
Abstract
Axial excitation confinement beyond the diffraction limit is crucial to the development of next-generation, super-resolution microscopy. STimulated Emission Depletion (STED) nanoscopy offers lateral super-resolution using a donut-beam depletion, but its axial resolution is still over 500 nm. Total internal reflection fluorescence microscopy is widely used for single-molecule localization, but its ability to detect molecules is limited to within the evanescent field of ~100 nm from the cell attachment surface. We find here that the axial thickness of the point spread function (PSF) during confocal excitation can be easily improved to 110 nm by replacing the microscopy slide with a mirror. The interference of the local electromagnetic field confined the confocal PSF to a 110-nm spot axially, which enables axial super-resolution with all laser-scanning microscopes. Axial sectioning can be obtained with wavelength modulation or by controlling the spacer between the mirror and the specimen. With no additional complexity, the mirror-assisted excitation confinement enhanced the axial resolution six-fold and the lateral resolution two-fold for STED, which together achieved 19-nm resolution to resolve the inner rim of a nuclear pore complex and to discriminate the contents of 120 nm viral filaments. The ability to increase the lateral resolution and decrease the thickness of an axial section using mirror-enhanced STED without increasing the laser power is of great importance for imaging biological specimens, which cannot tolerate high laser power.
Similar content being viewed by others

Introduction
The confinement of excitation light is the key for optical microscopy to resolve sub-cellular fine structures against their neighboring background1, 2, 3. To give a few classic examples, laser scanning confocal microscopy offers higher resolution than wide-field microscopy because it uses a tightly focused excitation beam and pinhole detection to reject out-of-focus background light; stimulated emission depletion (STED) microscopy offers lateral resolution beyond the optical diffraction limit because it employs a donut-beam confined excitation to project an optically saturated virtual pinhole4, 5, and 3D super-resolution can be attained through further axial modulation6; total internal reflection fluorescence (TIRF) microscopy is suitable for single-molecule localization microscopy because it takes advantage of the evanescent field to image a superficial layer of the biological cell with an imaging depth of ~100 nm, attaining sharper features of the specimen2.
Optical interference has been used to enhance the axial resolution by generating different types of excitation modulation in wide-field fluorescence microscopy (Supplementary Fig. S1 and Supplementary Table S1)7, 8, 9, 10, 11, 12, 13. Bailey et al.7 proposed standing-wave excitation microscopy based on mirror reflection, but because wide-field illumination was used, the interference layer was mixed with the out-of-focus region in detection. No experimental data using mirror reflection were obtained until the I5M microscopy14, where two opposing objectives were used to interfere two coherent incident beams and the fluorescent signal. Most recently, by employing a low numerical aperture (NA) objective, a mirror, and a plan-convex lens to form Newton rings, standing-wave multiplanar excitation was demonstrated for axial imaging9.
Interference of confocal excitation light during 4Pi microscopy has been demonstrated with an axial resolution of ~100 nm (ref. 15), but it requires the precise alignment of two diffraction-limited focal spots by two opposite microscope setups. Similarly, the isotropic focusing approach utilizes a spatial light modulator (SLM) to modulate the wave front and generate two focal spots axially, and a mirror is placed in between to form the constructive interference10, 16, 17. The position of the mirror is critical to generate effective interference of the two ‘real’ focal spots; therefore, the alignment precision of the SLM-Mirror setup is required to be <50 nm, whereas the 4Pi setup requires alignment precision <100 nm.
Other techniques have been explored to achieve axial super-resolution. Using the STED process, an angle-STED, based on a tilted mirror, has been built to generate angled super-resolution excitation (Supplementary Fig. S1j)11, similar to that of reflected light sheet microscopy18. Axial localization of fluorescent molecules with spectrally self-interference microscopy, by mirror reflection and a large spacer (>10λ)8 or a Langmuir–Blodgett thin film as spacer with metal-induced energy transfer has been demonstrated for axial optical nanosectioning12. Most recently, the fluorescence lifetimes have been found to correlate highly with the distance of a fluorophore from a gold mirror because the nanoplasmonics interactions and quenching/enhancement effects are highly sensitive to the distance, which has been employed for constructing a novel type of optical nanoscopy to image the membranes of live cells with super-resolution on a gold mirror surface13.
