
Abstract
Despite enormous advances, management of multiple myeloma (MM) remains challenging. Multiple factors impact the decision to treat or which regimen to use at MM relapse/progression. Recent major randomized controlled trials (RCTs) showed widely varying progression-free survivals (PFS), ranging from a median of 4 months (MM-003) to 23.6 months (ASPIRE). Based on these RCTs, next-generation proteasome inhibitors (carfilzomib and ixazomib), next-generation immunomodulatory agent (pomalidomide), and monoclonal antibodies (elotuzumab and daratumumab) were approved for relapsed and refractory MM. Daratumumab, targeting CD38, has multiple mechanisms of action including modulation of the immunosuppressive bone marrow micro-environment. In addition to the remarkable single agent activity in refractory MM, daratumumab produced deep responses and superior PFS in MM when combined with lenalidomide/dexamethasone, or bortezomib/dexamethasone. Other anti-CD38 antibodies, such as isatuximab and MOR202, are undergoing assessment. Elotuzumab, targeting SLAMF7, yielded superior response rates and PFS when combined with lenalidomide/dexamethasone. New combinations of these next generation novel agents and/or antibodies are undergoing clinical trials. Venetoclax, an oral BH3 mimetic inhibiting BCL2, showed single agent activity in MM with t(11;14), and is being studied in combination with bortezomib/dexamethasone. Selinexor, an Exportin-1 inhibitor, yielded promising results in quad- or penta-refractory MM including patients resistant to daratumumab. Pembrolizumab, an anti-PD1 check-point inhibitor, is being tested in combination with lenalidomide/dexamethasone or pomalidomide/dexamethasone. Chimeric antigen receptor-T cells targeting B-cell maturation antigen have yielded deep responses in RRMM. Finally, salvage autologous stem cell transplantation (ASCT) remains an important treatment in MM relapsing/progressing after a first ASCT. Herein, the clinical trial data of these agents are summarized, cautious interpretation of RCTs highlighted, and algorithm for salvage treatment of relapse/refractory MM proposed.
Similar content being viewed by others

Introduction
Despite enormous advances with the advent of proteasome inhibitors (PI) and immunomodulatory agents (IMiDs), management of multiple myeloma (MM) remains challenging, and relapse of MM and disease progression is common even after achievement of a complete remission.1 Moreover, relapsed/progressive MM acquires additional mutation or genetic alterations that render the disease more resistant, leading to progressively shorter durations of remission or response to each salvage therapy, and the ultimate development of relapsed/refractory MM (RRMM).2
Factors impacting decisions to treat and the choice of salvage regimens
While treatment is often indicated at relapse/progression of MM, a number of factors need to be considered before embarking on therapy. The first question is whether it is a true MM relapse. This is particularly important with more effective induction regimens, often followed by autologous stem cell transplantation (ASCT), rendering a deep response (>very good partial response) in the majority of transplant-eligible MM patients.3 However, it was well recognized that patients with a deep response may develop oligoclonal reconstitution, characterized by the transient presence of a monoclonal gammopathy of a different isotype (Figure 1) but occasionally with the same isotype.4, 5, 6 While oligoclonal reconstitution may result in a measurable M-protein, it is often transient, lasting for a period of weeks to up to a year.5, 7 Therefore, recognition of oligoclonal reconstitution is important in order not to over-treat the patients and expose them to unnecessary toxicity, especially when it may be associated with a good prognosis.8

Oligoclonal reconstitution (OCR) illustrated by the change of monoclonal IgAL at diagnosis (Dx) to OCR of IgGk+IgGL+IgMk during complete remission.
