
Abstract
The role of the intestinal epithelium as part of the physical barrier to infection is well established alongside its central roles in food absorption, sensing nutrients, and water balance. Nematodes are one of the most common types of pathogen to dwell in the intestine. This article reviews recent data that have identified crucial roles for intestinal epithelial cells in sensing these kinds of pathogens and initiating innate responses, which qualitatively influence adaptive immune responses against them. Moreover, it is now clear that the epithelium itself—in addition to the cells that lie within it—are key to many of the protective mechanisms that result in expulsion of these large multicellular parasites from the intestine. An understanding of the IEC and intraepithelial leukocyte response is crucial to both development of mucosal vaccines, and the mechanisms that underlie the emerging use of intestinal dwelling helminths for therapeutic treatments of inflammatory and autoimmune disease.
Similar content being viewed by others

Introduction
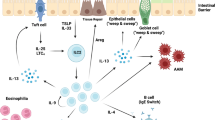
Nematode parasites that inhabit the mammalian gastrointestinal tract remain one of the most prevalent groups of infectious microorganisms of humans.1 In geographical areas in which nematode parasites are endemic, immunity to infection in previously exposed individuals is associated with expression of T-helper type-2 (TH2) cytokines, while persistent heavy infections can result in overproduction of proinflammatory cytokines and development of severe intestinal inflammation.2, 3, 4 and 5 Murine models of intestinal nematode infection provide a powerful in vivo tool to interrogate the cellular and molecular basis for resistance or susceptibility to infection. Over the last 15 years, studies on a number of murine nematode infection models have shown that CD4+ TH1 cells expressing IFN-γ promote parasite persistence and host susceptibility, while resistance to infection is dependent on CD4+ TH2 cells and TH2-associated cytokines, including IL-4, IL-9, IL-13, IL-25, and IL-336, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 and 17 (Figure 1).

TH cell-dependent regulation of resistance and susceptibility to intestinal nematode parasites. Following exposure to intestinal nematode parasites, intestinal epithelial cells express an array of immunoregulatory cytokines, including TSLP and IL-33. These factors can license intestinal DC responses potentially through regulation of their homing properties, their expression of surface molecules, including Notch ligands, and their production of proinflammatory cells. Naïve T cells are activated in the draining mesenteric lymph nodes and depending on the mouse strain, differentiate into either host protective Th2 cells that express IL-4, IL-5, IL-9, IL-13, and IL-25, or non-protective TH1 cells that express IFN-γ.
Despite these advances in delineating cytokine regulation of host resistance and susceptibility, the questions of how protective anti-nematode TH2 cytokine responses are initiated and what effector mechanisms TH2 cytokines elicit to mediate host protection have remained two areas of intense research. This review will focus on recent studies that have identified intestinal epithelial cells (IECs) as a key cell population in anti-nematode immune responses. First, we will discuss the influence of IECs on the recognition of nematode parasites and initiation of innate immune responses that govern development of adaptive CD4+ TH2-cell responses required for host protective immunity. Second, we will highlight recent findings that identify a role for TH2 cytokine-dependent regulation of the epithelial tissue—including IEC proliferation, differen-tiation and turnover—in expulsion of nematode parasites. Finally, we will highlight recent clinical studies suggesting that exposure to nematode parasites can be used in the treat-ment of chronic inflammatory diseases and discuss how IEC responses to nematode parasites may underlie these therapeutic effects.
Intestinal epithelial cells and initiation of anti-nematode immune responses
Intestinal epithelial cells: sentinels in barrier immunity
IECs exhibit numerous physical and biochemical adaptations to maintain barrier function, including expression of elaborate tight junctions, actin-rich microvillar extensions, and secretion of a mucin and polysaccharide-rich glycocalyx.18 In addition, IECs can express a battery of antimicrobial peptides, including defensins, cathelicidins, and calprotectins.19 Most of these peptides are rich in hydrophobic and basic residues that confer amphiphilic properties, resulting in broad-spectrum antimicrobial properties. In addition to barrier function, there are numerous reports suggesting that IECs can influence innate and adaptive immune cell function. IECs express germ-line-encoded pattern-recognition receptors, including Toll-like receptors (TLRs) and intracellular Nod-like receptors that enable microbial recognition.20, 21 and 22 Ligation of TLRs and Nod-like receptors results in activation of innate immune responses, including induction of the expression of proinflammatory cytokines and chemokines that are essential components of the anti-pathogen response. IECs express a wide range of cytokines and chemo-kines, including tumor-necrosis factor (TNF), transforming growth factor-β (TGF-β), IL-1, IL-6, IL-7, IL-8, IL-10, Monokine Induced by Interferon Gamma (MIG), IFN-inducible T cell α chemoattractant (ITAC), MIP-3α, CXCL9, CXCL10, CXCL11, and fracktalkine that can promote the recruitment and/or activation of immune cells. They also express major histocompatibility complex class I and class II molecules and all the machinery required for antigen processing and presentation.23 Although their capacity to act as antigen-presenting cells remains controversial,24, 25 and 26 expression of these immune response genes supports their role as integral components of innate and adaptive anti-pathogen response in the intestinal microenvironment.
Activation of IECs following exposure to viral or bacterial pathogens has been well documented (reviewed by Shibolet and Podolsky,27 Gewirtz,28 Abreu et al.,29 and Kelly and Conway30). For instance, following infection with the Gram-negative, enteropathogenic bacterium, Salmonella typhimurium, components of the flagellum bind to TLR5 on IECs and activate nuclear factor-κB (NF-κB).31, 32 and 33 In addition, transcription of multiple immune response genes is initiated, including cytokines and chemokines, which recruit dendritic cells (DCs) and other inflammatory cells, and initiate innate and adaptive immunity. The importance of IECs in the in vivo response to intestinal bacteria was highlighted in studies using bone marrow chimeric mice in which the non-hematopoietic cell compartment, including IECs, were specifically targeted. For example, IECs deficient in single immunoglobulin domain-containing IL-1R-related protein, a negative regulator of TLR/IL-1R signaling, were more susceptible to intestinal inflammation driven by inappropriate immune responses to commensal bacteria.34, 35 and 36 In addition, deletion of TLR4, Nod1, or MyD88 within non-hemato-poietic cells resulted in impaired control of bacterial infections, implicating IEC-mediated recognition of bacteria as a key component of protective immunity in the gastrointestinal microenvironment.37, 38 and 39
How do IECs respond to intestinal nematode infection?
The role of IECs in innate recognition of bacterial pathogens and development of TH1 or TH17 cell-dependent immunity is becoming clear. As discussed above, immunity to nematode parasites requires development of pathogen-specific TH2 cytokine responses in the gut-associated lymphoid tissues. However, whether IECs become activated following exposure to nematode parasites, and their potential influence on nematode-induced TH2 cytokine responses and host protective immunity, has received less attention.
Two murine models of intestinal nematode infection, Trichuris muris and Trichinella spiralis, have been used to dissect the response of IECs following exposure to infection. Although their biologies differ, these parasites share a number of common features, including the capacity to invade IECs and elicit polarized TH2 cytokine responses in resistant strains (see Box 1; Figure 2). In vitro studies demonstrated that IECs become activated following exposure to nematode parasites or their products. For example, following exposure to T. muris-derived antigens, IEC lines exhibit activation of the NF-κB pathway and expression of major histocompatibility complex class II, that is under the control of NF-κB signaling (Figure 3). IEC-intrinsic NF-κB activation was also observed in vivo following T. muris infection and this was associated with elevated expression of NF-κB-dependent thymic stromal lymphopoetin (TSLP)(see below).40 IECs are also a potent source of IL-33, which has been termed a “Th2 accelerator”.41 IL-33 mRNA is upregulated early following T. muris infection and exogenous IL-33 delivery upregulates TSLP expression in the intestine of mice.16 Similar activation of IECs has been observed in the T. spiralis model. A panel of human IEC lines was activated following in vitro infection with T. spiralis. Bile-activated infective larvae successfully migrated into IEC monolayers and provoked expression of IL-1β, IL-8, and epithelial neutrophil-activating peptide 78.42 Interestingly, there was no expression of TNF or TGF-β following invasion by T. spiralis, suggesting that IECs are selectively activated following nematode infection. A key question regarding these findings is how nematode parasites activate IECs. It is possible that analogous to studies with bacterial and viral pathogens, IECs possess pattern-recognition machinery such as TLRs or Nod-like receptors, which could specifically recognize molecular motifs common to nematode parasites. Although helminth-derived antigens have been shown to signal through TLRs43, 44 and 45 and activate NF-κB,45, 46 no nematode-specific pattern recognition receptors have been identified to date. It is interesting to note that MyD88-null mice generate strong protective type-2 responses to T. muris, suggesting that this signaling pathway is not critical to protection.47 TRIF-null mice also remain resistant to T. muris infection (JL Pennock, unpublished observations). However, mucosal epithelial cells express other innate recognition receptors, including C-type lectins such as dectins, that recognize β-glucans that are components of fungal pathogens,48, 49, 50 and 51 although their functions in innate recognition of nematode parasites in vivo has not been examined. Alternatively, nematode parasites may indirectly activate IECs via induction of the expression of endogenous activating factors such as TNF, or through physical damage resulting from the burrowing action of the parasites that could elicit stress responses. Notwithstanding this, it appears that IECs exhibit elaborate, and perhaps specific, patterns of activation following exposure to nematode parasites.