In this work, we report a highly practical approach to immediately enable laser scanning confocal microscopy and STED nanoscopy to achieve axial super-resolution and to resolve structures, such as the nuclear pore complex, within the interior of cells (see Figure 1a and Supplementary Scheme S1). We found that when a mirror is placed behind the specimen, the interference between the excitation point spread function (PSF) and its reflection creates an axially narrowed PSF away from the mirror surface, ~110 nm in dimension. Therefore, working in the far-field regime, this mirror-enhanced axial-narrowing super-resolution (MEANS) technique enables a more than six-fold higher axial resolution than the optical diffraction limit. Our MEANS approach is equivalent to a virtual 4Pi configuration, but it relaxes the alignment precision and does not require wave front modulation, as in isotropic focusing. With the axial sectioning capability and two-fold improvement of the MEANS-STED super-resolution, the inner ring structure of the nuclear pore complex, as well as the tubular structure of a viral filamentous virion, has been successfully revealed with optical microscopy for the first time. Because biological specimens and the fluorescent dye can be photodamaged by high laser power, it is of particular significance that MEANS-STED has improved both the lateral and the axial resolution without increasing the laser power.

Schematic diagram of MEANS (a) and correlative images of a single cell acquired in TIRF (b) and MEANS (c) microscopy imaging modes. MEANS microscopy can be easily realized with a confocal microscope and takes advantage of axial interference between the incident and reflected electromagnetic field to generate an axially confined PSF ~100 nm above the reflective mirror surface. The merged image (d) of TIRF (green) and MEANS (red) shows that the MEANS approach complements the oblique illumination-based TIRF modality by optically sectioning a cell at a different axial layer which, in this case, is close to the mirror surface. The sample here is a Vero cell immunostained for tubulin using an AlexaFluor-488 secondary antibody. Scale bar=10 μm.
Materials and methods
MEANS sample preparation
In MEANS microscopy, a piece of mirror is employed in place of the microscope slide to provide the interference. Custom-made, first-surface mirrors were used (China Daheng Group, Beijing, China). The mirror should be a first-surface mirror, with a protective SiO2 coating, and an adjustable thickness (50, 100, 150 and 200 nm were tested), so that the constructive interference with a high NA objective can occur within the specimen. Due to the existence of the silica layer, cells can grow normally on the mirror surface. A coverslip can be applied to seal the specimen. As shown in Supplementary Scheme 1b, we have a custom-made mirror holder that is the same size as a microscope slide so that the mirror-backed specimen can be placed easily on any commercial confocal microscope.
Cellular sample preparation
Vero cells (CCL-81, ATCC, Manassas, VA, USA) were maintained in high-glucose DMEM (Lonza, Basel, Switzerland) with 10% fetal bovine serum (GE Healthcare Life Sciences HyClone, Logan, UT, USA), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). The human respiratory syncytial virus (hRSV) strain A2 (VR-1544, ATCC, Manassas, VA, USA) was propagated in HEp-2 cells (CCL-23, ATCC) at a titer of 1 × 106 p.f.u. ml−1 Cells were plated the day before infection at 25% confluency. Cells were infected by removing the media, washing with phosphate-buffered saline (PBS) (without Ca2+ and Mg2+ ions, Lonza, Basel, Switzerland), adding virus at a multiplicity of infection (MOI) of 1, and incubating the cells for 1 h at 37 °C. After adsorption, fresh medium was added to the inoculum. The cells were grown on top of a first-surface mirror coated with an SiO2 protective layer.
Vero cells were fixed with either 4% paraformaldehyde (VWR, Radnor, PA, USA) in PBS for 10 min at room temperature and then permeabilized with 0.2% Triton X-100 (for nuclear pore complex immunofluorescent staining, VWR) or with 100% ice-cold methanol for 10 min at −20 °C and then permeabilized with 100% ice-cold acetone for 2 min at −20 °C (for microtubule immunofluorescent staining, VWR). Nonspecific antibody binding was blocked with 5% bovine serum albumin (EMD Millipore, Darmstadt, Germany) in PBS for 30 min at 37 °C. The cells were then incubated with a primary antibody for 30 min at 37 °C, washed twice in PBS, incubated with a secondary antibody for 30 min at 37 °C, washed twice in PBS and mounted in a mixture of Mowiol 4-88 (Sigma Aldrich, St Louis, MO, USA) and DABCO (VWR)19.
The primary antibodies used were rabbit anti-alpha tubulin (polyclonal IgG, catalog: ab18251, Abcam, Cambridge, MA, USA) and mouse anti-nuclear pore complex (NPC) proteins that contain FXFG repeats (monoclonal IgG, catalog: ab24609, Abcam). The secondary antibodies used were goat anti-rabbit DyLight 650 (Thermo Fisher Scientific, Waltham, MA, USA) and donkey anti-mouse AlexaFluor 488 (Thermo Fisher Scientific).
