
Abstract
The intestinal epithelial cell (IEC) is increasingly recognized to play a prominent role as an important intermediary between the commensal microbiota and the intestinal immune system. Moreover, it is now recognized that intestinal inflammation in inflammatory bowel disease (IBD) may arise primarily from IEC dysfunction due to unresolved endoplasmic reticulum (ER) stress as a consequence of genetic disruption of X box binding protein-1 function. In addition to primary (genetic) abnormalities of the unfolded protein response, a variety of secondary (inflammation and environmental) factors are also likely to be important regulators of ER stress. ER stress pathways are also well known to regulate (and be regulated by) autophagy pathways. Therefore, the host's ability to manage ER stress is likely to be a major pathway in the pathogenesis of intestinal inflammation that arises primarily from the IEC. Herein we discuss ER stress in the IEC as both an originator and perpetuator of intestinal inflammation in IBD.
Similar content being viewed by others


Introduction
The epithelium, by virtue of its unique geography and topology, lays at the interface between the largest collection of commensal microbes and hematopoietic cells that are assembled in the human body.1 This relationship between the microbiota, epithelium, and hematopoietic system is of central importance to understanding mucosal homeostasis, on the one hand, and inflammation, on the other. Over the past several decades, it has become increasingly recognized that the epithelium can be an active participant in the immune system.1 Although it has been well known for some time that the epithelium can both respond to and secrete a wide variety of immune mediators, over the past several years it has become increasingly recognized that the epithelium can integrate apical signals from the lumenal microbiota and basal signals from the hematopoietic system.2 Important studies from the groups of Cerutti and colleagues,3 Rescigno and colleagues,4 and Artis and colleagues5 have, for example, shown how epithelial cells can respond to luminal microbial signals and condition dendritic cells and consequently lymphocytes to elicit immune functions in particular. For example, in response to a luminal roundworm, Trichuris muris, the epithelium has been shown in model systems to secrete thymic stromal lymphopoietin to block Thelper (Th1) pathways.5, 6 What has been less clear is whether epithelial cells can primarily direct and therefore initiate inflammatory processes such as those associated with human inflammatory bowel disease (IBD). An interesting example of this possibility is through an examination of the unfolded protein response (UPR) due to endoplasmic reticulum (ER) stress.
ER stress and the UPR
The UPR is a cellular stress mechanism that allows cells to cope with conditions of ER stress.7, 8 ER stress arises from conditions that cause the accumulation of misfolded or unfolded proteins within the ER. These conditions include homeostatic pathways involved in the secretion of proteins by highly secretory cells such as plasma cells,9, 10, 11 hepatocytes,12 pancreatic acinar cells,13 plasmacytoid dendritic cells,14 and, as recently described, Paneth cells and, to a lesser extent, goblet cells.15 As such, these cell types can be envisioned to be especially susceptible to environmental and/or primary genetic factors that promote ER stress. The range of environmental factors that could conceivably lead to ER stress of the epithelium are potentially numerous and include those associated with bacteria, metabolic factors, drug effects, hypoxia, and inflammation itself, among others.16 All of these factors have the common feature of promoting the secretory activity of highly secretory epithelial cells and/or disrupting the environment of the ER thus inducing stress on the protein quality control machinery. As such, ER stress has been increasingly recognized to be both a secondary consequence of neoplasia17 and inflammation of the gastrointestinal tract18, 19, 20 as well as a potential primary factor in the initiation of intestinal inflammation.15
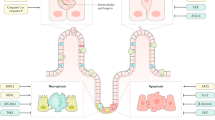