Intimate association of T. muris with host intestinal epithelial cells. Adult T. muris in infected mouse cecum. Mucosal surface of mouse cecum day 33 post infection (a). The posterior of multiple worms can be seen protruding out of the epithelial layer. Light micrograph of adult female T. muris day 42 post infection (b). Multiple transverse sections of anterior worm can be seen embedded within the epithelium at the crypt table, with transverse section of the posterior worm free in the lumen. Note eggs within the worm uterus (courtesy of N Humphreys). Scanning EM of adult worm highlighting syncytial epithelial tunnel (c).

Intestinal epithelial cells are activated following exposure to T. muris . The IEC line, HT29, was stimulated with either rTNF or T. muris antigen, and whole-cell or nuclear extracts prepared. Whole-cell IκBα degradation (a) and nuclear NF-κB1 localization (b) were demonstrated by western blotting. Immunofluorescence also demonstrated that T. muris antigen induced nuclear translocation of NF-κBp65 in HT29 cells (c). IECs were also activated in vivo following T. muris infection, as determined by major histocompatibility complex class II expression (d).
IECs license intestinal DC function following nematode infection
An in vivo role for IECs in regulating anti-nematode immune responses was recently shown using IEC-specific knockout mice. IEC-intrinsic NF-κB activation through the classical pathway was blocked by Villin-Cre-mediated deletion of the gene encoding IKKβ to create mice in which IKKβ was specifically deleted in IECs (ikkβΔIEC mice).40, 52 Naive ikkβΔIEC mice exhibited no basal defects in intestinal development or function; however, disruption of NF-κB activation exclusively within IECs resulted in defective development of TH2 cytokine responses and susceptibility to T. muris infection.40 While control mice developed protective TH2 cytokine responses, mesenteric lymph node cells isolated from infected ikkβΔIEC mice exhibited exaggerated production of parasite-specific IFN-γ and IL-17 that was associated with development of severe intestinal inflammation. Dysregulated CD4+ TH responses in T. muris-infected ikkβΔIEC mice was associated with aberrant DC responses in the gut-associated lymphoid tissues. Specifically, CD11c+ CD11b+ intestinal DCs exhibited heightened expression of TNF and IL-12/23p40,40 suggesting IECs have the capacity to license DC responses and maintain intestinal immune homeostasis following parasitic nematode infection.
Previous in vitro studies implicated IECs, through secretion of cytokines, in conditioning of DCs to limit their production of proinflammatory cytokines. For example, secretion of the immunoregulatory cytokine tslp by IECs has been shown to influence human DC responses.53 tslp is highly expressed in the epithelia at most mucosal surfaces, including the skin, airway, and the intestine. Within IECs, tslp mRNA is constitutively expressed and can be upregulated in response to a range of stimuli, including infection, inflammation, and tissue injury.53, 54, 55, 56 and 57 Related to intestinal nematode infection, T. muris infection of genetically resistant mouse strains results in a marked upregulation in TSLP expression in IECs.40In vitro studies by Rescigno and co-workers showed that IEC-derived TSLP could limit expression of IL-12 in human DCs and inhibit their capacity to promote TH1-cell differentiation. Moreover, IEC conditioning promoted IL-10 production by activated DCs and enhanced their capacity to promote regulatory and TH2 cytokine responses.53, 58 Consistent with these findings, TSLP-enriched culture supernatants from a murine IEC line could inhibit IL-12/23p40 expression in bone marrow-derived DCs.40 Deletion of TSLPR in mice resulted in constitutive over-expression of IL-12/23p40 expression in intestinal DCs and the inability to generate protective TH2 cytokine responses following exposure to T. muris.40 In addition to TSLP, IECs are known to express Notch receptors.59, 60 TH2 cytokine-dependent immunity to nematode parasites is dependent on the Notch pathway;61 however, whether there is cross-regulation between the Notch and TSLP pathways is at present unknown. Notwithstanding this, it is clear that IECs—in part via secretion of TSLP—play an essential role in regulating innate DC responses and development of protective TH2 cytokine responses following intestinal nematode infection. IECs can also directly influence multiple cell lineages, including macrophages, granulocytes, and lymphocytes. Given recent studies that implicate mast cells, basophils, and eosinophils in the innate responses that promote expression of TH2 cytokines,62, 63, 64, 65, 66 and 67 future analysis of how IECs interact with these cell populations, and perhaps govern their functions, following nematode infection could provide new insights into how anti-nematode responses are initiated and maintained.
The intestinal epithelium: an effector tissue in the expulsion of nematode parasites
In addition to influencing innate immune responses following intestinal nematode infection, recent studies have highlighted the role of IECs in the effector response required for expulsion of nematode parasites. The overarching view that protective immunity to gastrointestinal dwelling nematodes is specific in its induction, but non-specific in its action, is not new,68 but definition of the precise mechanisms involved have taken considerable time to be elucidated. The observations that resistance to intestine-dwelling nematodes was associated with many of the hallmarks of allergic disease, led to much work exploring the role of classical allergic mechanisms in worm expulsion; but it became clear that although many facets of allergic type responses were involved, they did not necessarily operate as they did in allergic responses and indeed, other mechanisms played important roles. In particular, the role of the intestinal epithelium as an integral component of the gut's array of immune effector mechanisms is only now beginning to be appreciated. As discussed above, it is now apparent that the intestinal epithelium is much more than a simple barrier in terms of host protective immunity. It is also becoming increasingly clear that it can function as an effector tissue under the regulation of the adaptive immune system. This is either via the cells that are present within the epithelial layer – including the intra epithelial leukocytes (IEL) or indeed the epithelial cells themselves. This is particularly true of pathogens inhabiting enterocytes for a part or all of their existence. Studies employing Trichuris spp. and Trichinella spp., together with those where the parasites inhabit other intestinal niches such as the intestinal lumen, are helping to define the role of the epithelium as an immune effector tissue, identifying mechanisms that may well operate against many types of intestinal pathogen.
Unusually for parasitic nematodes, T. spiralis has broad host specificity and readily infects laboratory rodents. Much work has been carried out on the mouse and it is clear that the intestinal phases of infection vary in length between inbred strains of mouse, although in almost all strains worms are expelled within 14–21 days post infection.69 The rate of worm expulsion in immunocompetent animals varies in relation to generation of adaptive immunity. Interestingly, the adult worms are not killed by the immune response. Surgical transplantation of adult parasites undergoing expulsion indicates they can recover and thrive until subsequent expulsion in the recipient.70 Indeed, it is a consistent observation for most intestinal-dwelling nematodes that worm loss is associated with alteration of habitat making the local environment unsuitable for optimum survival rather than a direct cytotoxic activity by immune cells or secreted molecules.