Clinically, some relapses occur in the form of symptomatic disease with the presence of hypercalcemia, renal impairment, anemia or bone disease (CRAB) or extramedullary disease, either in the form of plasma cell leukemia or extramedullary plasmacytoma, and warrant immediate treatment.9 However, in many cases, relapse/progression may be biochemical only, which is marked by reappearance or increase in the amount of measurable M-protein without symptoms. This biochemical relapse, defined as a 25% increase in M-protein with an absolute increase of 0.5 g/dl,10 does not warrant treatment. Moreover, in the case of biochemical relapse/progression, some progress in an indolent manner, while others demonstrate an aggressive pattern with rapid doubling of the M-protein.9 Therefore, based on the rate of rise or the absolute rise of paraprotein, treatment is recommended if there is a ‘significant’ biochemical relapse, which includes any of the following in two consecutive measurements within 2 months: (1) doubling of the M-component with an absolute value ⩾5 g/l, or (2) an increase in the absolute levels of serum M-protein by ⩾1 g/dl, or urine M-protein by ⩾500 mg/24 h, or involved FLC level by ⩾20 mg/dl (plus an abnormal FLC ratio).11, 12 Many factors affect the decision or choice of salvage therapy including age of the patient (for example, still eligible for a second ASCT?), duration of response to prior induction or ASCT, side-effects carried over from prior treatments (for example, peripheral neuropathy), co-morbid illnesses (caution with the use lenalidomide in renal impairment or carfilzomib in cardiac failure) performance status, or the presence of high-risk karyotypic aberrations such as del(17p), which would predict a short response to high-dose chemotherapy in the form of a second ASCT.13, 14 Finally, evidence from clinical trials, especially those from phase 3 randomized controlled trials (RCTs); influence the choice of salvage therapy. Herein, we discuss the most important clinical trials published in the last 2 years and some promising ongoing clinical trials.
Common features of RCTs in MM
In 2015−2016, multiple drugs were approved for the treatment of RRMM including carfilzomib, ixazomib, pomalidomide, panobinostat, elotuzumab and daratumumab, based on positive results from high-quality, phase 3 studies.15, 16, 17, 18, 19, 20, 21, 22 A common theme in the phase 3 RCT was that RRMM patients in the intervention arm were compared with a standard salvage regimen of lenalidomide/dexamethasone (Rd) or bortezomib/dexamethasone (Vd) in the majority but not all, hence randomized to ‘X’-Rd/Vd or Rd/Vd, hence a three-drug versus two-drug design. The primary end-point was often progression-free survival (PFS), while response rate (RR) and overall survival (OS) were secondary end-points. However, some studies also included minimal residual disease (MRD) as a secondary end-point.15, 18, 23 Over the last 2 years, a number of important clinical trials have been published that reshaped the current landscape of treatment of RRMM (Table 1). Important published RCTs of next-generation PI include ASPIRE15 which compared carfilzomib/lenalidomide/dexamethasone (KRd) with Rd; and TOURMALINE,16 which compared Ixazomib-Rd with Rd. MM-003 was an RCT that compared pomalidomide/low dose dexamethasone with high-dose dexamethasone.17 Important studies of monoclonal antibodies included daratumumab and elotuzumab.18, 20, 21, 22, 23 If one refers to v.3 2017 National Comprehensive Cancer Network (NCCN) guideline for patients with RRMM, there are a multitude of ‘category 1’ salvage agents/regimens including bortezomib/dexamethasone, lenalidomide-dexamethasone elotuzumab-lenalidomide-dexamethasone, ixazomib-lenalidomide-dexamethasone, carfilzomib-lenalidomide-dexamethasone, bortezomib/panobinostat/dexamethasone, pomalidomide/dexamethasone, daratumumab-lenalidomide-dexamethasone and bortezomib-lenalidomide-dexamethasone24 (Figure 2).

