
Abstract
The absolute requirement of the pulmonary immune system is to limit the inflammatory consequences of inhaled infectious agents while maintaining tolerance to harmless aeroallergens. This tolerance is maintained by a complex network of cells and molecules interacting with lung stromal cells. However, in some individuals there is a breakdown in tolerance to particles such as pollens, animal dander, or dust, resulting in the development of allergic pathology. Emerging evidence suggests that this breakdown in tolerance is influenced by the genetic background of individuals as well as environmental considerations such as early exposure to respiratory pathogens. Further understanding of the mechanisms used by the pulmonary immune system to maintain tolerance might result in exploitation of novel avenues for therapy to treat the growing number of chronic asthmatic patients.
Similar content being viewed by others


Main
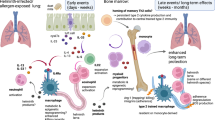
The lung is a unique mucosal site, with its huge surface area constantly being exposed to the external environment that contains vast amounts of antigenic material. Particles in the air that we breathe range from relatively large visible dust to very fine invisible particles that can penetrate deep within the lungs. Although these particles are potentially immunogenic, they do not usually represent a threat to the individual. Therefore, to prevent constant induction of de novo immune responses and development of memory effector cells that would precipitate chronic inflammation, a number of control mechanisms exist to promote immune tolerance. These pathways are vital because even a small amount of inflammation compromises respiratory function by limiting the surface area available for gas exchange. However, tolerance must be exquisitely controlled so that immune responses to pathogens are not compromised; thus, regulatory mechanisms must distinguish between harmless airborne particles and infectious agents. The complex network that makes this distinction comprises the pulmonary epithelium interacting with cells of the immune system, including regulatory T cells (Tregs), resident lung macrophages (Mφ), and cells of the innate immune system such as γδT cells (Figure 1).

Regulatory pathways in the lung maintain tolerance in the face of environmental exposure. Tolerance is maintained by a contribution from airway structural cells and their interaction with the complex network of immune cells and molecules. Loss of tolerance and development of inflammation is influenced by multiple factors including age, gender, obesity, infection history, atopic status, allergen exposure, nutrition (Vitamin D, A, E levels), which will affect the initiation and development of the allergic response.
How Pulmonary Immune System is Shaped Postnatally
Both the immune system and the lungs continue to develop after birth, and it is likely that early life events influence the pulmonary immune landscape. After birth, there is a rapid colonization of the neonatal skin and gut with microbial flora. Therefore, birth is characterized by a distinct pattern of innate immune molecules at the mucosal epithelia as well as the mobilization of acute-phase responses in the peripheral blood as a coordinated response aimed to limit infection while avoiding excessive inflammatory responses to microbial products.1 Murine studies have shown that expression of Toll-like receptor (TLR) 2 and TLR4 are undetectable in the immature fetus but increase several fold during prenatal development and after birth.2 Therefore, it is clear that early exposure to pathogens would affect TLR programming in the lung, but the influence of childhood infection on the specific development of allergic disease has been the matter of considerable debate. Although viral infection in early childhood is thought to be a risk factor for development of asthma, the “hygiene hypothesis” proposed that early childhood infections inhibit tendency to develop allergic disease,3 and hence early infection is actually protective. Epidemiologic evidence seems to support this hypothesis, as for those children living in developed countries, having several older siblings, early attendance at day care, and exposure to livestock are all associated with a lower incidence of allergic disease.4 However, most of the evidence relates to protection against atopy and atopic diseases rather than asthma itself. In addition, the evidence correlates bacterial infection or exposure with microbial products that would affect TLR programming in the lung. As the majority of the wheezing lower respiratory tract illnesses of childhood are caused by viruses, it has been postulated that early exposure to viruses has lasting effects on the shaping of pulmonary immune responses and are thus a risk factor for developing asthma.5, 6, 7
In addition to infection, other factors such as exposure to environmental pollution and dietary considerations,8, 9 including levels of zinc and vitamins D and E,10, 11 have recently been highlighted to influence the development of disease in early life. Increasingly, many of these factors have been implicated in the programming or development of regulatory pathways in early life. Irrespective of whether infection is beneficial or harmful for the development of asthma, it is clear that its influence is likely to be highly dependent on both the timing and nature of the infection and will have critical implications in the programming of the immune system. This programming is likely to take the form of regulation involving multiple cell types, including regulatory CD4+ T cells. However, direct evidence in patients is still lacking.
There is evidence that environmental factors may influence the neonatal immune system even before conception. Prenatal exposure to a farming environment influences innate immune patterning. Maternal exposure during pregnancy to an environment rich in microbial compounds was associated with higher levels of TLR2, TLR4, and CD14, implying that exposure might prevent sensitization of the children.12 Studies in mice have shown that exposure of mothers to endotoxin prevents subsequent allergen-induced sensitization and airway inflammation in the pups.13 Moreover, immunologic tolerance can be transferred from mother to her offspring if the mother is tolerized even before pregnancy, implying that even before conception the immune status of the mother is critical in defining the immune response of the offspring to allergens.14 Another environmental influence that affects a child's risk of developing asthma or atopy is the diet of the mother. Reduced maternal intake of vitamins D and E and zinc have all been associated with enhanced asthma symptoms in children,11, 15, 16 but a recent study has shown that dietary factors can also modify the risk of allergic airway disease through epigenetic mechanisms.8 Mice given a diet rich in methyl donors resulted in enhanced allergic airway disease that was inherited over multiple generations. This prenatal methyl-rich diet was postulated to promote DNA methylation and reduce transcriptional activity of genes associated with downregulation of allergic immune responses, such as Runx3.17 Prenatal maternal exposure to diets high in folates, vitamin B12, choline, and methionine—all of which provide methyl donors—as well as to cigarette smoke may repress gene transcription and promote asthma phenotypes.17 Although these studies have implicated regulatory factors such as interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) in the altered immune programming of the lung, there is little evidence that specific Treg populations are actually affected. In contrast, a recent elegant study examined whether exposure of lactating mice to an airborne allergen affected development of allergic airway disease in their progeny.18 Airborne antigens were transferred efficiently through breast milk and this transfer resulted in tolerance and protection from allergic asthma. Moreover, this breastfeeding-induced tolerance was dependent on the presence of TGF-β during lactation and was mediated by regulatory CD4+ T cells that signaled through TGF-β. These data provide a mechanism underlying tolerance provided by breastfeeding neonates and underpin the importance of maternal influences on the development of regulatory mechanisms in the neonate. Thus, there is an age-dependent maturation of the immune response after birth, but it is clear that the immune system is also shaped by events that may occur prenatally, and even preconception that may in turn affect how the immune system developed, and thus ultimately influence how the lungs respond to inhaled environmental allergens.
Maintenance of Pulmonary Immune Homeostasis
The pulmonary epithelium provides a barrier between the external inhaled environment and the internal tissues. The epithelium responds to microbes and noxious stimuli that overcome the mucociliary barrier and is thus vital for host defense. Within the epithelium, ciliated columnar, mucus-secreting goblet cells, and Clara cells that secrete surfactant adhere together to form a regulated, impermeable barrier due to the formation of tight junctions localized at the apical surface. This intercellular adhesion complex consists of interconnections of proteins and receptors, including ZO1-3, occludins, claudins, as well as transmembrane junctional adhesion molecules (β-catenin, E-cadherin, and Junctional adhesion molecules).19 During asthma, evidence shows that this barrier function is impaired, with disruption of tight junctions and increased epithelial permeability.20 Respiratory viruses, air pollutants, and proteolytically active allergens all have the capacity to inflict such damage on the epithelium, and are all associated with exacerbated clinical symptoms in asthma.