The intraepithelial mast cell in resistance
The mechanisms of immune-mediated worm expulsion of T. spiralis have been investigated extensively over many years (reviewed by Grencis71). Similar to other intestinal nematodes, worm expulsion is dependent upon generation of a strong CD4+ TH-cell response in the draining mesenteric lymph node. Cytokine analysis has shown that a dominant TH2 cytokine response is generated and influences the development of a variety of immune and immunopathological changes in the infected intestine and systemically. These include a strong peripheral IgE response, peripheral and intestinal eosinophilia, goblet cell hyper-plasia, villus atrophy and crypt hyperplasia, changes in intestinal muscle contractility, and mucosal mast cell hyperplasia. Dissection of the components of this broad type-2 response have highlighted the intraepithelial mast cell as a key player in worm expulsion.72 In mice and rats, the mast cell hyperplasia observed by traditional metachromatic staining occurs temporally alongside worm expulsion. Moreover, secretion of mucosal specific mast cell proteases (particularly mouse mast cell protease 1, MMCP1) into the intestinal tissue and circulation also occurs alongside worm expulsion.73, 74 Mast cell-deficient mice (WWv/WWv) show delayed worm expulsion,75 and depletion of mast cells in vivo in infected mice using anti-ckit or ant-stem cell factor antibody significantly delays worm expulsion.76, 77 The use of MMCP1-null mice convincingly demonstrated that this protease played a critical role in worm expulsion, with such animals exhibiting much delayed worm loss from the intestine.78T. spiralis-infected mice also show considerable loss of epithelial integrity during the intestinal phases of infection with increased permeability.79 Decreased intestinal epithelial cell resistance and sodium-linked glucose absorption is also observed following infection. All these changes have been shown to involve IL-4 and IL-13 operating through a STAT6-dependent pathway as have secretory response to mediators such as PGE2.80 The increased permeability is also associated with alteration in tight junction molecules such as occludin and the claudins.79, 81 An important role for mast cell proteases in this response was demonstrated by experiments using T. spiralis-infected MMCP1-null mice, which did not show increase in intestinal permeability, which was coincident with delayed worm expulsion.79
The intestinal mastocytosis observed during intestinal helminth infection is dependent upon the interplay between hemopoeitic growth factors such as stem cell factor and cytokines produced predominately from CD4+ TH2 cells, including IL-3, IL-4, and IL-9.77, 82, 83 and 84 Interestingly, stem cell factor plays a number of different roles, including promotion of proliferation and differentiation in addition to directing migration.85, 86 and 87 It is known that mast cell precursors are released from the bone marrow following T. spiralis infection and express the gut homing integrin αEβ7.87, 88 The chemokine CCL2 is expressed on the epithelium of nematode-infected mice and is known to induce migration of derived mucosal like mast cells89, 90 as is leukotriene –B4.91 Once in the epithelium, TGF-β1 is critical for activation of mast cell proteases, particularly MMCP1, and expression of the αE-subunit of αEβ7 integrin, which tethers them to the epithelium.92, 93 and 94 The inactive TGF-β1 latency-associated peptide can be activated by another epithelial expressed integrin, αVβ6. Interestingly αVβ6-null mice showed reduced migration of mast cells to the epithelium, which was coincident with delayed worm expulsion.92, 95 and 96 Thus, following infection movement of mast cells to the epithelium and activation by the epithelium is critical for their host protective role against nematode infection.
Although intestinal T. spiralis infection is associated with prominent IgE response and mast cells within the infected small intestine are IgE positive,97 the role of IgE in host protection, particularly during primary infections, is controversial. Worm expulsion is delayed considerably in animals lacking mast cells or MMCP1,78 although worm expulsion and mastocytosis are still evident in T. spiralis-infected animals that do not possess a functional high-affinity IgE receptor.71T. spiralis infection is also accompanied by a significant increase in intestinal muscle contractility, which is controlled by T cells secreting IL-4 and IL-13 through STAT6.98 Interestingly, IL-18, which is known to be produced in the gut, both in the epithelium and lamina propria mononuclear cells,99 has been shown to have definite, but varied, responses on intestinal mastocytosis induced by intestinal nematode infection;100. It is, thus, relatively easy to appreciate the crucial role of the epithelial cell/mast cell inter-action in the generation of intestinal inflammation, in rendering the interface between host and worm unsuitable for intestinal nematodes and affecting worm reproduction and survival. The altered environment in combination with the increased muscle contractility results ultimately in worm expulsion via the so-called “weep and sweep” response.
Macrophage–IEC interactions in expulsion of nematode parasites
It is also abundantly clear, however, that intestinal mast cell-driven inflammation is not effective against all species of intestine-dwelling nematodes at all stages of infection. Heligmosomoides polygyrus is a natural parasite of wild mice and animals become infected following ingestion of L3 larvae from the soil. Recently, Gause and co-workers101 have elegantly shown that alternatively activated macrophages (AAMacs) can effectively control H. polygyrus. This has, however, to date only been demonstrated during challenge or secondary infections. Following priming, in this case by an artificially abbreviated primary infection, AAMacs “trap” the larval stages of the parasite, which undergo a short developmental period in the submucosa prior to emerging into the intestinal lumen to develop into adults that live coiled around the villi of the small intestine. This mechanism is, therefore, well suited to parasites that are not particularly active (that is, during developmental stages) or spend time in the tissue, particularly outside of the intestinal epithelium, that is, at sites where recruitment of macrophages is possible. It would be of great interest to determine whether this mechanism of immunity operates after trickle infections (repeated low-dose infections, as experienced in nature).102 Normally, H. polygyrus infections are chronic in nature and a single primary infection lasts for many months in most inbred strains of mouse. It is noteworthy that a chronic primary infection of H. polygyrus is associated with downregulation of intestinal mast cell hyperplasia.103 Moreover, in mice, which have artificially elevated intestinal mast cell responses, such as those overexpressing the IL-9 gene, a primary infection of H. polygyrus is expelled efficiently,104 suggesting that if potent enough, mastocytosis may be effective in removing even lumen-dwelling parasites. Some strains of mouse naturally expel a primary infection of H. polygyrus (which can occur after several weeks),105 and it is unlikely that the AAMac mechanism described above plays a role against the lumen-dwelling stages of infection, although their contribution in other ways to parasite loss has not been explored. Equally, the influence of the cross talk between IECs and AAMacs on the expulsion of nematode parasites awaits investigation.
Intestinal epithelial cell differentiation in expulsion of nematode parasites: the role of goblet cells
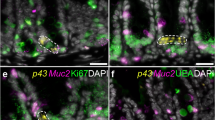
Each crypt within the intestine is an individual proliferative unit, with pluripotent stem cells located near the base of the crypt dividing by asymmetric division to give rise to a number of cell lineages, including Paneth cells, enteroendocrine cells, and goblet cells.106, 107 Goblet cells secrete mucus, trefoil peptides, and other bioactive molecules, and are a well-characterized signature of intestinal nematode infection. In the steady state, goblet cell-derived mucus is known to create a physical barrier and provide buffering function at the mucosal surfaces of the intestine. A number of studies have demonstrated that intestine-dwelling nematodes either do not establish, are impeded from doing so, or can be physically trapped in secreted mucus gel.108, 109 Mucins form the structural core of the secreted mucus gel, but can also be membrane bound. There are relatively little data concerning the functional role of mucins during intestinal nematode infection, although a number of studies have identified that Muc-2 (secreted) and Muc-3 (membrane bound) are upregulated in the small intestine following infection by T. spiralis.110, 111 Transcripts for Muc-2 are also upregulated in the large intestine following infection by T. muris112 as is the protein (see Figure 4). Glycosylation changes of mucins also occur following nematode infection, although mostly described through differential histological staining of goblet cells.113 How these changes relate to host protection is unclear, although infection of mice with the nematode Strongyloides venezuelensis suggests that recognition of carbohydrate moieties on IECs can profoundly influence where the nematodes establish themselves in the intestine.114

Elevated expression of goblet cell-derived MUC2 following exposure to T. muris. Muc 2 protein expression in the cecum of naïve BALB/c mice and on day 21 post infection. BALB/c mice normally expel their parasites by day 21 post infection. (courtesy of S Hussein).
Goblet cells are also the source of a number of other molecules involved in host defense, including intestinal trefoil factor-3.115 Its function is unclear, but a number of studies suggest a protective role in colitis116 and control by IL-4 and IL-13.117 It is also upregulated following infection by a number intestine-dwelling nematodes,111 but a role in host protection is undefined. It has been suggested that intestinal trefoil factor-3 may interact with Muc-2 in the protection of the mucosa against damage.116
Chloride channel, calcium-activated 3 (mCLCA3, Gob5)118 is also expressed exclusively by goblet cells and upregulated following infection by T. muris and T. spiralis,40, 111 and 112 although, again, its function is unclear. In a similar vein, intelectins were molecules originally described in mouse intestinal Paneth cells and mooted to have antimicrobial actions via their ability to bind a variety of non-mammalian sugars or through their ability to bind to lactoferrin and regulating its antimicrobial properties119 (reviewed by Nair et al.120). Interestingly, intelec-tin-1, which shows little change in BALB/c mice upon infection in the small intestine with T. spiralis, is upregulated following in BALB/c mice infected with T. muris around the time of worm expulsion.112, 120 A variant of intelectin-1 (intelectin-2), which is undetectable in naïve BALB/c mice, is strongly upregulated during T. spiralis infection and secreted into the mucus gel.121 Its protective role, if any, remains to be determined.
Goblet cells are also now known to be a source of one of the members of the resistin-like family of molecules, RELMβ (FIZZ2).120, 122, 123, 124 and 125 Expression is believed to be under the control of IL-13 and recent data have demonstrated it secretion into the intestinal lumen associated with worm loss following infection with Nippostrongylus brasiliensis, T. spiralis, and T. muris.123 Rapid expression of RELMβ is also a hallmark of TH2 memory responses following secondary challenge with T. muris.126 Interestingly, the site-specific expression transcription factor Cdx2, which is involved in intestine-specific expression of RELMβ in response to commensal bacteria, is not required for expression of RELMβ in response to intestinal nematode infection.124 The fact that RELMβ was highly upregulated in the intestine during the period of worm expulsion, implied that there may be a causal relationship. The functional activity of RELMβ has been hypothesized to act via interference with nematode sensing leading to “disorientation” of the worm, contributing to a reduced ability to maintain its niche within the intestine and reproduce effectively.120, 123 and 127 However, RELMβ-null mice are still able to expel T. muris (MG Nair and D Artis, unpublished observations), again highlighting the redundant nature of the effector responses mounted against these parasites.