Spin capture of RSV filaments on glass
To capture single hRSV filamentous virions on glass, hRSV A2 was propagated in HEp-2 cells at an MOI of 0.1. At 4 days post infection, the cell-associated and supernatant fractions were scraped, freeze-thawed and spun through 5 and 0.45 μm pore-size centrifugal filters (EMD Millipore) at 5000 × g and 4 °C for 4 and 1 min, respectively. The fraction between 0.45 and 5 μm in diameter was collected and immobilized onto a poly-l-lysine (Sigma Aldrich)-coated, first-surface mirror or cover glass by adsorption of 500 μl of filtered virus for 2 h at 4 °C. The immobilized virions were fixed using 4% paraformaldehyde and were immunofluorescently stained according to the aforementioned protocol. The antibodies used were anti-RSV F monocolonal (palivizumab, MedImmune, Gaithersburg, MD, USA) and anti-RSV N monoclonal (monoclonal IgG1, ab22501, Abcam). Coverslips were mounted in a mixture of Mowiol and DABCO (VWR)19.
MEANS-STED
MEANS-STED imaging was performed with a Leica TCS SP8 STED 3X system equipped with a white light laser as excitation and 592 and 660 nm for STED depletion. The HyD detector and 100 × oil-immersion objective (NA 1.4) were employed. Time-gated detection was also used20, in which the AlexaFluor-488 detection is delayed 0.5 ns, whereas the Cy5 detection is delayed 1 ns.
Results and discussion
MEANS and TIRF microscopy reveal complementary structural information
Supplementary Fig. S1 further illustrates the schematic difference comparing the MEANS technique with a variety of available axial-enhanced microscopy techniques. MEANS, similar to TIRF by providing wide-field microscopy with axial confinement and improved contrast, can confine the axial PSF depth of confocal microscopy to ~100 nm, with doubled signal intensity. Furthermore, MEANS complements the TIRF imaging modality by optically sectioning two different super-resolution layers: TIRF is responsible for imaging the sample layer close to the coverslip, whereas MEANS optically sections the layer that is close to the mirror (interior of the specimen). In Figure 1b–1d, we demonstrate a series of in situ correlative images of TIRF and MEANS from a single Vero cell. Whereas the layer near the coverslip can be imaged by TIRF (or HiLo mode21 because the refractive index of the medium is larger than the NA of the objective, Figure 1b), the MEANS imaging modality (Figure 1c) provides super-resolution information within the interior layer of the cells with a ~110 nm axial thickness but a distance of ~100 nm from the mirror surface (wavelength dependent), making MEANS more suitable for the study of problems that are not at the cell membrane but localized more deeply within the cell. When the MEANS image is overlaid with a complementary TIRF image (Figure 1d), the spatial relationship between the two images is clear (see also wide-field in Supplementary Fig. S2, in which both layers can be visualized simultaneously).
Theoretical simulation of the MEANS and MEANS-STED PSFs
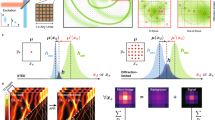
To predict the axial confinement of the PSF, we simulated the excitation electromagnetic field for multiple imaging techniques (Figure 2). Our simulation results of the electromagnetic field of the MEANS microscopy revealed that, the local maximum intensity for MEANS (Figure 2c) is approximately four times that of conventional confocal microscopy (Figure 2a) and approximately two times that of 4Pi (Figure 2b). Because the local maximum is a result of constructive interference, MEANS can generate local EM-field enhancement over a long focal distance (Supplementary Video S1), relieving the need for precise alignment of the mirror.

Theoretical simulation results of the focal intensity profiles of: (a) confocal excitation, (b) 4Pi excitation, (c) MEANS excitation, (d) STED depletion and (e) MEANS-STED depletion. The origin of the z-axis denotes the center of PSFs in a–c and the mirror position in d and e. Whereas confocal microscopy can generate a PSF with 700 nm axial thickness, 4Pi and MEANS can generate PSFs with ~110 nm axial thickness, benefitting from axial interference. Objectives with n=1.5, NA=1.4, and λex=488 nm and λdep=592 nm are used for simulation. The local maximum intensity for MEANS d is approximately four times that of conventional confocal microscopy a and two times that of 4Pi b because in 4Pi, the beam is split into two and then recombined, whereas MEANS takes full advantage of the incident intensity through reflection. Because the intensity of depletion is improved by 3.6-fold, close to two-fold resolution enhancement over conventional STED can be obtained for MEANS-STED, as shown in (f).