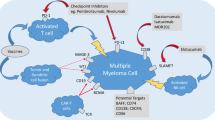
An array of salvage regimens for RRMM comprising proteasome inhibitor (PI), immunomodulatory agents (IMiD), anti-CD38 (daratumumab [Dara]) or anti-SLAMF7 (elotuzumab [Elo]) monoclonal antibodies, BCL2 inhibitor (BCL2i), exportin-1 inhibitor (XPO1-i), anti-PD1 check-point inhibitors or CAR-T cells. PI (bortezomib [V], carfilzomib [K] and ixazomib [Ixa]) and dexamethasone [Dex] induces intrinsic/mitochondrial apoptosis (via activation of caspase 9 through formation of the apoptosome) while IMiD (lenalidomide [R] and pomalidomide [P]) extrinsic apoptosis via activation of caspase 8. Low-dose dexamethasone (d) can be combined with either IMiD (Rd or Pd), or PI (Vd or Kd) to yield effective doublets, or both IMiD and PI to generate even more potent triplets including VRd, VPd, KRd, KPd and Ixa-Rd. Monoclonal antibody combinations targeting CD38 (Dara-Vd, Dara-Rd or Dara-Pd) or SLAMF7 (Elo-Rd) are effective salvage regimens. Venetoclax (VEN), or in combination with Vd, induces mitochondrial apoptosis. Selinexor (SEL)is an exportin-1 inhibitor, resulting in nuclear retention of tumor suppressor proteins (TSP). Check-point inhibitors (anti-PD1) such as pembrolizumab (Pem) or its combinations (Pem-Rd, Pem-Pd) unleash endogenous myeloma-specific cytotoxic T-cells (MM CTL) to induce cell-mediated myeloma cytotoxicity. Adoptive cell therapy with engineered autologous BCMA-specific CAR-T cells leads cell-mediated cytotoxicity.
Caution in interpretation of clinical trial data
The PFS data from these studies are widely different. For instance, median PFS was 26.3 months in the KRd arm of the ASPIRE study but only four months in the intervention arm of MM-003 using pomalidomide/dexamethasone.15, 17 While no two studies are directly comparable, the huge difference in PFS of these two studies could at least be partially accounted for by the patient populations in the trials. For instance, in MM-003, 95% of patients had ⩾3 lines of therapy, 82% were refractory to their last regimen, and 75% were double refractory to bortezomib and lenalidomide (Table 1). By contrast, in ASPIRE the median number of prior lines of therapy was two, and none were double refractory to bortezomib and lenalidomide since the control arm was Rd. Therefore, one has to be cautious in the comparison of results across clinical trials. Most RCTs (including ASPIRE, TOURMALINE, CASTOR, POLLUX and ELOQUENT-2) were conducted at an earlier state of relapse,15, 16, 17, 18, 20, 23 when the median number of prior lines of therapy was limited to one or two only, and few patients were refractory to their last regimen or double refractory to both bortezomib and lenalidomide (Table 1). By contrast, in MM-003, GEN501 and SIRIUS, patients had a median of 4–5 prior treatments, with >80% refractory to their last regimen and three quarters refractory to both bortezomib and lenalidomide. Therefore, meaningful comparison could only be made among trials comprising similar trial design, patient populations and similar control arms.25
Major clinical trials of next-generation PIs
Randomized controlled trials including ASPIRE and TOURMALINE were published in 2015 and 2016, demonstrating the superiority of the second-generation PIs (carfilzomib in ASPIRE and ixazomib in TOURMALINE) in combination with lenalidomide/dexamethasone [Rd] when compared to Rd.15, 16
Carfilzomib is an intravenous PI carrying an epoxyketone moiety, and functions as an irreversible inhibitor of the β5 chymotryptic subunit of the 20S proteasome. Ixazomib is an oral boronate PI with reversible inhibition of the proteasome. In contrast, bortezomib is a boronate PI with slowly reversible inhibition of the β5 subunit of the proteasome. In ASPIRE, RRMM patients were randomized to receive carfilzomib-Rd [KRd] or Rd only (Figure 2). Much higher response rates and a superior PFS were observed in the KRd arm compared with Rd (PFS: 26.3 versus 17.6 months, P<0.0001) In contrast to bortezomib, carfilzomib led to much less peripheral neuropathy but a higher rate of cardiopulmonary complications. Moreover, tumor lysis syndrome has occurred and hence a ramped-up schedule of carfilzomib has been adopted (20 mg/m2/d on D1/2, followed by 27 mg/m2/d on D8/9, D15/16 of cycle one and thereafter). Moreover, in an updated analysis, (KRd) has been shown to confer an OS benefit by reducing the risk of death by 21% over Rd alone, hence an almost 8-month survival prolongation (median OS: 48.3 months versus 40.4 months, HR=0.79) (https://www.amgen.com/media/news-releases/2017/07/second-phase-3-study-shows-kyprolis-carfilzomib-regimen-significantly-improves-overall-survival-in-patients-with-relapsed-multiple-myeloma/).