The epithelial escalator: elevated IEC turnover in expulsion of intestinal nematodes
One feature that is particularly consistent throughout all protective mechanisms against intestinal nematodes is the involvement of IL-13. As highlighted above, IL-13 regulates a number of epithelial responses, including goblet cell differentiation and maturation. Recent work involving the T. muris system has identified a role for IL-13 in a novel effector mechanism against intestinal nematodes mediated by IECs. As described above, infection of T. muris in the mouse (as with all species of Trichuris) is entirely confined to the epithelium of the cecum and colon (see Box 1). At all stages of the life cycle, the parasites must maintain their optimum position within the constantly moving epithelium, and avoid being shed into the intestinal lumen alongside the normal renewal of the intestinal enterocytes.
The majority of experimental studies on T. muris infection have used a moderate-to-large dose of eggs to follow infection. In most inbred strains of mouse, worms are expelled as larval stages, usually before the L3 stage that occurs between days 17–21 post infection. This response is dependent upon CD4+ T cells and a variety of studies have highlighted contributory roles for a variety of type-2 cytokines in the resistant response, including IL-4, IL-9, IL-10, IL-25, and IL-33, with dominant role for IL-13 (see above). Many type-2 immunopathological features accompany resistance, including elevated IgE levels, intestinal mastocytosis, goblet cell hyperplasia, and intestinal eosinophilia (reviewed by Cliffe and Grencis128). Worm expulsion proceeds normally in animals depleted of mast cells or eosinophils, and adoptive transfer of CD4+ T cells to severe combined immunodeficient mice is sufficient to induce worm expulsion, indicating that B-cell responses and antibody are not necessary in this system.129, 130, 131 and 132 Goblet cell hyperplasia has been correlated with resistance under the control of IL-13, and is concordant with the secretion of RELMβ as described above.123, 125
Another feature of T. muris infection is a change in the proli-ferative capacity of the intestinal epithelium.133 Profound changes in the rate of intestinal cell turnover occur in mice during the period of worm expulsion. In mice expelling their parasites, almost a doubling of the rate of turnover occurs, whereas in animals that do not expel their worms, only a slight elevation in turnover is apparent.134 Most notably, in IL-13-null mice (which do not expel) the rate of turnover is similar to that seen in naturally susceptible wild-type mice. In mouse strains that naturally do not expel their parasites, a type-1 cytokine response is elicited, characterized by production of IFN-γ, IL-12, and IL-18.13, 135, 136 and 137 The production of IFN-γ serves not only to counter-regulate the potential protective TH2 response, but also increases proliferation of the intestinal epithelium and, through induction of CXCL10 in the infected intestine, slows down the turnover rate.134 Accordingly, a slow-moving epithelium is established, presumably ideal for worm survival. Most notably, blocking CXCL10 in vivo raises the turnover rate, which causes worm expulsion, without changing the ongoing type-1 cytokine response normally seen in susceptible animals.134 In this way, the intestinal epithelium itself acts as an “epithelial escalator” moving the parasite out of its optimum niche and expelling it into the lumen (Figure 5). Interestingly, in animals naturally harboring a chronic infection, once the crypts in the infected intestine become very large, an increase in apoptotic cells is observed in the stem cell region of the cecum and colon. Again, this appears to be under immunological control of via TNF and IFN-γ.138

TH cytokines control intestinal epithelial cell proliferation, differentiation, and turnover. During T. muris infection in resistant mice, IL-13 released from TH2 cells or intraepithelial NK cells increase the turnover of cecal epithelium. Worms are unable to maintain their optimum niche within the epithelium and are extruded into the gut lumen along with epithelial cells as part of the normal tissue renewal—“the epithelial escalator”. In susceptible mice, IFN-γ released from TH1 cells (or NK cells) increases epithelial proliferation and through induction of CXCL10, slows down epithelial turnover and the parasite is provided with a slow-moving, growing epithelium, conducive to worm survival.
The epithelial escalator is under the control of IL-13, although whether this operates directly through epithelial cells themselves, or through other cell types such as myofibroblasts lying underneath the crypts, is unknown. Certainly, intestinal epithelial cells are known to express IL-4Rα.139 Also, TNF-α is involved in the protective response to T. muris infection,9 although its precise role remains to be defined. Recent work has suggested that TNF-α acts as a global enhancer of ongoing cytokine responses during infection.140, 141 Moreover, local cellular sources of IL-13 are yet to be defined precisely. It is known that CD4+ T cells capable of adoptively transferring immunity to severe combined immuno-deficient mice are required to home back to the intestine to mediate protection, and that blocking intestinal homing with antibodies prevents worm expulsion,142 and these are a likely source of the cytokine. Recent data have, however, highlighted other populations of cells within the epithelium of nematode infected mice that may, at least in part, fulfill this role. Following infection with the related nematode T. spiralis, a population of intraepithelial natural killer (NK) cells become capable of producing IL-13, which mediates a number of changes, including villus atrophy crypt hyperplasia and goblet cell hyperplasia. Interestingly, this cell population is also generated in athymic and severe combined immunodeficient mice and induces similar changes to those seen in wild-type mice.143 It is surprising that in wild-type animals other cell types (CD3+, CD4+, CD8+ cells) present in this compartment do not appear to express IL-13 following infection. The capacity to produce IL-13 is coincident with the expression of IL-4Rα on epithelial cells. Intra-epithelial leukocytes are particularly well placed to act upon the epithelium as it moves up and around this strategically positioned cell population. The mechanism of NK cell activation during intestinal nematode infection is presently unknown. With regards to the T. muris system, recent work by Gause and co-workers has observed that NK cells are present in the mesenteric lymph node following T. muris infection and that these cells are capable of producing IL-13.144 The role of these cells in immune protection to T. muris has been investigated by MR Hepworth et al. (unpublished observation) who has shown that NK cells play an increasingly important role as a source of protective IL-13 when the CD4+ T-cell compartment is limiting. These cells appear to be distinct from basophils, which have recently been described as a key cell type producing early IL-4 (and IL-13) and helps polarize responses toward the TH2 phenotype.65, 66 Thus, it appears that there are a variety of cell types producing IL-13 following intestinal nematode infection. A common feature of them all is that they act on IECs to induce a variety of immunological and physiological changes that ultimately are not conducive to worm survival. Taken together, studies in diverse nematode systems have demonstrated multiple TH2 cytokine-dependent effector mechanisms, including mastocytosis, eosinophilia, and recruitment of AAMacs. In addition, there are profound changes in IEC function, including altered permeability, proliferation, turnover, and differentiation. Therefore, as discussed above, parasite-specific CD4 T-cell responses elicit nonspecific “modular” type-2 responses (Figure 6). Depending on the parasite in question, distinct components of this modular response have differing degrees of effectiveness in precipitating worm expulsion.

TH2 cytokines control intestinal epithelial cell effector mechanisms against intestinal nematode parasites. TH2 cells release IL-4, IL-9 (and IL-3), which act in concert with stem cell factor and TGF-β1 to cause differentiation, maturation, and activation of intraepithelial mucosal mast cells, inducing release of mmcp-1. This alters tight junction integrity, leading to increased intestinal permeability. IL-13 from TH2 cells (and intraepithelial NK cells) induces goblet cell differentiation, expression of RELMβ, and is associated with mucus release into the gut lumen. IL-13 from TH2 cells increases epithelial cell turnover and in conjunction with IL-4, induces alternatively activated macrophages that can “entrap” parasites within the submucosa.
Another feature of many intestinal nematode infections is weight loss, particularly during the acute phases of infection. Intuitively, it is reasonable to suggest that this is an adaptive response for the host and the result of the combination of reduction in food intake (appetite suppression and avoidance of further infectious stages) and possibly cytokine mediated anorexia. TNF-α (cachectin) is known to be produced in response to nematode infection (see Figure 3),9, 140 and 141 although does not appear to play a significant role in the weight loss during the intestinal phase. Weight loss following infection is particularly evident following T. spiralis infection, with a period of hypophagia occurring during the period of worm expulsion. This is associated with an increase in enteroendocrine cell numbers in the epithelium of the small intestine and secretion of the satiety factor cholecysto-kinin into the circulation.145 Remarkably, this change is mediated through CD4+ T cells and secretion of IL-4 and IL-13. Again, this adds a further complexity to the battery of epithelial responses under the control of infection-induced immunity.
The consequences of intestinal nematode infection; regulation—a therapeutic opportunity?