The UPR initiates the mechanisms that are required to resolve ER stress. These include several adaptive mechanisms, which induce a temporary halt in protein translation as well as induction of pathways that lead to the expansion of the secretory pathway of cells. On sustained and unabated ER stress, programmed cell death is a final common pathway.7, 8 Three proximal effectors of the UPR exist in mammalian cells that sense the accumulation of misfolded proteins. These include eukaryotic inositol-requiring transmembrane kinase-endoribonuclease-1 (IRE1) that has two isoforms (IRE1α and IRE1β), pancreatic ER kinase (PERK) and activated transcription factor 6 (ATF6).7, 8 Under homeostatic conditions in the absence of significant protein folding abnormalities, IRE1, PERK, and ATF6 exist in an inactive state through association with a chaperone, immunoglobulin heavy chain-binding protein (BiP), which is also known as glucose-regulated protein 78 (grp78). Grp78 senses the presence of misfolded proteins and releases IRE1, PERK, and ATF6 from inhibition resulting in transcriptional programs, which allow a cell to cope with such stress (Figure 1). The cellular survival or death pathways that are controlled by IRE1, PERK, and ATF6 are mediated by downstream transcription factors. When released from grp78, PERK is autophosphorylated and phosphorylates and thus inhibits eukaryotic translation-initiation factor 2-α (eIF2-α), resulting in the cessation of translation. The mRNA encoding activating transcription factor 4 (ATF4) as well as ATF5 are, however, resistant to eIF2-α inhibition allowing for ATF4 (and ATF5) production and activation of their own transcriptional programs. Similarly, on release from grp78-mediated suppression, ATF6 is mobilized from the ER to the Golgi apparatus where its cytoplasmic tail is subject to cleavage by site 1 and site 2 proteases (S1P and S2P), resulting in release of the ATF6 cytoplasmic tail (ATF6-N), which is transcriptionally active. ATF4 and ATF6-N control cell death largely through the transcription factor CHOP (CCAAT/enhancer-binding homologous protein), during the chronic phases of ER stress.

Multiple proximal effectors of ER stress response. Please see text for specific details. Adapted from reference.7 The transcriptional program of each factor are in the yellow areas.
The most evolutionarily conserved of these three pathways is the kinase-endoribonuclease known as the ubiquitously expressed IRE1α and its intestinal epithelial specific isoform, IRE1β. Activation through oligomerization and autophosphorylation on release from grp78 results in the excision of a 26-base pair fragment from the mRNA of unspliced XBP1 (XBP1u) by an unconventional splicing event that generates a spliced isoform of XBP1 with an altered reading frame (XBP1s).21, 22, 23 In contrast to XBP1u, which is transcriptionally inert, XBP1s is a potent inducer of a subset of UPR target genes. XBP1s is required for ER expansion and the development and survival of a variety of secretory cells as previously noted as well as the adaptation of cells to a variety of stressful tissue environments such as that associated with hypoxia and calcium and glucose deprivation, among others.
XBP1, paneth cells and intestinal inflammation
The existence of an important link between cell-intrinsic ER stress and organ-specific inflammation has recently been reported within the intestines.15 Specifically, conditional deletion of XBP1 in the intestinal epithelium of mice was shown to result in the spontaneous development of intestinal inflammation in the small intestine that presented with hallmarks of human IBD including crypt abscesses, leukocytic infiltration, and frank ulcerations. Remarkably, even the deletion of one XBP1 allele was sufficient to induce spontaneous enteritis in a significant portion of the animals. Moreover, hypomorphic XBP1 with the loss of one allele also led to Paneth cell dysfunction and an increased susceptibility to DSS colitis and deletion of both alleles resulted in apoptotic cell death of Paneth cells. Paneth cells are one of the four lineages of epithelial cells differentiating from intestinal epithelial stem cells that reside at the base of the crypt.24 Paneth cells secrete abundant amounts of antimicrobial peptides (α-defensins or cryptdins) and hence are predicted to regulate the complex intestinal microbiota and/or its adherence to the intestinal epithelium.25 Indeed, NOD2−/−, CD1d−/−, and MyD88−/− mice all display structural or functional alterations of Paneth cells together with an alteration of the composition of the intestinal microbiota or increased adherence of the microbiota with the intestinal epithelium.25, 26, 27 Moreover, Paneth cells may also be considered as an inflammatory cell type capable of secreting numerous pro-inflammatory mediators including tumor necrosis factor (TNF), leptin, and adiponectin.28 Moreover, several primary human genetic risk factors have recently been appreciated to affect Paneth cell function including NOD227, 29 and ATG16L1,30 which are associated with intracellular bacterial sensing31, 32 and autophagy,30, 33 respectively. It is therefore interesting that mice that are hypomorphic in XBP1 function,15 similar to NOD2-deficient mice,27 also show a marked defect in handling an oral model pathogen (Listeria monocytogenes). These data predict that XBP1 and ER stress pathways through their effect on the antimicrobial activity of the epithelium may have a substantial impact on the composition of the intestinal microbiota and thus influence susceptibility to IBD.