Demonstration of optical sectioning using MEANS and excitation scanning
Because the distance between the mirror and the MEANS-enhanced layer is linearly proportional to the excitation wavelength, we can realize optical sectioning within the specimen with MEANS through excitation wavelength modulation (MEANS-excitation scanning optical nanosectioning, MEANS-ESON), which has been experimentally verified using MEANS to axially section fluorescent nanodiamonds by scanning the excitation wavelength and taking advantage of the broad absorption spectrum of the nitrogen vacancy centers within nanodiamonds. A laser scanning confocal system (Leica TCS SP8) equipped with a white light laser and a plan-apochromatic objective (Leica HC PL APO CS2, 63x/1.40 OIL) has been used. As shown in Figure 3, three scanning results using different excitation bands resulted in different layers of nanodiamond nanoparticles (pseudocolor is used to indicate different layers of nanoparticles scanned by different excitation color). If the nanodiamonds are in the same layer, then the color should be identical. However, from the arrows, we can see that some of the nanodiamonds are yellow as the dominant color (which is a combination of R and G), whereas others are strongly blue (which locates mainly in the B layer, closer to the mirror).

Imaging of nanodiamond particles embedded in agarose with different excitation wavelengths. Excitation with longer wavelength results in a further PSF relative to the mirror. In the lower RGB image, the fluorescence excited by 470–490, 550–570 and 650–670 nm is mapped to the B, G and R channels, respectively. Arrows show beads with different colors, which indicates that they are at different depths and are differentiated by MEANS-excitation scan optical nanosectioning (MEANS-ESON). Scale bar=5 μm.
Furthermore, taking advantage of the virtual PSF, MEANS modality has only one sidelobe with which to generate images with sharper contrast, whereas the PSF of 4Pi has two symmetric sidelobes22, which requires deconvolution for data representation. In our MEANS approach, after passing through the confocal pinhole, the only sidelobe is largely suppressed, making the image very sharp.
Verification of the axial confinement thickness
To verify the improved axial PSF confinement of 110 nm, we prepared a phantom specimen by embedding 20 nm fluorescent beads inside low-concentration agarose, which was then evaluated by MEANS confocal microscopy. As shown in Supplementary Fig. S3, a three-fold enhancement in the signal-to-background ratio (SBR) was achieved at the 13–14-μm layer primarily because with the decrease of the PSF, the fluorescent signal increases proportionally due to the higher excitation energy confinement. We have also simulated the effect of the integrated energy enhancement (detection photon number) for MEANS with different confocal pinhole sizes, and the simulation further validated our experimental results.
Dual-color MEANS microscopy
To further verify the above simulation results and to demonstrate the robustness of the MEANS modality, we applied the MEANS approach on a commercial laser scanning confocal microscope (single- and two-photon excitation) as well as a spinning-disk microscope. We acquired a series of images along the z-direction of the microtubules of a Vero cell grown on the mirror substrate. The images for confocal, two-photon and spinning disk approaches are shown in Supplementary Figs S4–S6, respectively. The dual-color imaging of microtubules and NPCs in a Vero cell is shown in Figure 4. In these figures, image stacks are acquired by moving the objective toward the specimen. According to our simulation result (Supplementary Video S1), the position of the MEANS confinement focal spot is fixed ~100 nm above the mirror surface during mirror translation. The layers from 2–2.5 μm in Figure 4 and Supplementary Fig. S4 belong to the MEANS modality region and show almost identical images due to the fixed MEANS focal spot. The optical sectioning layers above this 0.5 μm region belong to the conventional confocal microscopy modality. These quantitative imaging results indicate that the MEANS-confocal modality has a tolerant region of ~0.4 μm to section an axially confined thin layer of the cell, 100 nm from the mirror, which is consistent with our simulation result (Supplementary Video S1). Comparing the MEANS image (Figure 4f) to the confocal images (Figure 4a–4c), the MEANS with excitation intensities enhanced by interference can significantly enhance the image SNR through its increased fluorescence signal intensity, which suggests that the conventional axial resolution measurement, in which the specimen translates relative to the objective will not work in MEANS because the PSF does not move with the objective.

Confocal and MEANS image of the dual-stained Vero cell. The microtubules of the cell are stained with Dylight 650 (pseudo-colored red), and the nuclear pore complex of the cell is stained with Alexa 488 (pseudo-colored green). (a–h) Image series taken from a confocal microscope (Olympus FV1200) with axial step of 0.5 μm. As observed, MEANS forms at 2.5 μm depth in f, in which the nuclear pore proteins are shown clearly as grains. Scale bar=5 μm.