Recently, a head-to-head comparison of carfilzomib with bortezomib has been conducted in the ENDEAVOR trial, in which patients with RRMM were randomized to receive carfilzomib/dexamethasone (Kd)(N=464) or bortezomib/dexamethasone (Vd)(N=465)19 (Figure 2). In this study, patients with prior exposure but who were not refractory to bortezomib or carfilzomib were eligible. Patients receiving Kd had a higher response rate (RR)/⩾complete remission (CR)/very good partial response (VGPR) rates (RR/⩾CR/⩾VGPR rates: 77%/13%/54% versus 63%/29%/6%, P<0.0001) and superior PFS than those receiving Vd (median PFS: 18.7 months versus 9.4 months, HR: 0.53, P<0.0001). An updated report of ENDEAVOR study showed that Kd provided 7.6 months median OS benefit (median OS: 47.6 months versus 40.0 months Vd; HR 0.791; P=0.010).26 If the subset of patients with prior exposure to bortezomib is excluded and only bortezomib-naïve RRMM patients are analyzed, the RR and PFS of the Kd arm remained superior to that of the Vd arm.27 Therefore, carfilzomib appears to be a more potent PI than bortezomib in this setting.
In TOURMALINE,16 RRMM patients were randomized to receive ixazomib (Ixa)-Rd (N=360) or Rd only (N=362). Compared to the Rd control, the Ixa-Rd arm led to superior response rates (RR/⩾CR/⩾VGPR: 78%/14%/48% versus 72%/7%/39%) and PFS (20.6 months versus 14.7 months, HR: 0.74, P=0.01) (Figure 2). Moreover, in patients with high-risk cytogenetics, Ixa-Rd resulted in a superior PFS (21.4 months) than placebo-Rd (9.7 months, HR: 0.543), comparable to PFS of 20.6 months in standard-risk treated by Ixa-Rd.28 Therefore, the adverse impact of high-risk cytogenetics has been abolished by Ixa-Rd.
Major clinical trials of next-generation IMiD
Pomalidomide, a third-generation IMiD more potent than lenalidomide, has been shown to yield superior RR and PFS compared to high-dose dexamethasone in the MM-003 study.17 In MM-003, ‘refractory’ MM patients were randomized to receive oral pomalidomide (4 mg/d D1-D21) and low dose dexamethasone (40 mg weekly), Pd, at 4-weekly intervals versus high-dose dexamethasone (40 mg/d D1-4, D9-12, D17-20). RR/⩾CR/⩾VGPR rates were 31%/1%/6% in the Pd arm and 10%/0%/<1% in the control arm, and median PFS was 4.0 months and 1.9 months (HR: 0.48; P<0.0001). Recently, a randomized phase 2 study reported a higher response rate (65% versus 39%, P=0.035) and superior median PFS (9.5 months versus 4.4 months) in patients receiving pomalidomide, cyclophosphamide, dexamethasone (PCd) than Pd.29 Moreover, a phase 3 study (MM-007) comparing the efficacy and safety of pomalidomide-bortezomib-dexamethasone (Pom-Vd) with bortezomib-dexamethasone (Vd) is ongoing in RRMM.30
Major clinical trials of HDAC inhibitor
In MM, resistance to bortezomib has been attributed at least in part to activation of the histone deacetylase (HDAC) 6-dependent aggresome pathway that leads to catabolism of excessive unfolded proteins, thereby precluding induction of cellular apoptosis by cellular stress inherent with excessive unfolded protein upon proteasome inhibition.31, 32, 33 Panobinostat is an oral pan-HDAC inhibitor that produces synergistic antimyeloma effects when combined with bortezomib and dexamethasone. A randomized, placebo-controlled, double-blind phase 3 trial comparing panobinostat, bortezomib and dexamethasone with placebo, bortezomib and dexamethasone has been conducted in patients with RRMM,34 which yielded a median PFS of 11.99 months in the panobinostat combination arm and 8.08 months in the placebo control arm (HR: 0.63, P<0.0001). Response rate was not different between the arms but CR or near CR rates were higher in the panobinostat combination (27.6% versus 15.7%, P<0.0006). However, grade 3/4 thrombocytopenia (67%), diarrhea (26%) and asthenia (24%) occurred more frequently in the panobinostat arm. A selective HDAC 6 inhibitor ricolinostat has been studied in combination with bortezomib and dexamethasone in RRMM.35 Compared to panobinostat, patients receiving ricolinostat at 160/240 mg daily together with bortezomib/dexamethasone (N=20) had comparable RR/VGPR rates (37%/11%), but much less grade 3/4 thrombocytopenia (20%), asthenia (5%) and diarrhea (5%).