Apart from providing a physical barrier, the pulmonary epithelium is immunologically active and is pivotal in the development of immune responses in the lung. Pulmonary epithelial cells are able to secrete a wide range of cytokines and chemokines, but physically they are in intimate contact with the immune system, and there is growing evidence that they are also able to direct immune responses. Epithelial cells sense microbes through pattern recognition receptors (PRRs) that include TLRs,21 NOD-like receptors,22 protease-activated receptors (PARs) 1–4,23 and C-type lectins24 that recognize pathogen-associated molecular patterns from viruses, bacteria, fungi, protozoa, and multicellular parasites. Although PRR-triggered immune responses provide a critical antimicrobial monitoring system, activation of these receptors represents a critical link between homeostasis mucosal injury, and it is now clear that many allergens have the potential to modify the epithelial barrier through PRRs. They can also secrete a range of antimicrobial mediators including lysozyme, defensins, collectins, complement components, and other mediators that can break immunological tolerance. Many of the molecules secreted by epithelial cells in response to danger have the ability to regulate immune reactions and recruit cells of the innate and adaptive immune system. Therefore, the initiation and maintenance of inflammation at epithelial surfaces is induced by local mechanisms that have marked effects on the outcome of any immune response. Newly identified, epithelial-derived mediators, including IL-25, IL-33, and thymic stromal lymphopoietin, are critical upstream effectors of allergic sensitivity, triggered by TLR and PAR-receptor signaling, facilitating the breakdown of tolerance to allergens. Interestingly, IL-33 is capable of driving allergic inflammation even in the absence of IL-425 and promotes Th2 cell differentiation by programming the function of dendritic cells (DCs).26 IL-25 exacerbates airway hyperreactivity (AHR) in the absence of Th2 cytokines.27 The expression of thymic stromal lymphopoietin is induced by various stimuli including TLR,28, 29 PAR-2 triggering,30 and house dust mite (HDM) allergen,19 preceding DC infiltration.31 This group of innate proallergic cytokines is able to promote allergen-specific Th2 responses through DC recruitment and activation, but in addition drive pathology directly, even in the absence of Th2 cytokines.
The lung is equipped with a sophisticated network of DCs that serve as sentinels for pulmonary immune responses. Multiple subsets of DCs operate within the lung to control immune responses.32 Classical DCs are resident within the lung at steady state and coexist with plasmacytoid DCs that are thought to promote tolerance to inhaled antigens.33 During inflammatory episodes there is recruitment of both inflammatory DCs and interferon-producing killer DCs. Recent studies have determined that interaction between the DC community within the lung and pulmonary epithelial cells is vital for DC activation and migration to lymph nodes.34 Epithelial tight junctions regulate sampling of the airways by DCs, but this barrier can be breached through the proteolytic action of enzymatically active allergens such as Dermatophagoides pteronissinus (Der p) within HDM. Moreover, most allergens contain TLR agonists that also results in DC activation through TLRs on the epithelial surface. The functional importance of epithelial cell TLR signaling was recently explored using a series of bone marrow chimeric mice in which TLR4 was absent from either the stromal cells in the lung or the immune cells. Hammad et al.35 showed that it was the expression of TLR4 on the pulmonary epithelium that was vital to trigger DC activation, migration, and thus initiate an allergic response. Interestingly, Der p-2, a major allergen of HDM, has been shown to share functional homology with the lipopolysaccharide-binding adaptor MD-2, thus offering the possibility of facilitating TLR4 signaling to maximize allergic responses.36 Moreover, although experimental induction of asthma in TLR4-deficient mice led to diminished antigen-specific Th2 response, absence of the TLR adaptor molecule myeloid differentiation primary response gene 88 (MyD88) led to an additional reduction in the antigen-specific Th17 cells, suggesting that a combination of PRR signals may promote distinct allergic phenotypes.37 Stimulation of the respiratory epithelium by potent immunogenic β-glucan moieties contained within HDM has been shown to initiate the allergic cascade through ligation of non-TLRs, most likely dectin receptor-1 on epithelial cells, resulting in CCL20 (chemokine (C-C motif) ligand 20)-dependent recruitment of antigen-presenting DCs to the lung resulting in initiation of the allergic cascade.38 It is thought that the intrinsic protease activity associated with many allergens drives allergic responses through the cleavage of PAR-2 on the surface of epithelial cells.39, 40 For example, it has been shown that the HDM allergen Der p-1-induced cytokine release from respiratory epithelial cells is, in part, mediated by the activation of PAR-2.41 Collectively, these studies highlight the importance of interaction and cooperation between structural cells and immune cells in controlling lung immune responses.
Macrophages in the airways are long-lived residents of the airways that are involved in steady-state homeostasis. After activation, macrophages can be broadly classified in two main groups: classically activated macrophages (or M1), whose prototypical activating stimuli are interferon-γ and lipopolysaccharide, and alternatively activated macrophages (or M2), further subdivided in M2a (after exposure to IL-4 or IL-13), M2b (immune complexes in combination with IL-1β or lipopolysaccharide), and M2c (IL-10, TGF-β, or glucocorticoids).42 M1 show potent microbicidal properties and promote strong IL-12-mediated Th1 responses, whereas M2 support Th2-associated effector functions. Beyond infection, M2 polarized macrophages have a role in the resolution of inflammation through high endocytic clearance capacities and trophic factor synthesis, accompanied by reduced proinflammatory cytokine secretion.43 Alveolar macrophages (AMφs) are the predominant immune effector cells resident in both alveolar spaces and conducting airways. AMφs function to respond to inhaled antigens, but are also needed to be able to limit excessive inflammation to preserve pulmonary function. Therefore, it is thought that they have dual pro- and anti-inflammatory functions. Recent evidence suggests that subpopulations of macrophages account for this dual role. Lung interstitial macrophages (IMφs) can be distinguished from AMφs by their unique capacity to inhibit DC maturation after TLR stimulation, thus preventing sensitization to aeroallergen.44 Until recently, it was largely unknown how the lung environment instructs AMφs to suppress innate and adaptive immunity. Their close proximity with the epithelial surface of the lung implied potential influence for the epithelial cells on AMφs; nevertheless, only anecdotal studies were published regarding this.45 It is now clear that maintaining the hyporesponsive state of AMφs in the absence of antigen is achieved through several important lung-specific homeostatic pathways. In baseline conditions, AMφs closely adhere to epithelial cells through αvβ6, a TGF-β-inducible integrin.46 This tethering is rapidly lost during inflammation, permitting innate functions such as phagocytosis and secretion of proinflammatory cytokines.46, 47 Moreover, the inhibitory effect of AMφ is also mediated by binding of surfactant proteins to the inhibitory receptor signal regulatory protein-α.48 AMφs express high basal levels of the regulatory CD200 receptor (CD200R).49 Ligation of luminally expressed CD200 on the airway epithelium by CD200R on AMφs has a central role in homeostasis of the respiratory tract.49 These studies provide important insights into how macrophage innate immune function is closely regulated in the demanding environment of the lung.
Regulatory Cells
An absolute requirement of the pulmonary immune system is to maintain tolerance during constant antigenic exposure. Experimental systems have shown that delivery of simple proteins such as ovalbumin (OVA) promotes respiratory tolerance.50 Although intranasal instillation of OVA generated transient immunoglobulin E (IgE) responses, rechallenge of mice through the peritoneum resulted in significantly lower IgE responses compared with control mice.50 Similarly, repeated intranasal exposure of mice with low-dose OVA results in the development of antigen-specific Tregs, expressing membrane-bound TGF-β as well as Forkhead box P3 (FoxP3), a master regulator in the development and function of Tregs.51 A higher dose of OVA resulted in a CD4+ Treg population that suppressed allergic inflammation through secretion of IL-10.52 These Tregs represent an important mechanism of respiratory tolerance to inhaled allergens in both experimental animal systems and in asthmatic patients, and has been extensively reviewed recently.53 Major populations of Tregs studied in the context of pulmonary immune health include the naturally occurring thymus-derived CD4+FoxP3+ Tregs and peripherally antigen-induced adaptive CD4+ Treg cells, which may be either FoxP3 positive or negative.54
The importance of Treg populations in modulating allergic responses has been determined using murine models of allergic airway disease. Adoptive transfer of antigen-specific CD4+CD25+ Treg cells resulted in suppression of key features of the allergic response—including AHR, eosinophilic inflammation, and Th2 cytokine production.55 Moreover, transfer of these cells during a chronic allergic response also downregulated established airway inflammation and prevented the development of airway remodeling.56 Conversely, depletion of the CD4+CD25+ T-cell subset before allergen challenge was sufficient to enhance severity of allergic responses in the lung.57 Although antigen specificity is not a prerequisite of these cells,58 the ability to secrete IL-10 is, and they appear in the airways very rapidly after allergen challenge.59 Tregs have been found within the lung parenchyma and airway lumen as well as in the draining lymph nodes after inhaled allergen challenge (see Kearley et al.55 and Strickland et al.59). However, experiments designed to generate populations of CD4+IL-10+ Tregs in vivo showed that regulation (as determined by decreased antigen-specific T cells) occurred in the lung but not the draining lymph nodes (see Campbell et al.60). Appropriate localization of Tregs is important for effective function as suppressive activity may be through cell–cell contact as well as section of suppressor cytokines. The chemokines receptor CCR4 is essential for the recruitment of CD4+FoxP3+ Tregs in the lungs, and CCR4 deficiency in these cells results in severe lymphocytic infiltration of the lungs.61 Maintaining protective Treg activity in the lung is thought to be due to continued allergen exposure, as withdrawal of allergen results in a reduction in regulatory activity with subsequent resurgence of Th2-type pathology.59 It is likely that the traffic of Tregs and antigen-specific effector Th2 cells between the airway mucosa and the draining lymph node is critical for effective regulation, but further investigation is necessary to define mechanisms.