Moving the MEANS layer within the cell
To show the possibility of imaging at different depths into a cell, we grew Vero cells on mirrors coated with different thicknesses of silica. Figure 5 shows different optical sectioning layers of actin filaments within the Vero cells, achieved by changing the thickness of the silica coating from 50, 100, 150 and 200 nm. Actin microfilaments, one type of cytoskeletal element, are responsible for cell shape and cell motility. Figure 5c and 5d shows the optical sections of the MEANS layer as it is moved closer and closer to the cell surface membrane when the thickness of silica spacer is increased from 50 to 150 nm. By rigorously examining the 50 nm layers, we found that all images (typically shown in Figure 5b) were rich in actin filaments and bundles but with very little blank shadow of the nuclei. The images from the 100 and 150 nm layers (Figure 5c and 5d) show clear elliptical shadows of the nuclei free of the actin filaments inside. In addition, the margins of the cell are more apparent in Figure 5c and 5d compared with the 50 nm in Figure 5b. After further increasing the thickness of the silica spacer to 200 nm, the images contain both actin and nuclei (Figure 5e), similar to the control images taken by conventional confocal microscopy (Figure 5f), suggesting that the axial super-resolution power from the MEANS modality disappears when the silica spacer is more than 200 nm. In addition, MEANS is tolerant to small-angle misalignments of the mirror relative to the objective (Supplementary Fig. S7).

Imaging of the actin filaments with different thickness of silica spacer. Adjusting the spacer thickness can obtain the cross-sectional imaging of different layers of the cell specimen. For MEANS constructive interference, the maximum spacer thickness is 100–150 nm. The images are all of the same size. Scale bar=10 μm.
MEANS-STED microscopy of the NPC and viral filaments
Another significant advantage of the MEANS modality is its compatibility with STED microscopy. According to our simulation results (Figure 2d and 2e, and Supplementary Video S2), the local electromagnetic field for the vortex-modulated ‘doughnut’ STED PSF is axially confined to form a 163-nm MEANS-STED depletion PSF. Therefore, both the intensity of the donut-shaped depletion beam and the excitation beam23, 24, 25 have been enhanced by 3.6-fold within the MEANS region. Because the resolution of STED is dependent on the depletion intensity, which can be written as26, 27

where Idep is the depletion intensity and Isat is the saturation intensity of the molecule, the constructive interference in MEANS-STED mode provides close to two-fold resolution enhancement over conventional STED.
To verify our MEANS method in STED nanoscopy, we applied the MEANS approach in a commercial Leica TCS SP8 STED 3X super-resolution microscope. As a result of the MEANS-assisted STED (Figure 6a–6d), we achieved 19-nm resolution to visualize the inner rim of the NPC using a relatively low 592-nm depletion laser power of 60 mW measured at the back aperture of the objective. The NPC restricts the diffusion of molecules between the nucleoplasm to the cytoplasm and is composed of three concentric ring nucleoporins (Nups) held together by linker proteins. The inner channel is composed of the phenylalanine-glycine (FG) Nups28. Because of the small diameter of FG Nups (~50 nm), its ring structure has only been revealed by electron microscopy from isolated NPC complexes29, 30. Even when other super-resolution techniques, such as dSTORM, were utilized with localization precision of 20 nm, the ring structure was unresolvable31. After the inner channels (including Nup62, Nup95 and Nup110 proteins) of the NPCs were stained, MEANS in conjunction with a commercial STED system enabled us to record one of the first optical images of an FG Nup in a mammalian cell (Figure 6a–6d). The measured diameter relative to the size of the whole nuclear pore complex confirmed that the antibody was indeed bound to Nups in the center of the NPC29. The resolution of 19 nm is one of the new record of STED super-resolution in biological applications32, 33, 34, 35, 36. The resolution improvement is largely because two-fold resolution enhancement can be obtained without increasing the depletion laser power. Because biological specimens and fluorescent dyes are generally very sensitive to high power laser, this is a very important advance in order to dramatically improve the resolution without increasing the laser power. A comparison with conventional 3D STED can be found in Supplementary Fig. S8.

The NPC of a Vero cell (a–c) imaging using confocal (a: upper right) and MEANS-STED (a: lower bottom) modalities, and the hRSV viral filaments imaging via conventional STED (f–i) and MEANS-STED (l–o) modalities. MEANS-STED gives clear lateral resolution enhancement in a. (b) Magnification of the boxed area in a, in which the porous structure of the NPCs can be observed. (c) Magnified individual NPCs. (d) Plot of the intensity distribution along arrows in c. To demonstrate the advantage of the axial confinement of MEANS-STED, the hRSV filaments are imaged with conventional STED (on coverglass, f–i) and MEANS-STED (on mirror, l–o). The simulation of the convolution of conventional STED PSF vs MEANS-STED PSF with the filament structure is shown in (e) and (k), respectively. The central hollow structure is hard to visualize due to the uniform intensity distribution in e, but in k it can be observed. (i,o) Magnifications of the white box areas in h and n, respectively. The hollow structure of the hRSV-F can be visualized, taking advantage of the optical sectioning of MEANS-STED. (j, p) Intensity plots of the line indicated by the yellow arrows in i and o, respectively. Gaussian fitting of the data in j only shows one slope with width of 214 nm, whereas the fitting in p indicates that the width of the hRSV-F is ~56 nm, and the distance between the split peaks is ~119 nm. Scale bar=500 nm (f, l).