The aforementioned discussion clearly shows that intestinal nematodes regardless of where they live within the intestine, induce strong innate and adaptive response—they are not the “silent serpents” they were originally thought to be. The relationship between parasite and host is an active dynamic one irres-pective of whether the infection is acute or chronic. The host responds with the intention of removing the infection and gene-rates a coordinated type-2 response comprising of a variety of effector cells and molecules. It is now appreciated that this has to extend beyond the conventional “antibody and cells” response to be effective and is, in part, no doubt due to evolution of immune evasion mechanisms employed by the parasites. It would appear that the host cannot distinguish between the different species of nematode/helminth that infects per se, but rather, certain signatures associated with this type of pathogen are sensed and a generic or modular TH2 response is generated with the hope that one or more of the type-2 controlled effector mechanisms generated will be effective. The fact that under natural conditions protective immunity takes considerable time to develop, reflects the effectiveness of the immune evasion mechanisms employed by the parasite and the natural history of infection (that is, low repeated dose of infection). The potential for damage and inflammation following infection by these pathogens is, therefore, considerable and is evident in the well-documented pathology and disease that accompanies infections of this type as worldwide with estimates of 39 million DALYS associated with intestinal nematode infection.146
At first glance, therefore, it is somewhat paradoxical that intestine-dwelling nematodes are currently being assessed and used in clinical trials to control a variety of inflammatory diseases, including inflammatory bowel disease and some autoimmune conditions. Of particular relevance to the present review is the use of Trichuris suis as a therapeutic agent in the clinic.147, 148 and 149 As with all Trichuris species, T. suis inhabits the epithelial niche of the cecum and colon. The basis for its therapeutic use is that in order for the host and parasite to survive, part of the immune evasion strategy used by the parasite involves regulation of any potentially damaging intestinal pathology it induces. Indeed there is considerable evidence emerging from experimental studies of T. muris infection to show that without regulation, the colitis and inflammation that occurs following infection can be severe and even life threatening.150 The cytokine IL-10 is critical to this regulatory process and is coincident with an increase in the production of regulatory T cells (NE Humphreys, unpublished observations). A detailed treatise is beyond the scope of this review, but a number of studies have shown that T regulatory cells are induced following nematode infection and can control any potential pathology associated with infection through cytokines such as IL-10 or TGF-β.151 Moreover, these regulatory cells can influence unrelated pathological responses distant from the site of infection (Humphreys, unpublished observations). It is thus proposed that, as most species of intestinal nematodes are species specific, patent (chronic) infections of T. suis cannot establish in humans and only a limited and curtailed infection occurs—enough, however, to generate immunoregulatory mechanisms that can also control concurrent autoimmune or aberrant inflammation such as inflammatory bowel disease. As discussed above, IECs become activated following exposure to nematode parasites and secrete a wide range of bioactive molecules, including regulatory cytokines. Therefore, it likely that at least a portion of nematode-induced immunoregulation in the intestine is dependent on IECs, perhaps through interactions with DCs.
The literature reviewed above clearly shows that the host epithelium responds to nematode parasite infections, particularly by invasive species such as Trichuris, and a vigorous, coordinated response is generated against them. This includes a battery of effector mechanisms, which vary in effectiveness depending upon the species of the nematode. That if these responses are effective and the host expels their worms via an adaptive mediated immunity, then in common with responses to other pathogens, the host is primed, establishes immunological memory, and is able to expel future challenge infections. If, however, protective immunity is not established, by whatever means (infection dose, evasion mechanisms), then the potential for host damage is considerable. Chronic infection then ensues and to prevent potentially damaging immunopathology and inflammation, regulation is required, primarily for host survival. The influence of this parasite-induced immunoregulation is not parasite specific and can influence other ongoing immune responses, including those that can cause disease. How this fits in with the current helminth therapeutic regimes being used in the clinic is unclear. One either has to speculate that repeated abbreviated infections are sufficient to induce immunoregulation—which is counter to the majority of the literature, as it should induce resistance to future infection—or that some parasites establish themselves, lasting longer than anticipated. These would potentially induce pathology, which would subsequently lead to activation of the regulatory mechanisms highlighted above. Regardless of whatever scenario, it is clear that intestine-dwelling nematodes present a considerable immunological challenge to the host intestinal immune system. They are undoubtedly pathogens, not commensals, and as such will be recognized as antigenic and potentially dangerous. As such the aim of the body is to remove them as efficiently as possible. If this does not happen, then a precarious and dynamic relationship is established between the host and parasite, with the epithelium playing a crucial role in the etiology of the infection, ultimately directing the immune response and influencing the degree of disease that ensues.
Concluding remarks
In summary, it is clear IECs exhibit robust innate immune responses during intestinal nematode infection that can influence both the initiation and effector phases of the anti-parasite response. Given the increasing recognition of the role of IECs in eliciting and regulating intestinal immune responses against pathogens, new approaches harnessing the natural immuno-regulatory properties of IECs may offer greater success in the design of oral vaccines or worm-based immunotherapies.
Disclosure
The authors declare no conflict of interest.
References
WHO. 54th World Health Assembly: Schistosomiasis and Soil Transmitted Helminth Infections (item 13.3) ( WHO: Geneva, 2001 ).
MacDonald, T.T. et al. Immunoepidemiology of intestinal helminthic infections. 3. Mucosal macrophages and cytokine production in the colon of children with Trichuris trichiura dysentery. Trans. R Soc. Trop. Med. Hyg. 88, 265–268 (1994).
Grencis, R.K. & Cooper, E.S. Enterobius, trichuris, capillaria, and hookworm including ancylostoma caninum. Gastroenterol. Clin. North Am. 25, 579–597 (1996).
Jackson, J.A. et al. T helper cell type 2 responsiveness predicts future susceptibility to gastrointestinal nematodes in humans. J. Infect. Dis. 190, 1804–1811 (2004).
Jackson, J.A. et al. Cytokine response profiles predict species-specific infection patterns in human GI nematodes. Int. J. Parasitol. 34, 1237–1244 (2004).
Else, K.J., Finkelman, F.D., Maliszewski, C.R. & Grencis, R.K. Cytokine-mediated regulation of chronic intestinal helminth infection. J. Exp. Med. 179, 347–351 (1994).
Finkelman, F.D. et al. Cytokine regulation of host defense against parasitic gastrointestinal nematodes: lessons from studies with rodent models. Annu. Rev. Immunol. 15, 505–533 (1997).
Bancroft, A.J., McKenzie, A.N. & Grencis, R.K. A critical role for IL-13 in resistance to intestinal nematode infection. J. Immunol. 160, 3453–3461 (1998).
Artis, D., Humphreys, N.E., Bancroft, A.J., Rothwell, N.J., Potten, C.S. & Grencis, R.K. Tumor necrosis factor alpha is a critical component of interleukin 13-mediated protective T helper cell type 2 responses during helminth infection. J. Exp. Med. 190, 953–962 (1999).
Urban, J.F. Jr et al. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity 8, 255–264 (1998).
Else, K.J. & Finkelman, F.D. Intestinal nematode parasites, cytokines and effector mechanisms. Int. J. Parasitol. 28, 1145–1158 (1998).
Richard, M., Grencis, R.K., Humphreys, N.E., Renauld, J.C. & Van Snick, J. Anti-IL-9 vaccination prevents worm expulsion and blood eosinophilia in Trichuris muris-infected mice. Proc. Natl. Acad. Sci. USA 97, 767–772 (2000).
Helmby, H., Takeda, K., Akira, S. & Grencis, R.K. Interleukin (IL)-18 promotes the development of chronic gastrointestinal helminth infection by downregulating IL-13. J. Exp. Med. 194, 355–364 (2001).
Urban, J.F. Jr, Noben-Trauth, N., Schopf, L., Madden, K.B. & Finkelman, F.D. Cutting edge: IL-4 receptor expression by non-bone marrow-derived cells is required to expel gastrointestinal nematode parasites. J. Immunol. 167, 6078–6081 (2001).
Owyang, A.M. et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J. Exp. Med. 203, 843–849 (2006).
Humphreys, N.E., Xu, D., Hepworth, M.R., Liew, F.Y. & Grencis, R.K. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J. Immunol. 180, 2443–2449 (2008).
Anthony, R.M., Rutitzky, L.I., Urban, J.F. Jr, Stadecker, M.J. & Gause, W.C. Protective immune mechanisms in helminth infection. Nat. Rev. Immunol. 7, 975–987 (2007).
Shen, L. & Turner, J.R. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G577–G582 (2006).
Magalhaes, J.G., Tattoli, I. & Girardin, S.E. The intestinal epithelial barrier: how to distinguish between the microbial flora and pathogens. Semin. Immunol. 19, 106–115 (2007).
Janeway, C.A. Jr & Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 20, 197–216 (2002).
Fritz, J.H., Ferrero, R.L., Philpott, D.J. & Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7, 1250–1257 (2006).
Philpott, D.J. & Girardin, S.E. The role of Toll-like receptors and Nod proteins in bacterial infection. Mol. Immunol. 41, 1099–1108 (2004).
Hershberg, R.M. & Mayer, L.F. Antigen processing and presentation by intestinal epithelial cells—polarity and complexity. Immunol. Today 21, 123–128 (2000).