In contrast, to NOD2-deficient27 or ATG16L1-hypomorphic30 mice that do not exhibit spontaneous inflammation or increased sensitivity to DSS administration in the case of NOD2,27 XBP1−/− mice exhibit spontaneous intestinal inflammation within the small intestine.15 As Paneth cell loss alone does not result in spontaneous inflammation,34, 35 we have previously hypothesized that by virtue of its effects on both of the major drivers of IBD (i.e., the intestinal microbiota and immunological tone of the gut), XBP1 within the epithelium may have a unique function in the pathogenesis of IBD.36, 37 Indeed, hypomorphic function of XBP1 in the epithelium results in ER stress under physiologic conditions as manifest by an upregulation of grp78 and a profound induction of IRE1 activity.15 As a consequence of this, JNK15 activity is markedly increased resulting in an increased sensitivity of the stressed epithelial cell to bacterial ligands important to IBD such as flagellin38 and cytokine signals such as TNF. Consistent with these observations, it has been reported earlier that IRE1β−/− mice are also more susceptible to DSS colitis compared with wild-type controls.39 Whereas IRE1α is an ubiquitous isoform, IRE1β is an intestinal epithelial cell specific isoform of IRE139 emphasizing the importance of the IRE1-XBP1 pathway to homeostasis of the intestinal epithelium.
ER stress and goblet cells
In addition to Paneth cells, goblet cells are also among the most highly secretory cells within the intestinal epithelium and are responsible for the majority of mucous glycoproteins as well as soluble mediators involved in host defense such are RELMβ and trefoil factors.40, 41 Consistent with this, it has also been observed that loss of XBP1 function in intestinal epithelium leads to an ∼30% reduction in the number of goblet cells in the small intestine but interestingly not in the colon.15 This is also likely through induction of programmed cell death as observed with Paneth cells. The relation between goblet cells, ER stress, and inflammation is unclear although goblet cells or their products have been linked to inflammation. Muc2−/− mice have, for example, been observed to develop spontaneous colitis.42 Similarly, intestinal trefoil factor-deficient mice exhibit increased susceptibility to DSS colitis.43 However, given the absence of spontaneous inflammation in Math1-deficient,44 Gfi-deficient,45 and ITF-diptheria toxin transgenic46 mice that lack goblet cells, we have recently hypothesized that goblet cells that lack the ability to secrete mucus but retain the ability to secrete pro-inflammatory factors may promote inflammation.16 This is supported by observations that mice with abnormalities in MUC2 protein folding due to point mutations that affect protein structure (so-called Winnie and Eeyore mice) exhibit spontaneous inflammation in association with an exaggerated ER stress response.20 It is not known, however, whether the ER stress that is observed in these mice is due to the inflammation that results from the absence of functional mucins and/or primarily derived from ER stress in the goblet cells caused by the abnormally folded mucin proteins. However, both are possibilities given observations that ER stress can be detected in the colitis associated with IL-10 deficiency18 and in HLA-B27 transgenic rats.47, 48, 49 In the latter case, the HLA-B27 protein is particularly prone to misfolding,50 making it possible that the inflammation observed in these mice may primarily derive from an ER stress response as observed in XBP1−/− mice. Taken together, these data suggest that goblet cells may be both a target of ER stress in the context of inflammation or a direct participant in the development of inflammation as a consequence of ER stress.