The axial super-resolution achieved by the MEANS approach has further assisted STED for 3D super-resolution visualization of the sub-micron hollow structures. hRSV produces filamentous virions that are 100–200 nm in diameter and up to several microns in length. The distance between the centroid of the fusion (F) proteins was measured as ~120 nm by dSTORM37. To use this model system to test MEANS-STED, we immunofluorescently stained the F protein, which is in the virion membrane, and the nucleoprotein (N), which is bound to the genomic RNA inside the virion space. Without the mirror, despite using even higher STED depletion power (108 mW at 592 nm) than the MEANS-STED (72 mW), the conventional STED with the axial PSF of over 500 nm cannot resolve the F protein on the two opposing sides of the membrane (Figure 6f–6i) due to contaminating light from the F protein present on the top and bottom of the filament. With the MEANS implementation (Figure 6l–6o), a thin section in the middle of the filament is excited, and the top and bottom of the filament are not excited, allowing us to resolve the F proteins on opposing sides of the viral envelope with the N protein in the center.
These two model systems illustrate the benefits of the MEANS implementation. The NPC structure (a ring laying parallel to the mirror surface) benefits from increased lateral resolution. According to the Nyquist-Shannon sampling theorem, to resolve a ring structure of ~50 nm in diameter, a 25-nm imaging resolution is needed. For the hRSV filament (a cylinder lying parallel to the mirror surface), a very thin optical section (less than the diameter of the filament) is required to avoid exciting the entire surface of the filament envelope and to resolve two opposing sides. A more detailed explanation of the geometry of the viral filament can be found in Supplementary Fig. S9.
Because the axial light confinement occurs away from the metal surface, the MEANS method is immune to metal surface quenching13. The working distance for MEANS super-resolution can therefore be adjusted by customizing the thickness of the silica coating, which is a standard procedure for commercial protective mirrors. Growing the cell on a coated layer of silica rather than directly on the metal surface provides a more compatible environment for cell growth13. Further, with excitation wavelength tuning, MEANS can be extended to axial sectioning of the specimen through MEANS-ESON.
MEANS is compatible with a variety of confocal-based technologies, such as laser scanning, spinning disk and two-photon. In addition, MEANS-STED is broadly applicable to PSF engineering-based super-resolution techniques, including the pulsed or time-gated STED20, RESOLFT4, ground-state depletion, excitation-state absorption, saturation38, optical data storage39, upconversion40 and charge-state depletion41. Compared with the other imaging techniques based on the recollection of the signal through interference to confine the axial resolution, including 4Pi 15, I5M14 and wide-field standing-wave microscopy7 or mirrors combined with spatial light modulator10, 42, MEANS is much simpler and does not require precise alignment of the mirror. In addition, MEANS is compatible with conventional sample preparation procedures, which is advantageous over virtual imaging43.
Conclusion
It has been demonstrated that with axial confinement, such as TIRF, improved image contrast can be obtained. We found that through the straightforward use of a mirror beneath the specimen, we can generate an interference-enhanced layer away from the mirror reflective surface with a thickness of ~110 nm. A 3.6-fold local EM field enhancement can be obtained, resulting in an increased SBR. Although MEANS is very similar to the 4Pi approach, when using MEANS, the height of the interference-enhanced layer is constant during mirror translation of 0.4 μm, which largely relieves the requirement of ultra-precise system alignment. Furthermore, MEANS is compatible with a series of confocal modalities, such as two-photon, spinning disk and laser scanning. When MEANS is applied to STED, both the lateral and axial resolution can be improved simultaneously. The ring structure of the inner NPC structure (diameter of 53 nm) can be visualized with a resolution of ~19 nm. To the best of our knowledge, this resolution is among the best recorded for STED super-resolution in biological specimens. Taking advantage of the thin axial depth of MEANS, we can discern the tubular structure of the hRSV-F protein with a diameter of ~120 nm. The capability of achieving optical sectioning and high lateral resolution is particularly appealing to biological applications because the specimens are usually prone to strong light power.
Author contributions
HX and PX conceived the project and conducted the simulation, PX, DJ and PJS designed the experiments and managed the project, EA and PJS prepared the biological cell specimens, XY and PX performed the imaging experiments and data analysis. DJ, XY, HX, PJS and PX wrote the manuscript. All authors participated in discussion and editing of the manuscript.
References
Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006; 313: 1642–1645.
Axelrod D . Total internal reflection fluorescence microscopy in cell biology. Traffic 2001; 2: 764–774.
Orrit M . Nobel prize in chemistry: celebrating optical nanoscopy. Nat Photonics 2014; 8: 887–888.