Investigation of regulation in the lung has focused primarily on CD4+ T-cell subsets; however, a number of other cells have the capacity for regulatory potential—generally through the expression of antiinflammatory receptors such as CTLA-4 (cytotoxic T-lymphocyte antigen 4) or PD1 (programmed death 1), or secretion of suppressive cytokines. These include novel Th1,62 CD8+ T cells,63 natural killer (NK) cells,64 B cells,65 mast cells, 66 and several antigen presenting cell (APC) populations.67 One of the most unique lung resident cells implicated in maintaining airway tolerance are the γδT cells, a distinct intraepithelial T-cell lymphocyte lineage enriched within the respiratory epithelium.68 The primary immunoregulatory capacity of γδT cells is maintenance of normal airway tone, and is shown in studies of with γδT cell-deficient mice that are naturally hyperresponsive upon airway challenge.69 Although the effect is independent of the Th2 response, the mechanism remains undefined.69 Paradoxically, γδT cells have also been reported to promote allergic responses in the airways.70
Such conflicting observations raise the question of how γδT cells identify antigens. Although there are some reports of bonafide γδTCR ligands, stimulation independent of the TCR has also been reported.71, 72, 73 Indeed, mismatch antigen studies have shown that the regulatory role of airway γδT cells is antigen dependent but not antigen specific.74 Moreover, γδT cells express PRR and thus have the potential to respond to pathogen-associated molecular pattern contaminants contained within allergen preparations or through damage-associated molecular pattern molecules from the airway epithelium in the allergic microenvironment.73, 75
Residency of γδT cells at epithelial borders presents an interface with both the host tissue and environment.76 γδT Cells may therefore set the threshold of activation required to render the airway epithelium “pro-asthmatic,” granting permission or “instructing” epithelial-derived mediators that signal DC activation. This location also fosters interaction with accessory cells such as Mφ and DCs, providing a means to amplify and elicit a broad spectrum of γδT cell functions.77 Interactions between γδT cells and DCs has a bidirectional functional influence on each cell, promoting antigen presentation and shaping the subsequent effector responses.78 Interestingly, interactions between γδT cells and other resident leukocytes have an essential role in lung homeostasis.79 Improved understanding of pulmonary γδT cells and the complexities underlying their relationship with the epithelium and interplay with resident leukocytes holds potential to understand susceptibility to allergic disease in humans.
Regulatory γδT cell responses that affect T effector immunity coexist with other, more potent, tolerance mechanisms and hence their primary purpose might be to subvert some of the damaging effects of inflammation rather than the response itself. Indeed, protection of host tissue against the damaging side effects of the established inflammatory response has been reported to be a property of airway γδT cells,80 leading to the concept that γδT cells possess a dual functionality that is instructed by the environmental context, i.e., homeostasis vs. that of an allergic lung after allergen provocation. The importance of γδT cell plasticity is supported by the large body of literature showing both the beneficial and deleterious effects of γδT cells during infectious inflammation and adequate tissue repair.81, 82, 83
NK T (NKT) cells constitute a unique and evolutionarily conserved population of T cells that express both a TCR and NK receptors.84 The evolutionarily conserved invariant NKT cell subset recognizes glycolipid antigens such as α-galactosylceramide presented by the major histocompatibility complex class I-related glycoprotein CD1d.84 NKT cells are considered versatile immunomodulators as they possess the ability to rapidly produce large amounts of Th1 and Th2 cytokines in response to TCR engagement.85 There is currently debate over whether NKT cells have effector or suppressor function in the allergic airway. Self-glycolipid antigens that are normally concealed in the respiratory tract may be exposed or expressed when allergens enter the lungs, in which they are recognized by NKT cells, causing activation. Indeed, NKT cells are present in the lungs of asthmatic individuals and produce high levels of IL-4 and IL-13, which is thought to potentiate the Th2 response that drive the development of asthma.86 Interestingly, the secondary immune response to α-galactosylceramide when compared with the primary is blunted, inducing a state of long-term anergy and impairing their ability to transactivate other cells.87 Invariant NKT cells appear to be a double-edged sword, which enhance or suppress disease course depending on the immune condition of host, but this requires further clarification, particularly in asthmatic patients.88
Breakdown in Tolerance Leads to Allergic Inflammation
Imbalance in pulmonary homeostatic control mechanisms can lead to chronic inflammatory disease. A loss of tolerance to innocuous inhaled particles may result in development of allergic inflammation, culminating in asthma. Asthma is a heterogeneous chronic inflammatory disease of the airways associated with AHR as well as structural changes to the lung termed airway remodeling. Approximately 30 million people in Europe have asthma, with the total cost of asthma in Europe being approximately €17.7 billion per year, and productivity lost to poor asthma control is estimated at €9.8 billion per annum (European Respiratory Society, White Book, 2003).
A combination of genetic and environmental factors is thought to influence the decision of whether inflammation resolves or progresses. Considering the position of the pulmonary epithelium as the barrier between the external environment and the internal tissues, functioning of this mucosal surface is likely to be influenced by a range of environmental factors that will affect both barrier and immune functions. Components of the inhaled environment are able to active PRRs on the epithelial surface, such as dust and particulate matter, as well as respiratory viral infections.28 Respiratory viral infections, particularly respiratory syncytial virus and rhinovirus, are common infections but have been shown to be risk factors for development of asthma, particularly after serial infections in infants.89, 90, 91 Although both infections are relatively mild, they are associated with development of allergic symptoms and exacerbations in chronic asthmatics. Both viruses infect the respiratory epithelium that elicits an antiviral response, including the production of inflammatory cytokines such as type I and II interferons.92, 93
Epidemiological studies suggest that exposure to environmental pollutants (such as cigarette smoke and car exhaust) is a risk factor for development of allergic disease, particularly in babies and young children.94, 95, 96 Diesel exhaust particles have an adjuvant effect for allergen challenge in animals and human subjects.97, 98, 99, 100 Short-term, low-level exposure to diesel exhaust particles promoted acute airway inflammation and AHR in allergic mice.101
It is thought that these environmental “triggers” initiate a series of reactions starting with the epithelium.102 The epithelium signals to the immune system through danger signals, such as chemokines, cathelicidins, and defensins, and cytokines, such as thymic stromal lymphopoietin, IL-33, and IL-25. In addition, communication with cells of the underlying mesenchyme is thought to occur through cytokines such as TGF-β, resulting in morphological changes to lung structural cells, particularly the airway smooth muscle cells.
Although the molecular mechanisms underlying epithelium–immune interactions are not well understood, it is increasingly recognized that the underlying pulmonary commensal landscape is likely to affect immune homeostasis and, therefore the development and chronicity of allergic responses. Although this has yet to be investigated comprehensively, there is some evidence to implicate microbiota in asthma etiology.103 Asymptomatic neonates whose throats are colonized with Streptococcus pneumoniae, Haemophilus influenzae, or Moraxella catarrhalis are at increased risk for recurrent wheeze and asthma early in life.104 These same bacteria have consistently been associated with exacerbations of both asthma and chronic obstructive pulmonary disease.105, 106 The response of asthmatics to antibiotics also suggests the importance of acute and chronic bacterial infections in the pathogenesis of disease.107 Epidemiological research has consistently indicated that a rich microbial environment in early life confers protection against the development of asthma,108 suggesting the need to understand the extent and nature of normal airway flora.