Major clinical trials of monoclonal antibodies
Promising results were seen in studies of daratumumab, either as a single agent,21, 22 or in combination with bortezomib/dexamethasone (DVd) or lenalidomide/dexamethasone (DRd).18, 23 Daratumumab is a fully humanized IgG1 antibody targeting CD38, a transmembrane glycoprotein uniformly expressed at high levels in MM plasma cells, but at low levels in normal myeloid and lymphoid cells. Daratumumab has a multiple mechanisms of action.36 First, binding of daratumumab to CD38 of myeloma plasma cells directly induces apoptosis. Moreover, myeloma cell death is also mediated by complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis. Besides, daratumumab also modulates the immunosuppressive bone marrow microenvironment favorably by suppressing production of immunosuppressive molecules such as adenosine by inhibition of CD38 enzymatic activity on myeloma plasma cells. On the other hand, CD38 is also expressed on the immunosuppressive regulatory T-cells (Treg) and myeloid-derived suppressor cells which are hence also targeted and inhibited by daratumumab, thereby relieving repression of the myeloma-specific cytotoxic T cells in the bone marrow microenvironment.36
Clinically, significant single agent activity of daratumumab in myeloma has been demonstrated in two studies GEN501 (a phase 1–2 study) and SIRIUS.21, 22 In the phase 2 study, SIRIUS, patients with refractory MM with a median of five prior lines of therapy, 97% refractory to their last regimen and 85% refractory to both bortezomib and lenalidomide, were enrolled.21 A total of 106 patients received 16 mg/kg daratumumab (weekly × 8, then 2-weekly × 8, then 4-weekly thereafter). In this highly refractory cohort of MM, RR/⩾CR/⩾VGPR rate were 29.2%/2.8%/9.4%, with a median PFS of 3.7 months and OS of 17.5 months, demonstrating significant single agent activity of daratumumab in MM. The median time to first response was only one month, indicating a rapid onset of action. In the phase 1/2 GEN501 study,22 in which 80% of patients were refractory to their last regimen and 64% had MM double refractory to both bortezomib and lenalidomide), no maximum tolerated dose was identified in the dose-escalation phase. Moreover, in the dose-expansion phase, 42 patients with advanced MM and a median of four prior lines of therapy received intravenous once weekly daratumumab (16 mg/kg weekly for 8 doses over 2 months, 2-weekly for 8 doses over 4 months and then monthly thereafter until disease progression), The RR/⩾CR/⩾VGPR rates were 36%/6.7%/6.7% with a median PFS of 5.6 months. Finally, in a combined analysis of all MM patients receiving daratumumab 16 mg/kg (weekly × 8, 2-weekly × 8, and then 4 weekly thereafter until disease progression) in both GEN501 (N=42) and SIRIUS (N=106) clinical trials, the RR/⩾CR/⩾VGPR rates were 31%/4.7%/8.8% and median PFS was 4 months and OS 20.1 months.37 Interestingly, while median PFS was only 4 months, it translated into a prolonged median survival of 20 months.37
Building on these promising data from single agent daratumumab, subsequent trials have compared bortezomib/dexamethasone (Dara-Vd) or lenalidomide/dexamethasone (Dara-Rd) with or without daratumumab in phase 3 RCTs18, 23 (Figure 2). In the CASTOR study,18 patients with RRMM were randomized to receive Dara-Vd (N=251) or Vd (N=247). In the Dara-Vd arm, patients received daratumumab (weekly × 9, then every 3-weeks × 15, and then every 4-weeks thereafter until disease progression) in addition to eight 3-weekly cycles of bortezomib/dexamethasone (bortezomib 1.3 mg/m2 subcutaneously on D1,4,8,11 Q3W and dexamethasone 20 mg/d on D1/2, 4/5, 8/9, 11/12). Patients in the control Vd arm received eight cycles of Vd as described above. The CASTOR trial reported encouraging RR/⩾CR/⩾VGPR rates of 83%/19%/59% and the median PFS was not reached in the Dara-Vd arm compared with a median PFS of 7.2 months in the Vd control (HR: 0.39; P<0.0001). In the POLLUX study,23 patients with RRMM were randomized to receive Dara-Rd (N=286) or Rd only (N=283). In the Dara-Rd arm, patients received daratumumab at 16 mg/kg (weekly × 8, 2-weekly × 8, and 4-weekly thereafter) in addition to the regular 4-weekly cycles of Rd (lenalidomide 25 mg/d D1-D21, dexamethasone 40 mg weekly) until disease progression. The study yielded very promising results with RR/⩾CR/⩾VGPR rates of 93%/43%/78%, and an 18-month PFS of 78% (median not reached) compared to the 18-month PFS of 52% and median PFS of 18.4 months in the Rd control arm (HR: 0.037; P<0.0001).
