solution. Medium was changed every hour and supernatants containing LP cells were collected. After digestion, supernatants were pooled, spun at 400 g and enumerated with a Coulter Counter (Beckman Coulter, Fullerton, CA).
Flow cytometry. MLN and spleen-derived cells were excised, crushed with frosted slides, and were resuspended in phosphate-buffered saline and enumerated with a Coulter Counter. LP cells, MLNs, and spleen-derived cells (2 × 105 cells per well) or whole blood (10 μl per well) were seeded onto 96-well plates, centrifuged at 4 °C at 3,000 r.p.m. for 4 min, and washed with phosphate-buffered saline containing 5% serum and 0.09% sodium azide (fluorescence-activated cell sorting buffer). To assess differential monocyte/macrophage subsets, the cells were then incubated in the dark at 4 °C for 20 min in FcBlock (20 μg ml–1; BD Pharmingen, San Jose, CA), and then for an additional 20 min with fluorochrome-conjugated primary antibodies anti-F4/80-PE-Cy5 (5 μg ml–1, eBioscience, San Diego, CA), anti-CD11b-Alexa Fluor 700 (2 μg ml–1, eBioscience), anti-CD40-APC (3 μg ml–1, BD), anti-MHC II-PE (2 μg ml–1, eBioscience), anti-Ly6C-PerCP-Cy5.5 (2 μg ml–1, eBioscience), anti-TLR-4-PE-Cy7 (2 μg ml–1, eBioscience), and anti MCP-1-FITC (3 μg ml–1, BD). For lymphocyte assessment, cells were incubated with anti-CD4-FITC (3 μg ml–1; BD Pharmingen), anti-CD8-PerCp-Cy5.5 (2 μg ml–1, eBioscience), anti-CD3 PE-Cy5 (2 μg ml–1; BD Pharmingen), anti-CD25-APC (2 μg ml–1, BD), and anti-FoxP3-PE (2 μg ml–1, eBioscience) as previously shown.38 Flow results were computed with a BD LSR II flow cytometer and data analyses were performed with FACS Diva software (BD).
Quantitative real-time RT-PCR. Total RNA was isolated from colons using the RNA isolation Minikit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Total RNA (1 μg) was used to generate complementary DNA (cDNA) template using the iScript cDNA Synthesis Kit (Bio-Rad). The total reaction volume was 20 μl with the reaction incubated as follows in an MJ MiniCycler: 5 min at 25 °C, 30 min at 52 °C, 5 min at 85 °C, and hold at 4 °C. PCR was performed on the cDNA using Taq DNA polymerase (Invitrogen, Carlsbad, CA) and using previously described conditions.2 Each gene amplicon was purified with the MiniElute PCR Purification Kit (Qiagen) and quantitated on an agarose gel by using a DNA mass ladder (Promega, Madison, WI). These purified amplicons were used to optimize real-time RT-PCR conditions and to generate standard curves in the real-time PCR assay. Primer concentrations and annealing temperatures were optimized for the iCycler iQ system (Bio-Rad) for each set of primers using the system's gradient protocol. PCR efficiencies were maintained between 92 and 105% and correlation coefficients above 0.98 for each primer set during optimization and also during the real-time PCR of sample DNA.
cDNA concentrations for genes of interest were examined by real-time quantitative PCR using an iCycler IQ System and the iQ SYBR green supermix (Bio-Rad). A standard curve was generated for each gene using tenfold dilutions of purified amplicons starting at 5 pg of cDNA and used later to calculate the starting amount of target cDNA in the unknown samples. SYBR green I is a general double-stranded DNA intercalating dye and may therefore detect nonspecific products and primer/dimers in addition to the amplicon of interest. In order to determine the number of products synthesized during the real-time PCR, a melting curve analysis was performed on each product. Real-time PCR was used to measure the starting amount of nucleic acid of each unknown sample of cDNA on the same 96-well plate.
GeneChip hybridization. RNA from three of macrophage-specific PPAR-γ-deficient mice (PPAR-γ flfl Cre+) and three of control (Cre−) littermates was processed and labeled according to the standard target labeling protocols, and the samples were hybridized, stained, and scanned per standard Affymetrix protocols at the Virginia Bioinformatics Institute (VBI) core laboratory on Mouse 430 2.0 expression arrays (Affymetrix, Santa Clara, CA).