Bland, P.W. & Warren, L.G. Antigen presentation by epithelial cells of the rat small intestine. I. Kinetics, antigen specificity and blocking by anti-Ia antisera. Immunology 58, 1–7 (1986).
Kaiserlian, D., Vidal, K. & Revillard, J.P. Murine enterocytes can present soluble antigen to specific class II-restricted CD4+ T cells. Eur. J. Immunol. 19, 1513–1516 (1989).
Sanderson, I.R., Ouellette, A.J., Carter, E.A., Walker, W.A. & Harmatz, P.R. Differential regulation of B7 mRNA in enterocytes and lymphoid cells. Immunology 79, 434–438 (1993).
Shibolet, O. & Podolsky, D.K. TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1469–G1473 (2007).
Gewirtz, A.T. Intestinal epithelial toll-like receptors: to protect. And serve? Curr. Pharm. Des. 9, 1–5 (2003).
Abreu, M.T., Fukata, M. & Arditi, M. TLR signaling in the gut in health and disease. J. Immunol. 174, 4453–4460 (2005).
Kelly, D. & Conway, S. Bacterial modulation of mucosal innate immunity. Mol. Immunol. 42, 895–901 (2005).
Elewaut, D., DiDonato, J.A., Kim, J.M., Truong, F., Eckmann, L. & Kagnoff, M.F. NF-kappa B is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J. Immunol. 163, 1457–1466 (1999).
Gewirtz, A.T. et al. Salmonella typhimurium induces epithelial IL-8 expression via Ca(2+)-mediated activation of the NF-kappaB pathway. J. Clin. Invest. 105, 79–92 (2000).
Gewirtz, A.T., Navas, T.A., Lyons, S., Godowski, P.J. & Madara, J.L. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167, 1882–1885 (2001).
Wald, D. et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat. Immunol. 4, 920–927 (2003).
Garlanda, C. et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA 101, 3522–3526 (2004).
Garlanda, C. et al. Increased susceptibility to colitis-associated cancer of mice lacking TIR8, an inhibitory member of the interleukin-1 receptor family. Cancer Res. 67, 6017–6021 (2007).
Schilling, J.D., Martin, S.M., Hung, C.S., Lorenz, R.G. & Hultgren, S.J. Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 100, 4203–4208 (2003).
Brandl, K., Plitas, G., Schnabl, B., DeMatteo, R.P. & Pamer, E.G. MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J. Exp. Med. 204, 1891–1900 (2007).
Lebeis, S.L., Bommarius, B., Parkos, C.A., Sherman, M.A. & Kalman, D. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J. Immunol. 179, 566–577 (2007).
Zaph, C. et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature 446, 552–556 (2007).
Schmitz, J. et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490 (2005).
Li, C.K., Seth, R., Gray, T., Bayston, R., Mahida, Y.R. & Wakelin, D. Production of proinflammatory cytokines and inflammatory mediators in human intestinal epithelial cells after invasion by Trichinella spiralis. Infect. Immun. 66, 2200–2206 (1998).
Harnett, W., Deehan, M.R., Houston, K.M. & Harnett, M.M. Immunomodulatory properties of a phosphorylcholine-containing secreted filarial glycoprotein. Parasite Immunol. 21, 601–608 (1999).
Thomas, P.G. et al. Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J. Immunol. 171, 5837–5841 (2003).
Artis, D. et al. Dendritic cell-intrinsic expression of NF-kappaB1 is required to promote optimal Th2 cell differentiation. J. Immunol. 174, 7154–7159 (2005).
Artis, D. et al. Differential requirement for NF-kappa B family members in control of helminth infection and intestinal inflammation. J. Immunol. 169, 4481–4487 (2002).
Helmby, H. & Grencis, R.K. Essential role for TLR4 and MyD88 in the development of chronic intestinal nematode infection. Eur. J. Immunol. 33, 2974–2979 (2003).
Willment, J.A. & Brown, G.D. C-type lectin receptors in antifungal immunity. Trends Microbiol. 16, 27–32 (2008).
Pyz, E., Marshall, A.S., Gordon, S. & Brown, G.D. C-type lectin-like receptors on myeloid cells. Ann. Med. 38, 242–251 (2006).
Brown, G.D. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 6, 33–43 (2006).
Katsu, Y. & Iguchi, T. Tissue-specific expression of Clec2g in mice. Eur. J. Cell Biol. 85, 345–354 (2006).
Greten, F.R. et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296 (2004).
Rimoldi, M. et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat. Immunol. 6, 507–514 (2005).
Allakhverdi, Z. et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 204, 253–258 (2007).
Kato, A., Favoreto, S. Jr, Avila, P.C. & Schleimer, R.P. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 179, 1080–1087 (2007).
Li, M., Hener, P., Zhang, Z., Kato, S., Metzger, D. & Chambon, P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. USA 103, 11736–11741 (2006).
Lee, H.C. & Ziegler, S.F. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc. Natl. Acad. Sci. USA 104, 914–919 (2007).
Rimoldi, M., Chieppa, M., Vulcano, M., Allavena, P. & Rescigno, M. Intestinal epithelial cells control dendritic cell function. Ann. N Y Acad. Sci. 1029, 66–74 (2004).
Fre, S., Huyghe, M., Mourikis, P., Robine, S., Louvard, D. & Artavanis-Tsakonas, S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 435, 964–968 (2005).
van Es, J.H. et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435, 959–963 (2005).
Tu, L. et al. Notch signaling is an important regulator of type 2 immunity. J. Exp. Med. 202, 1037–1042 (2005).
Shinkai, K., Mohrs, M. & Locksley, R.M. Helper T cells regulate type-2 innate immunity in vivo. Nature 420, 825–829 (2002).
Min, B. et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J. Exp. Med. 200, 507–517 (2004).
Voehringer, D., van Rooijen, N. & Locksley, R.M. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J. Leukoc. Biol. 81, 1434–1444 (2007).
Voehringer, D., Reese, T.A., Huang, X., Shinkai, K. & Locksley, R.M. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J. Exp. Med. 203, 1435–1446 (2006).
Sokol, C.L., Barton, G.M., Farr, A.G. & Medzhitov, R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 9, 310–318 (2008).
Min, B. & Paul, W.E. Basophils and type 2 immunity. Curr. Opin. Hematol. 15, 59–63 (2008).
Wakelin, D. Immunity to intestinal parasites. Nature 273, 617–620 (1978).
Wassom, D.L., Wakelin, D., Brooks, B.O., Krco, C.J. & David, C.S. Genetic control of immunity to Trichinella spiralis infections of mice. Hypothesis to explain the role of H-2 genes in primary and challenge infections. Immunology 51, 625–631 (1984).
Kennedy, M.W. & Bruce, R.G. Reversibility of the effects of the host immune response on the intestinal phase of Trichinella spiralis in the mouse, following transplantation to a new host. Parasitology 82, 39–48 (1981).
Grencis, R.K. Th2-mediated host protective immunity to intestinal nematode infections. Philos. Trans. R Soc. Lond. B Biol. Sci. 352, 1377–1384 (1997).
Pennock, J.L. & Grencis, R.K. The mast cell and gut nematodes: damage and defence. Chem. Immunol. Allergy 90, 128–140 (2006).
Tuohy, M., Lammas, D.A., Wakelin, D., Huntley, J.F., Newlands, G.F. & Miller, H.R. Functional correlations between mucosal mast cell activity and immunity to Trichinella spiralis in high and low responder mice. Parasite Immunol. 12, 675–685 (1990).
Miller, H.R. Mucosal mast cells and the allergic response against nematode parasites. Vet. Immunol. Immunopathol. 54, 331–336 (1996).
Alizadeh, H. & Murrell, K.D. The intestinal mast cell response to Trichinella spiralis infection in mast cell-deficient w/wv mice. J. Parasitol. 70, 767–773 (1984).
Donaldson, L.E., Schmitt, E., Huntley, J.F., Newlands, G.F. & Grencis, R.K. A critical role for stem cell factor and c-kit in host protective immunity to an intestinal helminth. Int. Immunol. 8, 559–567 (1996).
Grencis, R.K., Else, K.J., Huntley, J.F. & Nishikawa, S.I. The in vivo role of stem cell factor (c-kit ligand) on mastocytosis and host protective immunity to the intestinal nematode Trichinella spiralis in mice. Parasite Immunol. 15, 55–59 (1993).
Knight, P.A., Wright, S.H., Lawrence, C.E., Paterson, Y.Y. & Miller, H.R. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J. Exp. Med. 192, 1849–1856 (2000).
McDermott, J.R., Bartram, R.E., Knight, P.A., Miller, H.R., Garrod, D.R. & Grencis, R.K. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc. Natl. Acad. Sci. USA 100, 7761–7766 (2003).