Hell SW . Far-field optical nanoscopy. Science 2007; 316: 1153–1158.
Hao X, Kuang CF, Gu ZT, Wang YF, Li S et al. From microscopy to nanoscopy via visible light. Light Sci Appl 2013; 2: e108, doi:10.1038/lsa.2013.64.
Schmidt R, Wurm CA, Jakobs S, Engelhardt J, Egner A et al. Spherical nanosized focal spot unravels the interior of cells. Nat Methods 2008; 5: 539–544.
Bailey B, Farkas DL, Taylor DL, Lanni F . Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature 1993; 366: 44–48.
Swan AK, Moiseev LA, Cantor CR, Davis B, Ippolito SB et al. Toward nanometer-scale resolution in fluorescence microscopy using spectral self-interference. IEEE J Select Top Quant Electron 2003; 9: 294–300.
Amor R, Mahajan S, Amos WB, McConnell G . Standing-wave-excited multiplanar fluorescence in a laser scanning microscope reveals 3D information on red blood cells. Sci Rep 2014; 4: 7359.
Mudry E, Le Moal E, Ferrand P, Chaumet PC, Sentenac A . Isotropic diffraction-limited focusing using a single objective lens. Phys Rev Lett 2010; 105: 203903.
Deguchi T, Koho S, Näreoja T, Hänninen P . Axial super-resolution by mirror-reflected stimulated emission depletion microscopy. Opt Rev 2014; 21: 389–394.
Elsayad K, Urich A, Tan PS, Nemethova M, Small JV et al. Spectrally coded optical nanosectioning (SpecON) with biocompatible metal–dielectric-coated substrates. Proc Natl Acad Sci USA 2013; 110: 20069–20074.
Chizhik AI, Rother J, Gregor I, Janshoff A, Enderlein J . Metal-induced energy transfer for live cell nanoscopy. Nat Photonics 2014; 8: 124–127.
Gustafsson M, Agard D, Sedat J . I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J Microsc 1999; 195: 10–16.
Hell SW, Lindek S, Cremer C, Stelzer EHK . Measurement of the 4Pi-confocal point spread function proves 75 nm axial resolution. Appl Phys Lett 1994; 64: 1335–1337.
Li S, Kuang CF, Hao X, Gu ZT, Liu X . Generation of a 3D isotropic hollow focal spot for single-objective stimulated emission depletion microscopy. J Opt 2012; 14: 085704.
You ST, Kuang CF, Rong ZH, Liu X, Ding ZH . Isotropic superresolution imaging for fluorescence emission difference microscopy. Appl Opt 2014; 53: 7838–7844.
Zhao ZW, Roy R, Gebhardt JCM, Suter DM, Chapman AR et al. Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy. Proc Natl Acad Sci USA 2014; 111: 681–686.
Wurm CA, Neumann D, Schmidt R, Egner A, Jakobs S . Sample preparation for STED microscopy. Methods Mol Biol 2010; 591: 185–199.
Vicidomini G, Moneron G, Han KY, Westphal V, Ta H et al. Sharper low-power STED nanoscopy by time gating. Nat Methods 2011; 8: 571–573.
Tokunaga M, Imamoto N, Sakata-Sogawa K . Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 2008; 5: 159–161.
Bewersdorf J, Schmidt R, Hell SW . Comparison of I5M and 4Pi-microscopy. J Microsc 2006; 222: 105–117.
Wilson T, Sheppard C . Theory and Practice of Scanning Optical Microscopy. London: Academic Press. 1984.
Watanabe T, Igasaki Y, Fukuchi N, Sakai M, Ishiuchi S et al. Formation of a doughnut laser beam for super-resolving microscopy using a phase spatial light modulator. Opt Eng 2004; 43: 1136–1143.
Gu M . Advanced Optical Imaging Theory vol. 75. Berlin: Springer Science & Business Media. 2000.
Xie H, Liu YJ, Santangelo PJ, Jin DY, Xi P . Analytical description of high-aperture STED resolution with 0-2π vortex phase modulation. J Opt Soc Am A 2013; 30: 1640–1645.
Westphal V, Hell SW . Nanoscale resolution in the focal plane of an optical microscope. Phys Rev Lett 2005; 94: 143903.
Alber F, Dokudovskaya S, Veenhoff LM, Zhang WZ, Kipper J et al. The molecular architecture of the nuclear pore complex. Nature 2007; 450: 695–701.
Guan T, Müller S, Klier G, Panté N, Blevitt JM et al. Structural analysis of the p62 complex, an assembly of O-linked glycoproteins that localizes near the central gated channel of the nuclear pore complex. Mol Biol Cell 1995; 6: 1591–1603.