While two RCTs should not be directly compared, it would appear that responses in the Dara-Rd arm of POLLUX (RR/⩾CR/⩾VGPR rates: 93%/43%/78%) were higher than that in the Dara-Vd arm of CASTOR (RR/⩾CR/⩾VGPR rates: 83%/19%/59%). Moreover, the 12-month PFS of POLLUX (83%) was higher than CASTOR (60.7%). In these two studies, there was no major difference in patient population in terms of the status of disease at enrolment (30% were refractory to their last treatment in Dara-Vd and 28% in Dara-Rd) and cytogenetic profiles with high-risk cytogenetics (22% of Dara-Vd and 19% of Dara-Rd).18, 23 However, per protocol, a total of 16 doses of daratumumab were used in Dara-Rd during induction, compared to a total of 14 doses in Dara-Vd. Moreover, in the maintenance phase, continual Rd in addition to monthly daratumumab was used until disease progression in DRd. In contrast, only single agent daratumumab was used after cycle 8 of Dara-Vd in CASTOR and no maintenance of bortezomib was given. Therefore, differences in dose intensity, especially with the use of additional Rd during maintenance in the Dara-Rd arm, may account, at least in part, for the difference in RR and PFS in Dara-Rd of POLLUX and Dara-Vd of CASTOR.
In an updated analysis of POLLUX and CASTOR, objective response rate and PFS were reported according to the cytogenetic risk status as detected by NGS, in which high-risk cytogenetic aberrations were those with t(4;14), t(14;16) or del(17p), and standard-risk those without.38 Addition of daratumumab to either lenalidomide/dexamethasone (Rd) or bortezomib/dexamethasone (Vd) yielded a superior PFS and a higher rate of deep responses (⩾CR or ⩾VGPR) in both high-risk and standard-risk subgroups than Rd/Vd only. In particular, the adverse impact of high-risk cytogenetics on PFS was mitigated by addition of daratumumab to either Rd or Vd. Moreover, at a sensitivity of one in 105, addition of daratumumab to Rd yielded a higher rate MRD negativity in both high-risk and standard-risk cytogenetic subgroups (HR subgroup: Dara-Rd 18% versus Rd 0%; P=0.0027; SR subgroup: Dara-Rd 30% versus Rd 10%; P=0.0001). Similarly, addition of daratumumab to Vd resulted in more frequent MRD negativity in both high-risk and standard-risk cytogenetic subgroups (high-risk subgroup: Dara-Rd 14% versus Rd 0%; P=0.0018; standard-risk subgroup: Dara-Rd 12% versus Rd 2%; P=0.0011). Therefore, addition of daratumumab to Rd or Vd improved treatment outcomes regardless of cytogenetic risk status in RRMM.38
Besides daratumumab, another anti-CD38 antibody isatuximab (also called SAR650984), a humanized IgG1 monoclonal antibody, has been studied in advanced MM. In an open-label, single-arm study, patients treated with isatuximab at ⩾10 mg/kg (n=74) had a median PFS of 3.65 months and a median OS of 18.63 months (95% CI, 15.7—not reached). The objective response rate was 24.3%, including those with high-risk cytogenetics.39 Another anti-CD38 antibody MOR202, a human combinatorial antibody library-derived human IgG1 CD38 monoclonal antibody, induces antibody-dependent cell-mediated cytotoxicity and antibody-dependent cellular phagocytosis, but not complement-dependent cytotoxicity, which is suspected to be a major cause of infusion-related reactions seen with other anti-CD38 antibodies. MOR202 is currently undergoing clinical trials in combination with lenalidomide or pomalidomide, and preliminary results revealed excellent tolerability with infusion-related reactions in only 10% of patients, all ⩽grade 2.40 This compared favorably with the much higher rates of infusion-related reaction, though mostly grade 1/2 and limited to the first infusion, occurring in 43–71% of patients treated with daratumumab monotherapy and 43% of patients receiving isatuximab monotherapy.41