Asthma frequently occurs during childhood, and studies have shown that most school-age atopic asthmatics develop symptoms and show reduced lung function during infancy. Importantly, these children are born with normal lung function, which declines significantly by 3 years, and is maximally reduced by age 6, persisting through adolescence into adulthood.109, 110 Biopsies from asthmatic children show evidence of inflammation and structural abnormalities.111 Although not every child with wheeze will develop asthma, recurrent wheeze during the first 3 years of life is considered a major risk factor, particularly if there is a parental history of asthma. This early development of allergic symptoms occurs while the immune system and the lungs are still developing. Indeed, a recent study in vitro determined that there were intrinsic biochemical and functional differences in cells obtained from children with asthma compared with those from healthy controls.112 Epithelial cells from asthmatic children showed a less mature phenotype, with augmented release of anti-inflammatory mediators concomitant with reduced levels of TGF-β1. These results support the argument that epithelial cells in the asthmatic lung are fundamentally abnormal, even in the absence of overt inflammation.
Manipulation of Immune Tolerance for Clinical Benefit
Manipulation of mucosal tolerance has long been recognized as a promising approach to prevent or treat allergic responses. Antigen-specific immunotherapy (IT) involves incremental delivery of the allergen to which an individual is sensitive, to effectively suppress the clinical symptoms of allergy. The therapy, normally delivered through either the subcutaneous or sublingual (SL) route, is effective in inducing long-term remission upon allergen re-exposure,113 reducing new sensitizations,114 and preventing progression to asthma.115, 116 Introduced over one century ago,117 IT is the only current treatment that can offer protection against allergen-induced complaints even for long periods after treatment is finished.118, 119 Despite the validated effectiveness of IT from abundant clinical experience, IT has not become a mainstay treatment for allergy. Moreover, although IT is beneficial for treatment of rhinitis118 and insect venom allergy,120 it is less effective in allergic asthma and in individuals with multiple sensitivities, and seldom results in complete alleviation of all symptoms.121
The beneficial effects of specific IT is presumed to be mediated through regulation of allergic inflammation because of the generation of tolerance (Figure 2). Reported immunological mechanisms of effective IT include downregulation of Th2 response122 and immune deviation,123 blocking antibody production.124, 125, 126 However, generation of Tregs is considered to be largely responsible for clinically successful IT, driving these immunological mechanisms such as production of IgG4.127, 128 Recent efforts to understand the underlying immunological mechanisms offer fresh hope to improve IT as an effective treatment for perennial asthma. In particular, the application of improved recent understanding of immune tolerance, particularly with regard to Tregs, has been the focus of several recent studies.129 Indeed, increased proportions of regulatory CD4+CD25+ T cells were found in grass pollen allergics after IT.130 Upon stimulation ex vivo, increased IL-10 production was observed, highlighting a mechanism by which Tregs might exert an effect to suppress Th2 cells. Moreover, although IL-10+ T cells and TGF-β+ T cells are not a feature of the normal nasal mucosa, local elevations in these Treg populations were observed in IT-treated seasonal hay fever patients.131 Interestingly, these increases were not detectable outside of the pollen season, suggesting that local mucosal contact with allergen is necessary for the observed clinical effect.131 In the same set of biopsies, IL-10 and TGF-β were also found colocalized with Mφs, B cells, and DCs, implying that alternative sources for these cytokines may exist. Clinical efficacy of IT is associated with a blunting of allergen-specific IgE responses. Jutel et al.129 illustrated a role for both IL-10 and TGF-β in the Treg response to the perennial allergen HDM after IT. Tregs producing IL-10 and/or TGF-β have the potential to suppress local allergen-specific T cells and redirect antibody class switching in favor of IgG4 (an IL-10 isotype switch factor)132 or IgA (TGF-β isotype switch factor).133 More recently, TGF-β was shown to be a key early mediator of allergen-specific immunosuppression observed after clinically effective HDM IT.134 IgA is a crucial component of mucosal barrier and it has been shown that IgA production is associated with oral tolerance.135 Moreover, transient IgA deficiency has been proposed as a risk factor for IgE sensitization in early life.136 Interestingly, mucosal induction of protective allergen-specific IgA antibodies after successful IT has recently been shown,137 providing evidence of additional regulatory changes locally within the oral mucosa. This increase in local IgA correlates with enhanced TGF-β expression within the nasal mucosa,138 likely from either APCs or Tregs; thus, IgA represents an important component of protolerogenic state induced by IT.137

Immunological mechanisms of allergen-specific immunotherapy. Immunotherapy improves the clinical parameters of allergy via effects on a variety of immune cells. Skewing from a Th2 response, decreasing the humoral IgE response, increasing immune regulatory cells and mediators, and class-switching to blocking antibodies reduce the number and activation of mast cells and eosinophils to the site of allergen exposure.
Animal models of allergic airway disease have been used to investigate the immune mechanisms underlying IT. These studies are important to address safety concerns regarding widespread clinical use of IT and provide a means for developing novel strategies to potentiate IT regimens. Van Oosterhout et al.139 successfully developed a mouse model of allergic airway disease, in which an IT schedule, adopted from standard clinical IT regimens, suppressed allergen-induced airway inflammation and AHR. Immunologically, this gave a long-term suppressive effect that resulted from reduced allergen-specific IgE and Th2 activity,140 similar to results that have been observed in human studies.118 Interestingly, induction of IL-10+ Tregs, rather than immune deviation to a Th1 response, were pivotal to the success of specific IT in this model. The major drawback of such in vivo models is the dependence on priming through the peritoneum with a Th2-deviating adjuvant and use of OVA as a surrogate allergen. Exposure to antigen leads to its uptake, processing, and presentation by APCs that drive the Treg response.141 DC induction of Tregs can occur through several mechanisms, including the production of IL-10 or TGF-β.142, 143 A natural mechanism that creates local T-cell tolerance in DCs is through expression of the cytosolic tryptophan converting enzyme, indoleamine 2,3-dioxygenase.144, 145 After crosslinking of FcɛR1, indoleamine 2,3-dioxygenase is expressed in DCs and regulates immune responses, both directly through depletion of tryptophan and immune modulation by tryptophan metabolites that promote tolerance.146 Inhibition of indoleamine 2,3-dioxygenase during IT partially abrogated the suppressive effect,147 illustrating that tryptophan metabolites have a partial role in mediating immune tolerance in vivo, augmenting the suppressive effects of IT during administration of allergen-specific IT but not during post-IT allergen challenge.147 In contrast, administration of tryptophan during IT did not inhibit IT efficacy, indicating that it is the formation of metabolites rather than the actual depletion of tryptophan that is important. Antigen-loaded IL-10+ Mφs have also been identified to have a crucial role in the amelioration of allergic inflammation and disease observed during IT.148 In a subsequent study by the same researchers, intravenous administration of allergen-loaded Mφs led to a migration of these cells to the spleen and induction of a local, allergen-specific memory lymphocyte response, suggesting the induction of allergen-suppressive Tregs.149 These observations suggests that resident Mφs are not involved in the induction of specific antigen tolerance, highlighting the administration of allergen-loaded macrophages as a potential therapy to induce indirect suppression of allergen-specific disease.