Microarray data analysis. Quality checking of array hybridizations was done by Bioconductor package “ArrayQualityMatrics.” All of six sample results passed quality control. Data input and subsequent steps were performed using the Bioconductor package “affy.”39 Raw microarray data obtained from CEL files were pre-processed by the gcRMA algorithm (GC Robust Multiarray Average) and then log2 transformed using Bioconductor package “gcrma.”40 Only probe sets with at least one present call across all the compared hybridizations was kept for downstream statistical analysis. All statistical analyses of the data were performed within R statistical environment, Version 2.9.0,41 using Bioconductor packages.40 A cutoff of P<0.01 and ≥2-fold change was used as the criterion for whether or not a gene was significantly modulated by a treatment. All the 124 significantly modulated genes were submitted to Ingenuity Pathways Analysis (www.ingenuity.com) for pathway analysis and to GO:termfinder for gene ontology analysis. The microarray data (both raw and normalized) have been submitted to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, data set: GSE23421).
Statistics. Data were analyzed as a completely randomized design. To determine the statistical significance of the model, analysis of variance was performed using the general linear model procedure of Statistical Analysis Software (SAS, Cary, NC), and probability value (P) of <0.05 was considered to be significant. When the model was significant, analysis of variance was followed by Fisher's protected least significant difference multiple comparison method.
References
Hanauer, S.B. & Present, D.H. The state of the art in the management of inflammatory bowel disease. Rev. Gastroenterol. Disord. 3, 81–92 (2003).
Bassaganya-Riera, J. et al. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology 127, 777–791 (2004).
Desreumaux, P. & Dubuquoy, L. PPARgamma agonists as a new class of effective treatment for ulcerative colitis. Inflamm. Bowel. Dis. 15, 959–960 (2009).
Lewis, J.D. et al. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology 134, 688–695 (2008).
Marcy, T.R., Britton, M.L. & Blevins, S.M. Second-generation thiazolidinediones and hepatotoxicity. Ann. Pharmacother. 38, 1419–1423 (2004).
Desvergne, B. & Wahli, W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 20, 649–688 (1999).
Bassaganya-Riera, J. et al. Conjugated linoleic acid ameliorates viral infectivity in a pig model of virally induced immunosuppression. J. Nutr. 133, 3204–3214 (2003).
Guri, A.J., Hontecillas, R. & Bassaganya-Riera, J. Abscisic acid ameliorates experimental IBD by downregulating cellular adhesion molecule expression and suppressing immune cell infiltration. Clin. Nutr. e-pub ahead of print 15 March 2010.
Guri, A.J., Evans, N.P., Hontecillas, R. & Bassaganya-Riera, J. T cell PPAR gamma is required for the anti-inflammatory efficacy of abscisic acid against experimental IBD. J. Nutr. Biochem. (in press) (2010).
Hontecillas, R. et al. Nutritional regulation of porcine bacterial-induced colitis by conjugated linoleic acid. J. Nutr. 132, 2019–2027 (2002).
Bassaganya-Riera, J. & Hontecillas, R. CLA and n-3 PUFA differentially modulate clinical activity and colonic PPAR-responsive gene expression in a pig model of experimental IBD. Clin. Nutr. 25, 454–465 (2006).
Hontecillas, R. & Bassaganya-Riera, J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J. Immunol. 178, 2940–2949 (2007).
Evans, N.P., Misyak, S.A., Schmelz, E.M., Guri, A.J., Hontecillas, R. & Bassaganya-Riera, J. Conjugated linoleic acid ameliorates inflammation-induced colorectal cancer in mice through activation of PPARgamma. J. Nutr. 140, 515–521 (2010).
Guri, A.J., Mohapatra, S.K., Horne, W.T. II, Hontecillas, R. & Bassaganya-Riera, J. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol. 10, 60 (2010).
Adachi, M. et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut 55, 1104–1113 (2006).
Mohapatra, S.K. et al. Immunoregulatory actions of epithelial cell PPAR gamma at the colonic mucosa of mice with experimental inflammatory bowel disease. PLoS One 5, e10215 (2010).
Wendelsdorf, K., Bassaganya-Riera, J., Hontecillas, R. & Eubank, S. Model of colonic inflammation: immune modulatory mechanisms in inflammatory bowel disease. J. Theor. Biol. 264, 1225–1239 (2010).
Shah, Y.M., Morimura, K. & Gonzalez, F.J. Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G657–G666 (2007).
Boismenu, R. & Havran, W.L. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science 266, 1253–1255 (1994).
Bassaganya-Riera, J. & Hontecillas, R. Dietary conjugated linoleic acid and n-3 polyunsaturated fatty acids in inflammatory bowel disease. Curr. Opin. Clin. Nutr. Metab. Care 13, 569–573 (2010).
Vowinkel, T. et al. CD40-CD40 ligand mediates the recruitment of leukocytes and platelets in the inflamed murine colon. Gastroenterology 132, 955–965 (2007).
Lapaque, N. et al. Differential inductions of TNF-alpha and IGTP, IIGP by structurally diverse classic and non-classic lipopolysaccharides. Cell. Microbiol. 8, 401–413 (2006).
Dubuquoy, L. et al. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology 124, 1265–1276 (2003).