ER stress and human IBD
Similar to rodents, studies in humans also suggest that ER stress can both be a primary cause of intestinal inflammation (e.g., as observed in XBP1−/− mice) or a consequence of inflammation (e.g., as observed in IL-10−/− mice). Studies by several investigators have shown evidence of ER stress based on increased grp78 expression and XBP1-splicing in a significant proportion of both UC and CD subjects, suggesting that ER stress is a common consequence of intestinal inflammation.15, 18, 20 In addition, polymorphisms have been detected in the XBP1 gene on chromosome 22 that are associated with increased risk for the development of both CD and UC.15 Linkage disequilibrium at the XBP1 locus is remarkably low, indicating that the XBP1 locus possesses a particularly complex genetic architecture such that multiple rare (private) single nucleotide polymorphisms (SNPs) might represent the basis for the locus association. Deep sequencing of >1,000 patients and controls indeed found three-fold more rare SNPs in CD and UC patients compared with healthy controls. Of the five rare non-synonymous (ns) SNPs discovered, four were only detectable in IBD patients but not controls, whereas the fifth occurred at equal frequency in CD, UC, and controls.15 Functional in vitro studies with constructs engineered to contain these SNPs revealed that the IBD-only nsSNP XBP1-constructs yielded decreased transactivation of XBP1-regulated UPR target genes and decreased activation of an UPR element reporter.15 In contrast, the non-IBD-associated nsSNP construct was indistinguishable from wild-type XBP1 constructs.15 Hence, the hypomorphic function of the IBD-associated rare nsSNP variants of XBP1 in vitro are consistent with the spontaneous intestinal inflammation observed in the mouse model as a consequence of Xbp1 deletion. Taken together, XBP1 might not only have a primary causative function in IBD in select patients harboring disease-associated variants within the XBP1 locus, but these mechanisms might also perpetuate inflammation originating from other causes, both genetic and environmental.
The relationship between ER stress pathways and other mechanisms for the development of IBD
Recent immunogenetic studies in IBD have identified a number of potential overarching mechanisms that distill into the inflammation characterized by CD and UC. These include pathways that are associated with innate immunity and specifically intracellular bacterial sensing through pattern recognition receptors (e.g., NOD2) and autophagy (e.g., ATG16L1 and IRGM).31, 51, 52 It is therefore interesting to consider the possibility that these pathways may indirectly or directly intersect with the UPR associated with ER stress. With regard to the former possibility, abnormal ER stress pathways may result in alterations of the microbial milieu15 and, as a corollary, abnormalities in the microbial milieu that may emanate from abnormalities in risk factors such as NOD2,53 or perhaps XBP1,15 may adversely affect ER stress pathways. It is now clear that microbes themselves can directly affect ER stress pathways and XBP1 in particular. Bacteria such as Streptomyces sp. are known to secrete small molecules such as macrocytic lactams (e.g., trierixin), which directly inhibit XBP1 activation.54 Similarly, Shiga toxigenic Escherichia coli secrete AB5 cytotoxins that activate ER stress pathways by direct cleavage of grp78.55 These and other examples show how there may be a bidirectional interaction between ER stress pathways within the epithelium and the microbial communities contained within the lumen, suggesting that different genotypes may converge phenotypically on ER stress pathways via effects on the intestinal microbiota.
Similarly, ER stress through IRE1-mediated JNK activation or PERK-mediated eIF2α phosphorylation may be an activator of autophagy based on studies in in vitro model systems.56, 57 Macroautophagy, which represents a lysosomal pathway involved in the turnover of cellular macromolecules and organelles that is involved in cellular homeostasis and anti-microbial defense, is now recognized as a major biologic pathway in the development of human CD. Polymorphisms in several genes associated with autophagy such as ATG16L1,51 IRGM52 and potentially LRRK258 have been identified as risk factors for the development of CD and interestingly, similar to XBP1, converge on the Paneth cell.31, 59 It can thus be predicted that ER stress mechanisms and autophagic pathways may interact at a functional level within the intestinal epithelium in vivo and may be synergistically related to inflammation.