IT has traditionally been administered through the subcutaneous route that permits both antigen-specific and long-lasting efficacy.118 However, widespread use of subcutaneous IT has been confounded by safety concerns regarding IgE-mediated side effects such as anaphylaxis.150 This imposes a rate-limiting effect and current research efforts have focused on alternatives strategies to improve the safety and efficacy. One such focus is the use of adjuvants and carriers to enhance the tolerogenic properties of DCs. In vitro cultures of human and murine cells have shown that delivery of IT sublingually in combination with vitamin D3 and dexamethasone enhanced suppression of AHR with an accompanying peripheral CD4+CD25+FoxP3+ Treg expansion to a significantly greater extent than allergen alone.151 Similarly, the potentiating effect of vitamin D on IT efficacy was also observed using an in vivo system in which improvements to experimentally induced allergic disease parameters were accompanied by elevated local IL-10 response and serum TGF-β, suggesting the induction of both Tr1 and Th3 regulatory populations.152
The sublingual route of IT (SLIT) is an attractive alternative method of administration. SLIT is used widely across Europe and is considered a safe and efficient treatment for respiratory allergies in both adults and children.153, 154 Furthermore, SLIT negates the requirement for specialist administration and inconvenience of repeated clinic attendance, thereby improving patient compliance.155 A recent study in patients with seasonal rhinitis confirmed a sustained clinical benefit of grass-pollen SLIT during 2 years of continuous treatment.156 Although meta-analyses support the clinical efficacy associated with SLIT,157 few clinical studies have included immunological analyses and a strong placebo effect has been reported.158 A recent randomized, double-blind placebo controlled trial of HDM SLIT showed induction of Tregs, IL-10, and TGF-β with concurrent suppression of allergen-specific T-cell proliferation.134 Although indicative of clinical efficacy, questions regarding optimal dose, efficacy, and safety remain. Improved understanding of the underlying mechanisms is therefore critical to better target allergen to the appropriate immune cells.
The local environment within the mouth is considered to have a degree of immune privilege, defaulting to induction of immunological tolerance while retaining the ability to mount effector responses.159 As such, there has been considerable interest in identifying lingual resident APCs and T cells to exploit this route for improved induction of allergen-specific tolerance. The lingual mucosa has a network of Langerhan-like cells with a relative paucity of effector cells compared with other mucosal sites. A recent in vivo study undertook an illuminating characterization of the lingual APC profile, identifying colocalization of CD11b+ myeloid cells with both effector and Tregs.160 However, a contribution from epithelial cells and monocytes to the natural tolerogenic capacity, as shown at other sites,131, 161 cannot be discounted. It is now important to functionally delineate the complex interactions between these cells to identify mechanisms that promote induction of tolerance over inflammation during the course of SLIT.
Another novel IT approach includes the development of peptide IT using CD+ T-cell epitopes to ameliorate antigen-specific inflammatory responses. Peptide IT has been the focus of intense clinical evaluation.162 One clinical study has shown induction of functional allergen-specific Tregs after peptide therapy.163 To reduce adverse reactions mediated by IgE crosslinking of mast cells, allergen-derived T-cell epitopes from the major cat allergen Fel d 1 were used. Intradermal delivery of these peptides to cat allergic patients resulted in bronchial hyporesponsiveness on rechallenge with allergen. Abrogation of symptoms in these patients was associated with a downregulation of Th2 responses to whole allergen and induction of allergen-specific T-regulatory pathways associated with increased IL-10.164 A mouse model designed to specifically mimic the human IT regimen determined that the peptide IT protocol resulted in the generation of CD4+IL-10+ Tregs, and that tolerance to subsequent rechallenge with allergen was dependent upon IL-10.60 Interestingly, these cells were only found in the lung and not in the draining lymph nodes, suggesting that these interactions take place primarily within the lungs. Further investigation of the mechanisms underlying tolerance after IT will likely improve the safety and efficacy of treatment and therefore would increase the suitability of IT for severe asthmatics.
Conclusions
Maintenance of immune homeostasis within the lung is mediated by complex interactions between the pulmonary immune system and lung stromal cells, primarily the lung epithelium. Preserving homeostasis involves discrimination between harmless inhaled particles and pathogens, but is vital as even a moderate degree of inflammation will compromise function. Breakdown in tolerance to innocuous aeroallergens occurs in susceptible individuals and results in asthma. Although it is recognized that development of asthma is dependent upon a variety of factors, including the genetic makeup of an individual as well as the environment in which we live, increasingly the importance of early life events, nutrition, and infection history all affect disease development. A further understanding of these factors will lead to improved therapies, particularly the ability to manipulate development of tolerance for patient benefit.
References
Levy, O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 7, 379–390 (2007).
Harju, K., Glumoff, V. & Hallman, M. Ontogeny of Toll-like receptors Tlr2 and Tlr4 in mice. Pediatr. Res. 49, 81–83 (2001).
Strachan, D.P. Hay fever, hygiene, and household size. BMJ 299, 1259–1260 (1989).
Wills-Karp, M., Santeliz, J. & Karp, C.L. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nat. Rev. Immunol. 1, 69–75 (2001).
Martinez, F.D. Viruses and atopic sensitization in the first years of life. Am. J. Respir. Crit. Care Med. 162, S95–S99 (2000).
Holt, P.G., Upham, J.W. & Sly, P.D. Contemporaneous maturation of immunologic and respiratory functions during early childhood: implications for development of asthma prevention strategies. J. Allergy Clin. Immunol. 116, 16–24 (2005).
Kusel, M.M. et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 119, 1105–1110 (2007).
Hollingsworth, J.W. et al. In utero supplementation with methyl donors enhances allergic airway disease in mice. J. Clin. Invest. 118, 3462–3469 (2008).
Patel, M.M. & Miller, R.L. Air pollution and childhood asthma: recent advances and future directions. Curr. Opin. Pediatr. 21, 235–242 (2009).
Litonjua, A.A. & Weiss, S.T. Is vitamin D deficiency to blame for the asthma epidemic? J. Allergy Clin. Immunol. 120, 1031–1035 (2007).
Devereux, G. Early life events in asthma--diet. Pediatr. Pulmonol. 42, 663–673 (2007).
Ege, M.J. et al. Prenatal farm exposure is related to the expression of receptors of the innate immunity and to atopic sensitization in school-age children. J. Allergy Clin. Immunol. 117, 817–823 (2006).
Gerhold, K. et al. Prenatal initiation of endotoxin airway exposure prevents subsequent allergen-induced sensitization and airway inflammation in mice. J. Allergy Clin. Immunol. 118, 666–673 (2006).
Polte, T., Hennig, C. & Hansen, G. Allergy prevention starts before conception: maternofetal transfer of tolerance protects against the development of asthma. J. Allergy Clin. Immunol. 122, 1022–1030 (2008).
Devereux, G. The increase in the prevalence of asthma and allergy: food for thought. Nat. Rev. Immunol. 6, 869–874 (2006).
Willers, S.M. et al. Maternal food consumption during pregnancy and the longitudinal development of childhood asthma. Am. J. Respir. Crit Care Med. 178, 124–131 (2008).
Miller, R.L. Prenatal maternal diet affects asthma risk in offspring. J. Clin. Invest. 118, 3265–3268 (2008).
Verhasselt, V. et al. Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat. Med. 14, 170–175 (2008).
Hammad, H. & Lambrecht, B.N. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat. Rev. Immunol. 8, 193–204 (2008).
Jiang, A. et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 27, 610–624 (2007).
Gribar, S.C., Richardson, W.M., Sodhi, C.P. & Hackam, D.J. No longer an innocent bystander: epithelial toll-like receptor signaling in the development of mucosal inflammation. Mol. Med. 14, 645–659 (2008).
Barrett, N.A. & Austen, K.F. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity 31, 425–437 (2009).
Lan, R.S., Stewart, G.A. & Henry, P.J. Role of protease-activated receptors in airway function: a target for therapeutic intervention? Pharmacol. Ther. 95, 239–257 (2002).
Haagsman, H.P., Hogenkamp, A., van, E.M. & Veldhuizen, E.J. Surfactant collectins and innate immunity. Neonatology 93, 288–294 (2008).
Kurowska-Stolarska, M. et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J. Immunol. 181, 4780–4790 (2008).
Rank, M.A. et al. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 123, 1047–1054 (2009).
Ballantyne, S.J. et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 120, 1324–1331 (2007).
Kato, A. & Schleimer, R.P. Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr. Opin. Immunol. 19, 711–720 (2007).
Lee, J.Y. et al. Murine thymic stromal lymphopoietin promotes the differentiation of regulatory T cells from thymic CD4(+)CD8(−)CD25(−) naive cells in a dendritic cell-independent manner. Immunol. Cell Biol. 86, 206–213 (2008).
Kouzaki, H., O'Grady, S.M., Lawrence, C.B. & Kita, H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J. Immunol. 183, 1427–1434 (2009).
Corrigan, C.J. et al. Early production of thymic stromal lymphopoietin precedes infiltration of dendritic cells expressing its receptor in allergen-induced late phase cutaneous responses in atopic subjects. Allergy 64, 1014–1022 (2009).