Grossman E, Medalia O, Zwerger M . Functional architecture of the nuclear pore complex. Annu Rev Biophys 2012; 41: 557–584.
Szymborska A, de Marco A, Daigle N, Cordes VC, Briggs JAG et al. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science 2013; 341: 655–658.
Donnert G, Keller J, Medda R, Andrei MA, Rizzoli SO et al. Macromolecular-scale resolution in biological fluorescence microscopy. Proc Natl Acad Sci USA 2006; 103: 11440–11445.
Wildanger D, Medda R, Kastrup L, Hell SW . A compact STED microscope providing 3D nanoscale resolution. J Microsc 2009; 236: 35–43.
Göttfert F, Wurm CA, Mueller V, Berning S, Cordes VC et al. Coaligned dual-channel STED nanoscopy and molecular diffusion analysis at 20 nm resolution. Biophys J 2013; 105: L01–L03.
Kolmakov K, Wurm CA, Meineke DNH, Göttfert F, Boyarskiy VP et al. Polar red‐emitting rhodamine dyes with reactive groups: synthesis, photophysical properties, and two‐color STED nanoscopy applications. Chemistry 2014; 20: 146–157.
Wurm CA, Kolmakov K, Göttfert F, Ta H, Bossi M et al. Novel red fluorophores with superior performance in STED microscopy. Opt Nanoscopy 2012; 1: 1–7.
Alonas E, Lifland AW, Gudheti M, Vanover D, Jung J et al. Combining single RNA sensitive probes with subdiffraction-limited and live-cell imaging enables the characterization of virus dynamics in cells. ACS Nano 2014; 8: 302–315.
Fujita K, Kobayashi M, Kawano S, Yamanaka M, Kawata S . High-resolution confocal microscopy by saturated excitation of fluorescence. Phys Rev Lett 2007; 99: 228105.
Gan ZS, Cao YY, Evans RA, Gu M . Three-dimensional deep sub-diffraction optical beam lithography with 9 nm feature size. Nat Commun 2013; 4: 2061.
Kolesov R, Reuter R, Xia KW, Stöhr R, Zappe A et al. Super-resolution upconversion microscopy of praseodymium-doped yttrium aluminum garnet nanoparticles. Phys Rev B 2011; 84: 153413.
Chen XD, Zou CL, Gong ZJ, Dong CH, Guo GC et al. Subdiffraction optical manipulation of the charge state of nitrogen vacancy center in diamond. Light Sci Appl 2015; 4: e230, doi:10.1038/lsa.2015.3.
Schnitzbauer J, McGorty R, Huang B . 4Pi fluorescence detection and 3D particle localization with a single objective. Opt Express 2013; 21: 19701–19708.
Wang ZB, Guo W, Li L, Luk'yanchuk B, Khan A et al. Optical virtual imaging at 50 nm lateral resolution with a white-light nanoscope. Nat Commun 2011; 2: 218.
Acknowledgements
The fluorescent nanodiamond sample was a gift from Huan-cheng Chang (Institute of Atomic and Molecular Sciences, Academia Sinica). We thank Jihong Liu from Leica (China) and Chunyan Shan (Peking University) for their help with the Leica TCS SP8 STED 3X, Weijia Yang (Wuhan YSL Photonics) for helpful discussion about the white light laser, and Xu Zhang (Tsinghua University) for helpful discussion about sample preparation. This research is supported by the National Instrument Development Special Program (2013YQ03065102), the ‘973’ Major State Basic Research Development Program of China (2011CB809101), the Natural Science Foundation of China (31327901, 61475010, 61428501) and the Australian Research Council Centre of Excellence for Nanoscale BioPhotonics (CE140100003). Support for E Alonas was provided by the National Institute of Health (GM094198 to PJS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Note: Supplementary Information for this article can be found on the Light: Science & Applications' website.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yang, X., Xie, H., Alonas, E. et al. Mirror-enhanced super-resolution microscopy. Light Sci Appl 5, e16134 (2016). https://doi.org/10.1038/lsa.2016.134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/lsa.2016.134
Keywords
This article is cited by
-
Light People: Professor Fan Wang
Light: Science & Applications (2023)
-
Axial localization and tracking of self-interference nanoparticles by lateral point spread functions
Nature Communications (2021)
-
Mirror-enhanced scanning light-field microscopy for long-term high-speed 3D imaging with isotropic resolution
Light: Science & Applications (2021)
-
Single-cell biomagnifier for optical nanoscopes and nanotweezers
Light: Science & Applications (2019)
-
Biocompatible carbon dots with low-saturation-intensity and high-photobleaching-resistance for STED nanoscopy imaging of the nucleolus and tunneling nanotubes in living cells
Nano Research (2019)