Conclusion and model
In summary, a proper ER stress response in the intestinal epithelium is likely to be necessary to maintain homeostasis. Hypomorphic XBP1 function specifically within the intestinal epithelium results in Paneth cell, and to a lesser extent, goblet cell dysfunction that is associated with spontaneous intestinal inflammation in a pathway that involves IRE1 activation of JNK, and possibly NFκB, and consequently, downstream inflammatory pathways. As the human XBP1 locus contains polymorphisms that function as genetic susceptibility factors for both forms of IBD, it is now evident that ER stress represents a new pathway that involves the intestinal epithelium and from which intestinal inflammation may emerge. This is highly significant given the strategic location of the epithelium between the two major factors that determine intestinal inflammation; the luminal microbiota and the immunologic tone of mucosal tissues in response to the microbiota.1 These studies suggest that XBP1 may regulate the ability of the intestinal epithelium to both regulate and sense the composition of luminal microbiota. ER stress may thus be a major determinant of luminal microbial epithelial interactions and inflammatory tone and an important factor in determining whether homeostasis versus inflammation will occur. The ability to amount an appropriate ER stress response within the intestinal epithelium may therefore be involved in both the initiation and/or perpetuation of mucosal inflammation as it pertains to IBD. In this model, it can be envisioned that a number of secondary factors of environmental and/or host origin that can compromise an appropriate ER stress response can promote intestinal inflammation. Such factors may include bacterial toxins,54, 55 metabolic (dietary) factors,7, 8 neurohumoral (stress) factors, immunologic factors associated with inflammation that promote (TNFα, hypoxia) or prevent (IL-10)18 ER stress as well as other factors60 (Figure 2). Moreover, it can be hypothesized that the genetically determined ability to respond to these challenges through ER stress-related genes such as XBP1-related pathways and their relationship with other genetically important risk factors such as autophagy,30, 51, 52, 61 mucin proteins,20 HLA-proteins such as HLA-B2748 or other ER stress pathway components19 among others will be an important contributing factor in the initiation and/or perpetuation of intestinal inflammation.

Model: primary (genetic) and secondary (environmental) factors can drive ER stress in the intestinal epithelium and consequently inflammation.
Conflict of interest
The authors declared no conflict of interest.
References
Blumberg, R.S. et al. Recent insights into the integration of the intestinal epithelium within the mucosal environment in health and disease. Mucosal. Immunol. 1, 330–334 (2008).
Artis, D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat. Rev. Immunol. 8, 411–420 (2008).
Xu, W. et al. Epithelial cells trigger frontline immunoglobulin class switching through a pathway regulated by the inhibitor SLPI. Nat. Immunol. 8, 294–303 (2007).
Rimoldi, M. et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat. Immunol. 6, 507–514 (2005).
Zaph, C. et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature 446, 552–556 (2007).
Saenz, S.A., Taylor, B.C. & Artis, D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 226, 172–190 (2008).
Todd, D.J., Lee, A.H. & Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 8, 663–674 (2008).
Zhang, K. & Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462 (2008).
Lee, A.H., Iwakoshi, N.N., Anderson, K.C. & Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 100, 9946–9951 (2003).
Iwakoshi, N.N. et al. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4, 321–329 (2003).
Reimold, A.M. et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300–307 (2001).
Ozcan, U. et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 (2004).
Lee, A.H., Chu, G.C., Iwakoshi, N.N. & Glimcher, L.H. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 24, 4368–4380 (2005).
Iwakoshi, N.N., Pypaert, M. & Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 204, 2267–2275 (2007).
Kaser, A. et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008).
Kaser, A. & Blumberg, R.S. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin. Immunol. 21, 156–163 (2009).
Ma, Y. & Hendershot, L.M. The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4, 966–977 (2004).
Shkoda, A. et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology 132, 190–207 (2007).
Brandl, K. et al. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc. Natl. Acad. Sci. USA 106, 3300–3305 (2009).
Heazlewood, C.K. et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS. Med. 5, e54 (2008).
Lee, K.P. et al. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100 (2008).
Korennykh, A.V. et al. The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 (2009).
Calfon, M. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002).
Radtke, F. & Clevers, H. Self-renewal and cancer of the gut: two sides of a coin. Science 307, 1904–1909 (2005).
Vaishnava, S., Behrendt, C.L., Ismail, A.S., Eckmann, L. & Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 105, 20858–20863 (2008).
Nieuwenhuis, E.E. et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J. Clin. Invest. 119, 1241–1250 (2009).
Kobayashi, K.S. et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005).
Keshav, S. Paneth cells: leukocyte-like mediators of innate immunity in the intestine. J. Leukoc. Biol. 80, 500–508 (2006).
Wehkamp, J. et al. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc. Natl. Acad. Sci. USA 102, 18129–18134 (2005).
Cadwell, K. et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 (2008).
Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 8, 458–466 (2008).
Franchi, L., Warner, N., Viani, K. & Nunez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128 (2009).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008).