Lambrecht, B.N. & Hammad, H. Lung dendritic cells: targets for therapy in allergic disease. Handb. Exp. Pharmacol. 99–114 (2009).
de Heer, H.J. et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 200, 89–98 (2004).
Jakubzick, C. et al. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J. Exp. Med. 205, 2839–2850 (2008).
Hammad, H. et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 15, 410–416 (2009).
Trompette, A. et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 457, 585–588 (2009).
Phipps, S. et al. Toll/IL-1 signaling is critical for house dust mite-specific helper T cell type 2 and type 17 [corrected] responses. Am. J. Respir. Crit. Care Med. 179, 883–893 (2009).
Nathan, A.T., Peterson, E.A., Chakir, J. & Wills-Karp, M. Innate immune responses of airway epithelium to house dust mite are mediated through beta-glucan-dependent pathways. J. Allergy Clin. Immunol. 123, 612–618 (2009).
Ebeling, C. et al. Proteinase-activated receptor 2 activation in the airways enhances antigen-mediated airway inflammation and airway hyperresponsiveness through different pathways. J. Allergy Clin. Immunol. 115, 623–630 (2005).
Takizawa, T. et al. Abrogation of bronchial eosinophilic inflammation and attenuated eotaxin content in protease-activated receptor 2-deficient mice. J. Pharmacol. Sci. 98, 99–102 (2005).
Kauffman, H., Tamm, M., Timmerman, J.A. & Borger, P. House dust mite major allergens Der p 1 and Der p 5 activate human airway-derived epithelial cells by protease-dependent and protease-independent mechanisms. Clin. Mol. Allergy 4, 5 (2006).
Mantovani, A., Sica, A. & Locati, M. Macrophage polarization comes of age. Immunity 23, 344–346 (2005).
Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4, 583–594 (2004).
Bedoret, D. et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Invest. 119, 3723–3738 (2009).
Sato, K. et al. Type II alveolar cells play roles in macrophage-mediated host innate resistance to pulmonary mycobacterial infections by producing proinflammatory cytokines. J. Infect. Dis. 185, 1139–1147 (2002).
Takabayshi, K. et al. Induction of a homeostatic circuit in lung tissue by microbial compounds. Immunity 24, 475–487 (2006).
Morris, D.G. et al. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 422, 169–173 (2003).
Gardai, S.J. et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115, 13–23 (2003).
Snelgrove, R.J. et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 9, 1074–1083 (2008).
Holt, P.G., Batty, J.E. & Turner, K.J. Inhibition of specific IgE responses in mice by pre-exposure to inhaled antigen. Immunology 42, 409–417 (1981).
Ostroukhova, M. et al. Tolerance induced by inhaled antigen involves CD4(+) T cells expressing membrane-bound TGF-beta and FOXP3. J. Clin. Invest. 114, 28–38 (2004).
Akbari, O. et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat. Med. 8, 1024–1032 (2002).
Lloyd, C.M. & Hawrylowicz, C.M. Regulatory T cells in asthma. Immunity 31, 438–449 (2009).
Hawrylowicz, C.M. & O'Garra, A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat. Rev. Immunol. 5, 271–283 (2005).
Kearley, J., Barker, J.E., Robinson, D.S. & Lloyd, C.M. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 202, 1539–1547 (2005).
Kearley, J., Robinson, D.S. & Lloyd, C.M. CD4+CD25+ regulatory T cells reverse established allergic airway inflammation and prevent airway remodeling. J. Allergy Clin. Immunol. 122, 617–624 (2008).
Lewkowich, I.P. et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J. Exp. Med. 202, 1549–1561 (2005).
Leech, MD, Benson, R.A., De Vries, A., Fitch, P.M. & Howie, S.E. Resolution of Der p1-induced allergic airway inflammation is dependent on CD4+CD25+Foxp3+ regulatory cells. J. Immunol. 179, 7050–7058 (2007). Ref Type: Generic.
Strickland, D.H. et al. Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. J. Exp. Med. 203, 2649–2660 (2006).
Campbell, J.D. et al. Peptide immunotherapy in allergic asthma generates IL-10-dependent immunological tolerance associated with linked epitope suppression. J. Exp. Med. 206, 1535–1547 (2009).
Sather, B.D. et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J. Exp. Med. 204, 1335–1347 (2007).
Stock, P. et al. Induction of T helper type 1-like regulatory cells that express Foxp3 and protect against airway hyper-reactivity. Nat. Immunol. 5, 1149–1156 (2004).
Stock, P. et al. CD8(+) T cells regulate immune responses in a murine model of allergen-induced sensitization and airway inflammation. Eur. J. Immunol. 34, 1817–1827 (2004).
Korsgren, M. et al. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J. Exp. Med. 189, 553–562 (1999).
Singh, A. et al. Regulatory role of B cells in a murine model of allergic airway disease. J. Immunol. 180, 7318–7326 (2008).
Moon, T.C. et al. Advances in mast cell biology: new understanding of heterogeneity and function. Mucosal Immunol. 3, 111–128 (2010).
Ozdemir, C., Akdis, M. & Akdis, C.A. T regulatory cells and their counterparts: masters of immune regulation. Clin. Exp. Allergy 39, 626–639 (2009).
Boismenu, R. & Havran, W.L. Gammadelta T cells in host defense and epithelial cell biology. Clin. Immunol. Immunopathol. 86, 121–133 (1998).
Lahn, M. et al. Negative regulation of airway responsiveness that is dependent on gammadelta T cells and independent of alphabeta T cells. Nat. Med. 5, 1150–1156 (1999).
Zuany-Amorim, C. et al. Requirement for gammadelta T cells in allergic airway inflammation. Science 280, 1265–1267 (1998).
Chien, Y.H. & Konigshofer, Y. Antigen recognition by gammadelta T cells. Immunol. Rev. 215, 46–58 (2007).
Crowley, M.P. et al. A population of murine gammadelta T cells that recognize an inducible MHC class Ib molecule. Science 287, 314–316 (2000).
Groh, V., Steinle, A., Bauer, S. & Spies, T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 279, 1737–1740 (1998).
Jin, N. et al. Mismatched antigen prepares gamma delta T cells for suppression of airway hyperresponsiveness. J. Immunol. 174, 2671–2679 (2005).
Pietschmann, K. et al. Toll-like receptor expression and function in subsets of human gammadelta T lymphocytes. Scand. J. Immunol. 70, 245–255 (2009).
Wands, J.M. et al. Distribution and leukocyte contacts of gammadelta T cells in the lung. J. Leukoc. Biol. 78, 1086–1096 (2005).
Scotet, E., Nedellec, S., Devilder, M.C., Allain, S. & Bonneville, M. Bridging innate and adaptive immunity through gammadelta T-dendritic cell crosstalk. Front Biosci. 13, 6872–6885 (2008).
Dieli, F. et al. Reciprocal stimulation of gammadelta T cells and dendritic cells during the anti-mycobacterial immune response. Eur. J. Immunol. 34, 3227–3235 (2004).
Kirby, A.C., Newton, D.J., Carding, S.R. & Kaye, P.M. Pulmonary dendritic cells and alveolar macrophages are regulated by gammadelta T cells during the resolution of S. pneumoniae-induced inflammation. J. Pathol. 212, 29–37 (2007).
Jameson, J. & Havran, W.L. Skin gammadelta T-cell functions in homeostasis and wound healing. Immunol. Rev. 215, 114–122 (2007).
Huber, S.A., Born, W. & O'Brien, R. Dual functions of murine gammadelta cells in inflammation and autoimmunity in coxsackievirus B3-induced myocarditis: role of Vgamma1+ and Vgamma4+ cells. Microbes. Infect. 7, 537–543 (2005).
Andrew, E.M. & Carding, S.R. Murine gammadelta T cells in infections: beneficial or deleterious? Microbes. Infect. 7, 529–536 (2005).
Carding, S.R. & Egan, P.J. Gammadelta T cells: functional plasticity and heterogeneity. Nat. Rev. Immunol. 2, 336–345 (2002).
Taniguchi, M., Seino, K. & Nakayama, T. The NKT cell system: bridging innate and acquired immunity. Nat. Immunol. 4, 1164–1165 (2003).