Odegaard, J.I. et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007).
Pascual, G. et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763 (2005).
Ricote, M. et al. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 95, 7614–7619 (1998).
Ricote, M., Li, A.C., Willson, T.M., Kelly, C.J. & Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391, 79–82 (1998).
Lewis, J.D. et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis. Am. J. Gastroenterol. 96, 3323–3328 (2001).
Sun, H. et al. Peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, suppresses CD40 expression and attenuates inflammatory responses after lithium pilocarpine-induced status epilepticus in rats. Int. J. Dev. Neurosci. 26, 505–515 (2008).
Hu, Q. et al. Peroxisome proliferator-activated receptor-gamma1 gene therapy attenuates atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Hum. Gene Ther. 19, 287–299 (2008).
Zhang, Y.J. et al. Effect of 15d-PGJ2 on the expression of CD40 and RANTES induced by IFN-gamma and TNF-alpha on renal tubular epithelial cells (HK-2). Am. J. Nephrol. 26, 356–362 (2006).
Liu, Z. et al. Hyperexpression of CD40 ligand (CD154) in inflammatory bowel disease and its contribution to pathogenic cytokine production. J. Immunol. 163, 4049–4057 (1999).
Battaglia, E., Biancone, L., Resegotti, A., Emanuelli, G., Fronda, G.R. & Camussi, G. Expression of CD40 and its ligand, CD40L, in intestinal lesions of Crohn's disease. Am. J. Gastroenterol. 94, 3279–3284 (1999).
Polese, L. et al. The role of CD40 in ulcerative colitis: histochemical analysis and clinical correlation. Eur. J. Gastroenterol. Hepatol. 14, 237–241 (2002).
Carlsen, H.S., Yamanaka, T., Scott, H., Rugtveit, J. & Brandtzaeg, P. The proportion of CD40+ mucosal macrophages is increased in inflammatory bowel disease whereas CD40 ligand (CD154)+ T cells are relatively decreased, suggesting differential modulation of these costimulatory molecules in human gut lamina propria. Inflamm. Bowel. Dis. 12, 1013–1024 (2006).
Borcherding, F. et al. The CD40-CD40L pathway contributes to the proinflammatory function of intestinal epithelial cells in inflammatory bowel disease. Am. J. Pathol. 176, 1816–1827 (2010).
Matsutani, T., Anantha Samy, T.S., Kang, S.C., Bland, K.I. & Chaudry, I.H. Mouse genetic background influences severity of immune responses following trauma-hemorrhage. Cytokine 30, 168–176 (2005).
Bassaganya-Riera, J., Misyak, S., Guri, A.J. & Hontecillas, R. PPAR gamma is highly expressed in F4/80(hi) adipose tissue macrophages and dampens adipose-tissue inflammation. Cell. Immunol. 258, 138–146 (2009).
Gautier, L., Cope, L., Bolstad, B.M. & Irizarry, R.A. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2004).
Gentleman, R.C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004).
Willems, F., Vollstedt, S. & Suter, M. Phenotype and function of neonatal DC. Eur. J. Immunol. 39, 26–35 (2009).
Acknowledgements
This study was supported by award number 5R01AT004308 of the National Center for Complementary and Alternative Medicine at the National Institutes of Health awarded to J.B.-R., European Commission grant number 224836, the Ramon y Cajal Program, the Virginia Bioinformatics Institute-Fralin CRI grants program to J.B.-R., NIAID Contract No. HHSN272200900040C to B.W.S., Contract No. HHSN2722020000561 to J.B.-R. and funds from the Nutritional Immunology and Molecular Medicine Laboratory.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Rights and permissions
About this article
Cite this article
Hontecillas, R., Horne, W., Climent, M. et al. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunol 4, 304–313 (2011). https://doi.org/10.1038/mi.2010.75
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.75
This article is cited by
-
Artificial intelligence-rationalized balanced PPARα/γ dual agonism resets dysregulated macrophage processes in inflammatory bowel disease
Communications Biology (2022)
-
Peroxisome proliferator-activator receptor γ and psoriasis, molecular and cellular biochemistry
Molecular and Cellular Biochemistry (2022)
-
Systems pharmacology approach uncovers Ligustilide attenuates experimental colitis in mice by inhibiting PPARγ-mediated inflammation pathways
Cell Biology and Toxicology (2021)
-
Inhibition of the activation of γδT17 cells through PPARγ–PTEN/Akt/GSK3β/NFAT pathway contributes to the anti-colitis effect of madecassic acid
Cell Death & Disease (2020)
-
Identification of new regulatory genes through expression pattern analysis of a global RNA-seq dataset from a Helicobacter pylori co-culture system
Scientific Reports (2020)

{kind=link}
{kind=link}
{kind=link}