Madden, K.B. et al. Enteric nematodes induce stereotypic STAT6-dependent alterations in intestinal epithelial cell function. J. Immunol. 172, 5616–5621 (2004).
Scudamore, C.L., Jepson, M.A., Hirst, B.H. & Miller, H.R. The rat mucosal mast cell chymase RMCP-II, alters epithelial cell monolayer permeability in association with altered distribution of the tight junction proteins ZO-1 and occludin. Eur. J. Cell Biol. 75, 321–330 (1998).
Abe, T., Ochiai, H., Minamishima, Y. & Nawa, Y. Induction of intestinal mastocytosis in nude mice by repeated injection of interleukin-3. Int. Arch. Allergy Appl. Immunol. 86, 356–358 (1988).
Madden, K.B., Urban, J.F. Jr, Ziltener, H.J., Schrader, J.W., Finkelman, F.D. & Katona, I.M. Antibodies to IL-3 and IL-4 suppress helminth-induced intestinal mastocytosis. J. Immunol. 147, 1387–1391 (1991).
Faulkner, H., Humphreys, N., Renauld, J.C., Van Snick, J. & Grencis, R. Interleukin-9 is involved in host protective immunity to intestinal nematode infection. Eur. J. Immunol. 27, 2536–2540 (1997).
Tsuji, K., Zsebo, K.M. & Ogawa, M. Murine mast cell colony formation supported by IL-3, IL-4, and recombinant rat stem cell factor, ligand for c-kit. J. Cell Physiol. 148, 362–369 (1991).
Tan, B.L. et al. Genetic evidence for convergence of c-Kit- and alpha4 integrin-mediated signals on class IA PI-3kinase and the Rac pathway in regulating integrin-directed migration in mast cells. Blood 101, 4725–4732 (2003).
Pennock, J.L. & Grencis, R.K. In vivo exit of c-kit+/CD49d(hi)/beta7+ mucosal mast cell precursors from the bone marrow following infection with the intestinal nematode Trichinella spiralis. Blood 103, 2655–2660 (2004).
Artis, D. et al. Beta7 integrin-deficient mice: delayed leukocyte recruitment and attenuated protective immunity in the small intestine during enteric helminth infection. Eur. J. Immunol. 30, 1656–1664 (2000).
Taub, D. et al. Bone marrow-derived murine mast cells migrate, but do not degranulate, in response to chemokines. J. Immunol. 154, 2393–2402 (1995).
Rosbottom, A., Scudamore, C.L., von der Mark, H., Thornton, E.M., Wright, S.H. & Miller, H.R. TGF-beta 1 regulates adhesion of mucosal mast cell homologues to laminin-1 through expression of integrin alpha 7. J. Immunol. 169, 5689–5695 (2002).
Weller, C.L. et al. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J. Exp. Med. 201, 1961–1971 (2005).
Brown, J.K. et al. Expression of integrin-alphaE by mucosal mast cells in the intestinal epithelium and its absence in nematode-infected mice lacking the transforming growth factor-beta1-activating integrin alphavbeta6. Am. J. Pathol. 165, 95–106 (2004).
Arizono, N. et al. Infection of Nippostrongylus brasiliensis induces development of mucosal-type but not connective tissue-type mast cells in genetically mast cell-deficient Ws/Ws rats. Blood 81, 2572–2578 (1993).
Pemberton, A.D., Brown, J.K., Wright, S.H., Knight, P.A. & Miller, H.R. The proteome of mouse mucosal mast cell homologues: the role of transforming growth factor beta1. Proteomics 6, 623–631 (2006).
Munger, J.S. et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 (1999).
Ludlow, A. et al. Characterization of integrin beta6 and thrombospondin-1 double-null mice. J. Cell Mol. Med. 9, 421–437 (2005).
Alizadeh, H., Urban, J.F. Jr, Katona, I.M. & Finkelman, F.D. Cells containing IgE in the intestinal mucosa of mice infected with the nematode parasite Trichinella spiralis are predominantly of a mast cell lineage. J. Immunol. 137, 2555–2560 (1986).
Akiho, H., Blennerhassett, P., Deng, Y. & Collins, S.M. Role of IL-4, IL-13, and STAT6 in inflammation-induced hypercontractility of murine smooth muscle cells. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G226–G232 (2002).
Pizarro, T.T. et al. IL-18, a novel immunoregulatory cytokine, is upregulated in Crohn's disease: expression and localization in intestinal mucosal cells. J. Immunol. 162, 6829–6835 (1999).
Helmby, H. & Grencis, R.K. IL-18 regulates intestinal mastocytosis and Th2 cytokine production independently of IFN-gamma during Trichinella spiralis infection. J. Immunol. 169, 2553–2560 (2002).
Anthony, R.M. et al. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat. Med. 12, 955–960 (2006).
Brailsford, T.J. & Behnke, J.M. The dynamics of trickle infections with Heligmosomoides polygyrus in syngeneic strains of mice. Int. J. Parasitol. 22, 351–359 (1992).
Behnke, J.M., Wahid, F.N., Grencis, R.K., Else, K.J., Ben-Smith, A.W. & Goyal, P.K. Immunological relationships during primary infection with Heligmosomoides polygyrus (Nematospiroides dubius): downregulation of specific cytokine secretion (IL-9 and IL-10) correlates with poor mastocytosis and chronic survival of adult worms. Parasite Immunol. 15, 415–421 (1993).
Hayes, K.S., Bancroft, A.J. & Grencis, R.K. Immune-mediated regulation of chronic intestinal nematode infection. Immunol. Rev. 201, 75–88 (2004).
Wahid, F.N., Robinson, M. & Behnke, J.M. Immunological relationships during primary infection with Heligmosomoides polygyrus (Nematospiroides dubius): expulsion of adult worms from fast responder syngeneic and hybrid strains of mice. Parasitology 98 (Part 3), 459–469 (1989).
Potten, C.S. Stem cells in gastrointestinal epithelium: numbers, characteristics and death. Philos. Trans. R Soc. Lond. B Biol. Sci. 353, 821–830 (1998).
Marshman, E., Booth, C. & Potten, C.S. The intestinal epithelial stem cell. Bioessays 24, 91–98 (2002).
Newlands, G.F., Gibson, S., Knox, D.P., Grencis, R., Wakelin, D. & Miller, H.R. Characterization and mast cell origin of a chymotrypsin-like proteinase isolated from intestines of mice infected with Trichinella spiralis. Immunology 62, 629–634 (1987).
Miller, H.R., Huntley, J.F. & Wallace, G.R. Immune exclusion and mucus trapping during the rapid expulsion of Nippostrongylus brasiliensis from primed rats. Immunology 44, 419–429 (1981).
Shekels, L.L. et al. Coordinated Muc2 and Muc3 mucin gene expression in Trichinella spiralis infection in wild-type and cytokine-deficient mice. Dig. Dis. Sci. 46, 1757–1764 (2001).
Knight, P.A., Pemberton, A.D., Robertson, K.A., Roy, D.J., Wright, S.H. & Miller, H.R. Expression profiling reveals novel innate and inflammatory responses in the jejunal epithelial compartment during infection with Trichinella spiralis. Infect. Immun. 72, 6076–6086 (2004).
Datta, R., deSchoolmeester, M.L., Hedeler, C., Paton, N.W., Brass, A.M. & Else, K.J. Identification of novel genes in intestinal tissue that are regulated after infection with an intestinal nematode parasite. Infect. Immun. 73, 4025–4033 (2005).
Karlsson, N.G., Olson, F.J., Jovall, P.A., Andersch, Y., Enerback, L. & Hansson, G.C. Identification of transient glycosylation alterations of sialylated mucin oligosaccharides during infection by the rat intestinal parasite Nippostrongylus brasiliensis. Biochem J. 350 (Part 3), 805–814 (2000).
Maruyama, H. et al. Strongyloides venezuelensis: longitudinal distribution of adult worms in the host intestine is influenced by mucosal sulfated carbohydrates. Exp. Parasitol. 100, 179–185 (2002).
Otto, W.R. & Thim, L. Trefoil factor family-interacting proteins. Cell Mol. Life Sci. 62, 2939–2946 (2005).
Mashimo, H., Wu, D.C., Podolsky, D.K. & Fishman, M.C. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274, 262–265 (1996).
Blanchard, C. et al. IL-4 and IL-13 upregulate intestinal trefoil factor expression: requirement for STAT6 and de novo protein synthesis. J. Immunol. 172, 3775–3783 (2004).
Komiya, T., Tanigawa, Y. & Hirohashi, S. Cloning and identification of the gene gob-5, which is expressed in intestinal goblet cells in mice. Biochem. Biophys. Res. Commun. 255, 347–351 (1999).
Komiya, T., Tanigawa, Y. & Hirohashi, S. Cloning of the novel gene intelectin, which is expressed in intestinal Paneth cells in mice. Biochem. Biophys. Res. Commun. 251, 759–762 (1998).