Bendelac, A., Savage, P.B. & Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 25, 297–336 (2007).
Akbari, O. et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat. Med. 9, 582–588 (2003).
Parekh, V.V. et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J. Clin. Invest. 115, 2572–2583 (2005).
Matangkasombut, P. et al. Natural killer T cells in the lungs of patients with asthma. J. Allergy Clin. Immunol. 123, 1181–1185 (2009).
Singh, A.M., Moore, P.E., Gern, J.E., Lemanske, R.F. Jr . & Hartert, T.V. Bronchiolitis to asthma: a review and call for studies of gene-virus interactions in asthma causation. Am. J. Respir. Crit. Care Med. 175, 108–119 (2007).
Lemanske, J. & BUSSE, W.W. 6. Asthma: factors underlying inception, exacerbation, and disease progression. J. Allergy Clin. Immunol. 117, S456–S461 (2006).
Jackson, D.J. et al. Wheezing rhinovirus illnesses in early life predict asthma development in high risk children. Am. J. Respir. Crit. Care Med. 178 667–672 (2008).
Pala, P. et al. Enhanced IL-4 responses in children with a history of respiratory syncytial virus bronchiolitis in infancy. Eur. Respir. J. 20, 376–382 (2002).
Behera, A.K., Kumar, M., Lockey, R.F. & Mohapatra, S.S. 2′–5′ Oligoadenylate synthetase plays a critical role in interferon-gamma inhibition of respiratory syncytial virus infection of human epithelial cells. J. Biol. Chem. 277, 25601–25608 (2002).
Svartengren, M., Strand, V., Bylin, G., Jarup, L. & Pershagen, G. Short-term exposure to air pollution in a road tunnel enhances the asthmatic response to allergen. Eur. Respir. J. 15, 716–724 (2000).
McCreanor, J. et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N. Engl. J. Med. 357, 2348–2358 (2007).
Morgenstern, V. et al. Atopic diseases, allergic sensitization, and exposure to traffic-related air pollution in children. Am. J. Respir. Crit. Care Med. 177, 1331–1337 (2008).
Dong, C.C. et al. Effect of diesel exhaust particles on allergic reactions and airway responsiveness in ovalbumin-sensitized brown Norway rats. Toxicol. Sci. 88, 202–212 (2005).
Sagai, M., Furuyama, A. & Ichinose, T. Biological effects of diesel exhaust particles (DEP). III. Pathogenesis of asthma like symptoms in mice. Free Radic. Biol. Med. 21, 199–209 (1996).
de Haar, C.,, Hassing, I.,, Bol, M.,, Bleumink, R.,, Pieters, R. Ultrafine carbon black particles cause early airway inflammation and have adjuvant activity in a mouse allergic airway disease model. Toxicol Sci. 87, 409–418 (2005).
Diaz-Sanchez, D., Tsien, A., Fleming, J. & Saxon, A. Combined diesel exhaust particulate and ragweed allergen challenge markedly enhances human in vivo nasal ragweed-specific IgE and skews cytokine production to a T helper cell 2-type pattern. J. Immunol. 158, 2406–2413 (1997).
Hao, M., Comier, S., Wang, M., Lee, J.J. & Nel, A. Diesel exhaust particles exert acute effects on airway inflammation and function in murine allergen provocation models. J. Allergy Clin. Immunol. 112, 905–914 (2003).
Holgate, S.T., Roberts, G., Arshad, H.S., Howarth, P.H. & Davies, D.E. The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc. Am. Thorac. Soc. 6, 655–659 (2009).
Cookson, W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat. Rev. Immunol. 4, 978–988 (2004).
Bisgaard, H. et al. Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 357, 1487–1495 (2007).
Kraft, M. The role of bacterial infections in asthma. Clin. Chest Med. 21, 301–313 (2000).
Sethi, S., Evans, N., Grant, B.J. & Murphy, T.F. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N. Engl. J. Med. 347, 465–471 (2002).
Blasi, F., Johnston, S.L. The role of antibiotics in asthma. Int. J. Anti. Age. 29, 485–493 (2007).
Eder W, Ege MJ, von Mutius E. The asthma epidemic. N. Engl. J. Med. 355, 2226–35 (2006).
Phelan, P.D., Robertson, C.F. & Olinsky, A. The Melbourne Asthma Study: 1964–1999. J. Allergy Clin. Immunol. 109, 189–194 (2002).
Morgan, W.J. et al. Outcome of asthma and wheezing in the first 6 years of life: follow-up through adolescence. Am. J. Respir. Crit. Care Med. 172, 1253–1258 (2005).
Saglani, S. et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am. J. Respir. Crit Care Med. 176, 858–864 (2007).
Kicic, A., Sutanto, E.N., Stevens, P.T., Knight, D.A. & Stick, S.M. Intrinsic biochemical and functional differences in bronchial epithelial cells of children with asthma. Am. J. Respir. Crit. Care Med. 174, 1110–1118 (2006).
Till, S.J., Francis, J.N., Nouri-Aria, K. & Durham, S.R. Mechanisms of immunotherapy. J. Allergy Clin. Immunol. 113, 1025–1034 (2004).
Pajno, G.B., Barberio, G., De, L.F., Morabito, L. & Parmiani, S. Prevention of new sensitizations in asthmatic children monosensitized to house dust mite by specific immunotherapy. A six-year follow-up study. Clin. Exp. Allergy 31, 1392–1397 (2001).
Moller, C. et al. Pollen immunotherapy reduces the development of asthma in children with seasonal rhinoconjunctivitis (the PAT-study). J. Allergy Clin. Immunol. 109, 251–256 (2002).
Jacobsen, L. et al. Specific immunotherapy has long-term preventive effect of seasonal and perennial asthma: 10-year follow-up on the PAT study. Allergy 62, 943–948 (2007).
Kim, D.S. & Drake-Lee, A.B. Allergen immunotherapy in ENT: historical perspective. J. Laryngol. Otol. 117, 940–945 (2003).
Durham, S.R. et al. Long-term clinical efficacy of grass-pollen immunotherapy. N. Engl. J. Med. 341, 468–475 (1999).
Valenta, R. The future of antigen-specific immunotherapy of allergy. Nat. Rev. Immunol. 2, 446–453 (2002).
Bilo, M.B. et al. The VISYT trial: venom immunotherapy safety and tolerability with purified vs. nonpurified extracts. Ann. Allergy Asthma Immunol. 103, 57–61 (2009).
Jacobsen, L. & Valovirta, E. How strong is the evidence that immunotherapy in children prevents the progression of allergy and asthma? Curr. Opin. Allergy Clin. Immunol. 7, 556–560 (2007).
Ebner, C. et al. Immunological changes during specific immunotherapy of grass pollen allergy: reduced lymphoproliferative responses to allergen and shift from TH2 to TH1 in T-cell clones specific for Phl p 1, a major grass pollen allergen. Clin. Exp. Allergy 27, 1007–1015 (1997).
Gardner, L.M., Spyroglou, L., O'Hehir, R.E. & Rolland, J.M. Increased allergen concentration enhances IFN-gamma production by allergic donor T cells expressing a peripheral tissue trafficking phenotype. Allergy 59, 1308–1317 (2004).
Bellinghausen, I. et al. Insect venom immunotherapy induces interleukin-10 production and a Th2-to-Th1 shift, and changes surface marker expression in venom-allergic subjects. Eur. J. Immunol. 27, 1131–1139 (1997).
Wachholz, P.A., Soni, N.K., Till, S.J. & Durham, S.R. Inhibition of allergen-IgE binding to B cells by IgG antibodies after grass pollen immunotherapy. J. Allergy Clin. Immunol. 112, 915–922 (2003).
Wachholz, P.A. & Durham, S.R. Induction of “blocking” IgG antibodies during immunotherapy. Clin. Exp. Allergy 33, 1171–1174 (2003).
Akdis, M. & Akdis, C.A. Therapeutic manipulation of immune tolerance in allergic disease. Nat. Rev. Drug Discov. 8, 645–660 (2009).
Francis, J.N. et al. Grass pollen immunotherapy: IL-10 induction and suppression of late responses precedes IgG4 inhibitory antibody activity. J. Allergy Clin. Immunol. 121, 1120–1125 (2008).