Garabedian, E.M., Roberts, L.J., McNevin, M.S. & Gordon, J.I. Examining the role of Paneth cells in the small intestine by lineage ablation in transgenic mice. J. Biol. Chem. 272, 23729–23740 (1997).
Wilson, C.L. et al. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 286, 113–117 (1999).
Ma, A. Unresolved ER stress inflames the intestine. Cell 134, 724–725 (2008).
Clevers, H. Inflammatory bowel disease, stress, and the endoplasmic reticulum. N. Engl. J. Med. 360, 726–727 (2009).
Lodes, M.J. et al. Bacterial flagellin is a dominant antigen in Crohn disease. J. Clin. Invest. 113, 1296–1306 (2004).
Bertolotti, A. et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Invest. 107, 585–593 (2001).
Linden, S.K., Sutton, P., Karlsson, N.G., Korolik, V. & McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal. Immunol. 1, 183–197 (2008).
McVay, L.D. et al. Absence of bacterially induced RELMbeta reduces injury in the dextran sodium sulfate model of colitis. J. Clin. Invest. 116, 2914–2923 (2006).
Van der Sluis, M. et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129 (2006).
Mashimo, H., Wu, D.C., Podolsky, D.K. & Fishman, M.C. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274, 262–265 (1996).
Yang, Q., Bermingham, N.A., Finegold, M.J. & Zoghbi, H.Y. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294, 2155–2158 (2001).
Shroyer, N.F., Wallis, D., Venken, K.J., Bellen, H.J. & Zoghbi, H.Y. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes Dev. 19, 2412–2417 (2005).
Itoh, H., Beck, P.L., Inoue, N., Xavier, R. & Podolsky, D.K. A paradoxical reduction in susceptibility to colonic injury upon targeted transgenic ablation of goblet cells. J. Clin. Invest. 104, 1539–1547 (1999).
Hammer, R.E., Maika, S.D., Richardson, J.A., Tang, J.P. & Taurog, J.D. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell 63, 1099–1112 (1990).
Turner, M.J. et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J. Immunol. 175, 2438–2448 (2005).
Tran, T.M. et al. Additional human beta2-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum. 54, 1317–1327 (2006).
Tran, T.M. et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J. Immunol. 172, 5110–5119 (2004).
Hampe, J. et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211 (2007).
Parkes, M. et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 39, 830–832 (2007).
Frank, D.N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 104, 13780–13785 (2007).
Tashiro, E. et al. Trierixin, a novel Inhibitor of ER stress-induced XBP1 activation from Streptomyces sp. 1. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. (Tokyo) 60, 547–553 (2007).
Paton, A.W. et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443, 548–552 (2006).
Ogata, M. et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231 (2006).
Yorimitsu, T., Nair, U., Yang, Z. & Klionsky, D.J. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299–30304 (2006).
Barrett, J.C. et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 40, 955–962 (2008).
Kaser, A. & Blumberg, R.S. Paneth cells and inflammation dance together in Crohn's disease. Cell. Res. 18, 1160–1162 (2008).
Yorimitsu, T. & Klionsky, D.J. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 3, 160–162 (2007).
Kuballa, P., Huett, A., Rioux, J.D., Daly, M.J. & Xavier, R.J. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS. ONE 3, e3391 (2008).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Kaser, A., Blumberg, R. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol 3, 11–16 (2010). https://doi.org/10.1038/mi.2009.122
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2009.122
This article is cited by
-
Pterostilbene attenuates intrauterine growth retardation-induced colon inflammation in piglets by modulating endoplasmic reticulum stress and autophagy
Journal of Animal Science and Biotechnology (2022)
-
Hyperglycemia-triggered ATF6-CHOP pathway aggravates acute inflammatory liver injury by β-catenin signaling
Cell Death Discovery (2022)
-
Age-dependent involvement of gut mast cells and histamine in post-stroke inflammation
Journal of Neuroinflammation (2020)
-
MicroRNA-674-5p induced by HIF-1α targets XBP-1 in intestinal epithelial cell injury during endotoxemia
Cell Death Discovery (2020)
-
lncRNA H19 sponging miR-93 to regulate inflammation in retinal epithelial cells under hyperglycemia via XBP1s
Inflammation Research (2020)