Nair, M.G., Guild, K.J. & Artis, D. Novel effector molecules in type 2 inflammation: lessons drawn from helminth infection and allergy. J. Immunol. 177, 1393–1399 (2006).
Pemberton, A.D. et al. Innate BALB/c enteric epithelial responses to Trichinella spiralis: inducible expression of a novel goblet cell lectin, intelectin-2, and its natural deletion in C57BL/10 mice. J. Immunol. 173, 1894–1901 (2004).
He, W. et al. Bacterial colonization leads to the colonic secretion of RELMbeta/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 125, 1388–1397 (2003).
Artis, D. et al. RELM{beta}/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA 101, 13596–13600 (2004).
Wang, M.L. et al. Regulation of RELM/FIZZ isoform expression by Cdx2 in response to innate and adaptive immune stimulation in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G1074–G1083 (2005).
Artis, D. New weapons in the war on worms: identification of putative mechanisms of immune-mediated expulsion of gastrointestinal nematodes. Int. J. Parasitol. 36, 723–733 (2006).
Zaph, C., Rook, K.A., Goldschmidt, M., Mohrs, M., Scott, P. & Artis, D. Persistence and function of central and effector memory CD4+ T cells following infection with a gastrointestinal helminth. J. Immunol. 177, 511–518 (2006).
Artis, D. et al. The IL-27 receptor (WSX-1) is an inhibitor of innate and adaptive elements of type 2 immunity. J. Immunol. 173, 5626–5634 (2004).
Cliffe, L.J. & Grencis, R.K. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv. Parasitol. 57, 255–307 (2004).
Betts, J., deSchoolmeester, M.L. & Else, K.J. Trichuris muris: CD4+ T cell-mediated protection in reconstituted SCID mice. Parasitology 121 (Part 6), 631–637 (2000).
Else, K.J. & Grencis, R.K. Antibody-independent effector mechanisms in resistance to the intestinal nematode parasite Trichuris muris. Infect. Immun. 64, 2950–2954 (1996).
Betts, C.J. & Else, K.J. Mast cells, eosinophils and antibody-mediated cellular cytotoxicity are not critical in resistance to Trichuris muris. Parasite Immunol. 21, 45–52 (1999).
Koyama, K. & Ito, Y. Mucosal mast cell responses are not required for protection against infection with the murine nematode parasite Trichuris muris. Parasite Immunol. 22, 13–20 (2000).
Artis, D., Potten, C.S., Else, K.J., Finkelman, F.D. & Grencis, R.K. Trichuris muris: host intestinal epithelial cell hyperproliferation during chronic infection is regulated by interferon-gamma. Exp. Parasitol. 92, 144–153 (1999).
Cliffe, L.J., Humphreys, N.E., Lane, T.E., Potten, C.S., Booth, C. & Grencis, R.K. Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science 308, 1463–1465 (2005).
Else, K.J., Hultner, L. & Grencis, R.K. Cellular immune responses to the murine nematode parasite Trichuris muris. II. Differential induction of TH-cell subsets in resistant versus susceptible mice. Immunology 75, 232–237 (1992).
Bancroft, A.J., Else, K.J., Sypek, J.P. & Grencis, R.K. Interleukin-12 promotes a chronic intestinal nematode infection. Eur. J. Immunol. 27, 866–870 (1997).
Bancroft, A.J., Humphreys, N.E., Worthington, J.J., Yoshida, H. & Grencis, R.K. WSX-1: a key role in induction of chronic intestinal nematode infection. J. Immunol. 172, 7635–7641 (2004).
Cliffe, L.J., Potten, C.S., Booth, C.E. & Grencis, R.K. An increase in epithelial cell apoptosis is associated with chronic intestinal nematode infection. Infect. Immun. 75, 1556–1564 (2007).
Morimoto, M. et al. Functional importance of regional differences in localized gene expression of receptors for IL-13 in murine gut. J. Immunol. 176, 491–495 (2006).
Hayes, K.S., Bancroft, A.J. & Grencis, R.K. The role of TNF-alpha in Trichuris muris infection I: influence of TNF-alpha receptor usage, gender and IL-13. Parasite Immunol. 29, 575–582 (2007).
Hayes, K.S., Bancroft, A.J. & Grencis, R.K. The role of TNF-alpha in Trichuris muris infection II: global enhancement of ongoing Th1 or Th2 responses. Parasite Immunol. 29, 583–594 (2007).
Bell, L.V. & Else, K.J. Mechanisms of leucocyte recruitment to the inflamed large intestine: redundancy in integrin and addressin usage. Parasite Immunol. 30, 163–170 (2008).
Zhao, A. et al. Dependence of IL-4, IL-13, and nematode-induced alterations in murine small intestinal smooth muscle contractility on Stat6 and enteric nerves. J. Immunol. 171, 948–954 (2003).
Liu, Q. et al. IL-18 stimulates IL-13-mediated IFN-gamma-sensitive host resistance in vivo. Eur. J. Immunol. 36, 1187–1198 (2006).
McDermott, J.R., Leslie, F.C., D’Amato, M., Thompson, D.G., Grencis, R.K. & McLaughlin, J.T. Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut 55, 492–497 (2006).
Chan, M.S. The global burden of intestinal nematode infections—fifty years on. Parasitol. Today 13, 438–443 (1997).
Summers, R.W., Elliott, D.E. & Weinstock, J.V. Is there a role for helminths in the therapy of inflammatory bowel disease? Nat. Clin. Pract. Gastroenterol. Hepatol. 2, 62–63 (2005).
Summers, R.W., Elliott, D.E., Urban, J.F. Jr, Thompson, R. & Weinstock, J.V. Trichuris suis therapy in Crohn's disease. Gut 54, 87–90 (2005).
Elliott, D.E., Summers, R.W. & Weinstock, J.V. Helminths and the modulation of mucosal inflammation. Curr. Opin. Gastroenterol. 21, 51–58 (2005).
Schopf, L.R., Hoffmann, K.F., Cheever, A.W., Urban, J.F. Jr & Wynn, T.A. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J. Immunol. 168, 2383–2392 (2002).
Maizels, R. Regulation of the immune system in metazoan parasite infections. Novartis Found. Symp. 281, 192–204 (2007), discussion 204–199.
Wakelin, D. Acquired immunity to Trichuris muris in the albino laboratory mouse. Parasitology 57, 515–524 (1967).
Wakelin, D. The development of the early larval stages of Trichuris muris in the albino laboratory mouse. J. Helminthol. 43, 427–436 (1969).
Wakelin, D. Studies on the immunity of albino mice to Trichuris muris. Suppression of immunity by cortisone acetate. Parasitology 60, 229–237 (1970).
Lee, T.D. & Wright, K.A. The morphology of the attachment and probable feeding site of the nematode Trichuris muris (Schrank, 1788) Hall, 1916. Can. J. Zool. 56, 1889–1905 (1978).
Tilney, L.G., Connelly, P.S., Guild, G.M., Vranich, K.A. & Artis, D. Adaptation of a nematode parasite to living within the mammalian epithelium. J. Exp. Zool. A Comp. Exp. Biol. 303, 927–945 (2005).
Wright, K.A. Trichinella spiralis: an intracellular parasite in the intestinal phase. J. Parasitol. 65, 441–445 (1979).
Despommier, D.D. Trichinella spiralis: the worm that would be virus. Parasitol. Today 6, 193–196 (1990).
Acknowledgements
Work in the Artis laboratory was supported by the National Institutes of Health (A161570 and AI074878), the Pilot Feasibility Program of the National Institute of Diabetes and Digestive Kidney Diseases (NIDDK DK50306), the Irvington Institute Fellowship Program of the Cancer Research Institute, the Crohn's and Colitis Foundation of America's William and Shelby Modell Family Foundation Research Award, the UPenn Research Foundation Award, and the UPenn Center for Infectious Diseases Pilot Grant. Work in the Grencis laboratory was supported by the Wellcome Trust, the Medical Research Council of the UK, and the Biotechnology and Biological Sciences Research Council. We thank Jackie Perrigoue and Meera Nair for comments on the paper and apologize to colleagues whose work could not be discussed due to space constraints.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Artis, D., Grencis, R. The intestinal epithelium: sensors to effectors in nematode infection. Mucosal Immunol 1, 252–264 (2008). https://doi.org/10.1038/mi.2008.21
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2008.21
This article is cited by
-
Impact of parasitic infection on human gut ecology and immune regulations
Translational Medicine Communications (2021)
-
Bovine neutrophils form extracellular traps in response to the gastrointestinal parasite Ostertagia ostertagi
Scientific Reports (2018)
-
Goblet cells: multifaceted players in immunity at mucosal surfaces
Mucosal Immunology (2018)
-
Intraperitoneal administration of the anti-IL-23 antibody prevents the establishment of intestinal nematodes in mice
Scientific Reports (2018)
-
Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity
Mucosal Immunology (2018)