Jutel, M. et al. IL-10 and TGF-beta cooperate in the regulatory T cell response to mucosal allergens in normal immunity and specific immunotherapy. Eur. J. Immunol. 33, 1205–1214 (2003).
Francis, J.N., Till, S.J. & Durham, S.R. Induction of IL-10+CD4+CD25+ T cells by grass pollen immunotherapy. J. Allergy Clin. Immunol. 111, 1255–1261 (2003).
Nouri-Aria, K.T. et al. Grass pollen immunotherapy induces mucosal and peripheral IL-10 responses and blocking IgG activity. J. Immunol. 172, 3252–3259 (2004).
Jeannin, P., Lecoanet, S., Delneste, Y., Gauchat, J.F. & Bonnefoy, J.Y. IgE versus IgG4 production can be differentially regulated by IL-10. J. Immunol. 160, 3555–3561 (1998).
Stavnezer, J. & Kang, J. The surprising discovery that TGF beta specifically induces the IgA class switch. J. Immunol. 182, 5–7 (2009).
O'Hehir, R.E. et al. House dust mite sublingual immunotherapy: the role for transforming growth factor-beta and functional regulatory T cells. Am. J. Respir. Crit Care Med. 180, 936–947 (2009).
Challacombe, S.J. & Tomasi, T.B. Jr . Systemic tolerance and secretory immunity after oral immunization. J. Exp. Med. 152, 1459–1472 (1980).
Taylor, B. et al. Transient IgA deficiency and pathogenesis of infantile atopy. Lancet 2, 111–113 (1973).
Scadding, G.W. et al. Sublingual grass pollen immunotherapy is associated with increases in sublingual Foxp3-expressing cells and elevated allergen-specific immunoglobulin G4, immunoglobulin A and serum inhibitory activity for immunoglobulin E-facilitated allergen binding to B cells. Clin. Exp. Allergy (2010).
Pilette, C. et al. Grass pollen immunotherapy induces an allergen-specific IgA2 antibody response associated with mucosal TGF-beta expression. J. Immunol. 178, 4658–4666 (2007).
van Oosterhout, A.J. et al. Allergen immunotherapy inhibits airway eosinophilia and hyperresponsiveness associated with decreased IL-4 production by lymphocytes in a murine model of allergic asthma. Am. J. Respir. Cell Mol. Biol. 19, 622–628 (1998).
Vissers, J.L. et al. Allergen immunotherapy induces a suppressive memory response mediated by IL-10 in a mouse asthma model. J. Allergy Clin. Immunol. 113, 1204–1210 (2004).
Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 3, 984–993 (2003).
Akbari, O., DeKruyff, R.H. & Umetsu, D.T. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2, 725–731 (2001).
Weiner, H.L. The mucosal milieu creates tolerogenic dendritic cells and T(R)1 and T(H)3 regulatory cells. Nat. Immunol. 2, 671–672 (2001).
Fallarino, F. et al. Murine plasmacytoid dendritic cells initiate the immunosuppressive pathway of tryptophan catabolism in response to CD200 receptor engagement. J. Immunol. 173, 3748–3754 (2004).
Belladonna, M.L. et al. Kynurenine pathway enzymes in dendritic cells initiate tolerogenesis in the absence of functional IDO. J. Immunol. 177, 130–137 (2006).
Munn, D.H. et al. Potential regulatory function of human dendritic cells expressing indoleamine 2, 3-dioxygenase. Science 297, 1867–1870 (2002).
Taher, Y.A. et al. Indoleamine 2, 3-dioxygenase-dependent tryptophan metabolites contribute to tolerance induction during allergen immunotherapy in a mouse model. J. Allergy Clin. Immunol. 121, 983–991 (2008).
Janssen, E.M., Wauben, M.H., Nijkamp, F.P., van, E.W. & van Oosterhout, A.J. Immunomodulatory effects of antigen-pulsed macrophages in a murine model of allergic asthma. Am. J. Respir. Cell Mol. Biol. 27, 257–264 (2002).
Janssen, E.M., van Oosterhout, A.J., Nijkamp, F.P., van, E.W. & Wauben, M.H. The efficacy of immunotherapy in an experimental murine model of allergic asthma is related to the strength and site of T cell activation during immunotherapy. J. Immunol. 165, 7207–7214 (2000).
Pajno, G.B. Sublingual immunotherapy: the optimism and the issues. J. Allergy Clin. Immunol. 119, 796–801 (2007).
Van, O.L. et al. IL-10-inducing adjuvants enhance sublingual immunotherapy efficacy in a murine asthma model. Int. Arch. Allergy Immunol. 145, 152–162 (2008).
Taher, Y.A., van Esch, B.C., Hofman, G.A., Henricks, P.A. & van Oosterhout, A.J. 1alpha, 25-dihydroxyvitamin D3 potentiates the beneficial effects of allergen immunotherapy in a mouse model of allergic asthma: role for IL-10 and TGF-beta. J. Immunol. 180, 5211–5221 (2008).
Passalacqua, G., Guerra, L., Pasquali, M., Lombardi, C. & Canonica, G.W. Efficacy and safety of sublingual immunotherapy. Ann. Allergy Asthma Immunol. 93, 3–12 (2004).
Pajno, G.B. et al. Sublingual immunotherapy abrogates seasonal bronchial hyperresponsiveness in children with Parietaria-induced respiratory allergy: a randomized controlled trial. Allergy 59, 883–887 (2004).
Valovirta, E., Passalacqua, G. & Canonica, W.G. Non-injection routes for immunotherapy of allergic diseases. Clin. Allergy Immunol. 18, 607–623 (2004).
Dahl, R. et al. Sublingual grass allergen tablet immunotherapy provides sustained clinical benefit with progressive immunologic changes over 2 years. J. Allergy Clin. Immunol. 121, 512–518 (2008).
Compalati, E., Penagos, M., Tarantini, F., Passalacqua, G. & Canonica, G.W. Specific immunotherapy for respiratory allergy: state of the art according to current meta-analyses. Ann. Allergy Asthma Immunol. 102, 22–28 (2009).
Pipet, A., Botturi, K., Pinot, D., Vervloet, D. & Magnan, A. Allergen-specific immunotherapy in allergic rhinitis and asthma. Mechanisms and proof of efficacy. Respir. Med. 103, 800–812 (2009).
Cuburu, N. et al. Sublingual immunization induces broad-based systemic and mucosal immune responses in mice. Vaccine 25, 8598–8610 (2007).
Mascarell, L. et al. Mapping of the lingual immune system reveals the presence of both regulatory and effector CD4+ T cells. Clin. Exp. Allergy 39, 1910–1919 (2009).
Calderon, M.A. et al. Allergen injection immunotherapy for seasonal allergic rhinitis. Cochrane Database Syst. Rev. 1, CD001936 (2007).
Larche, M. & Wraith, D.C. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat. Med. 11, S69–S76 (2005).
Haselden, B.M., Kay, A.B. & Larche, M. Immunoglobulin E-independent major histocompatibility complex-restricted T cell peptide epitope-induced late asthmatic reactions. J. Exp. Med. 189, 1885–1894 (1999).
Verhoef, A., Alexander, C., Kay, A.B. & Larche, M. T cell epitope immunotherapy induces a CD4+ T cell population with regulatory activity. PLoS Med. 2, e78 (2005).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article
Cite this article
Lloyd, C., Murdoch, J. Tolerizing allergic responses in the lung. Mucosal Immunol 3, 334–344 (2010). https://doi.org/10.1038/mi.2010.19
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.19
This article is cited by
-
Allergy immunotherapy restores airway epithelial barrier dysfunction through suppressing IL-25 -induced endoplasmic reticulum stress in asthma
Scientific Reports (2018)
-
CD8α+β− and CD8α+β+ plasmacytoid dendritic cells induce Foxp3+ regulatory T cells and prevent the induction of airway hyper-reactivity
Mucosal Immunology (2012)
-
Epicutaneous immunization with DNP‐BSA induces CD4+ CD25+ Treg cells that inhibit Tc1‐mediated CS
Immunology & Cell Biology (2012)
-
Innate immunity in the human lung: pathogen recognition and lung disease
Cell and Tissue Research (2011)