Elotuzumab is a humanized IgG1 monoclonal antibody targeting signaling lymphocytic activation molecule family member 7 (SLAMF7), a member of the SLAM family which belongs to the immunoglobulin receptor superfamily. SLAMF7 is densely expressed on myeloma plasma cells and natural killer cells.42 In contrast to daratumumab that demonstrates significant single agent activity in MM, binding of elotuzumab to SLAMF7 of myeloma plasma cells does not induce tumor cytotoxicity.42 On the other hand, binding of elotuzumab to NK cells leads to activation of NK cells, resulting in antibody-dependent cell-mediated cytotoxicity of elotuzumab-bound myeloma plasma cells. In the phase 3 study informed by preclinical studies,43 ELOQUENT-2 in which RRMM patients were randomized to receive intravenous elotuzumab (10 mg/kg weekly × 8, then every 2 weeks thereafter) or placebo with Rd (lenalidomide 25 mg/d D1-D21 and dexamethasone 40 mg weekly) at 4-weekly intervals, RR/⩾CR/⩾VGPR rates were 79%/4%/33% with a superior median PFS of 19.4 months in the Elo-Rd arm and 14.9 months in the Rd arm (HR: 0.7; P<0.001)20 (Figure 2). With an extended follow-up at 4 years, there was a sustained 29% reduction of progression/death (HR: 0.71; 95% CI: 0.59–0.86) in addition to a trend of OS benefit (4-year OS: 50% versus 43%; HR: 0.78; 95% CI: 0.63–0.96) in ELo-Rd compared with Rd.44 As the survival benefit of both PFS and OS were apparent early in the trial, and persisted thereafter, this is the first immune-oncology study supporting a sustained survival benefit associated with addition of elotuzumab to Rd.
Phase1/2 clinical trials arising from the published RCTs
Apart from these RCTs, other permutations of the newer generation novel PI and IMiDs have been studied. In lenalidomide-refractory MM patients, bortezomib has been combined with pomalidomide/dexamethasone (VPd), and carfilzomib with pomalidomide/dexamethasone (KPd) (Figure 2). Lacy et al. reported results for the use of VPd (N=47) to treat patients with lenalidomide-refractory MM and observed impressive RR/⩾CR/⩾VGPR rates (85%/19%/44.7%) and a median PFS of 10.7 months.45 In a phase 1 study, KPD showed RR/⩾CR/⩾VGPR rates of 50%/0%/16%.46 Most recently, a phase 1/2 study of KPd (N=64), using twice weekly carfilzomib at 27 mg/m2 in combination with pomaldomide and dexamethasone, yielded a promising RR/⩾CR/⩾VGPR rates (84%/19%/52%) and median PFS of 16.8 months and 2-year OS of 77%.47
Daratumumab has also been studied in combination with pomalidomide/dexamethasone (Dara-Pd)48, 49 (Figure 2). Dara-Pd was studied in 103 advanced RRMM, who had a median of four prior lines of therapy, and were refractory to their last treatment.50 Eighty-nine percent of patients were refractory to lenalidomide, 71% were refractory to bortezomib, and 64% of patients to both bortezomib and lenalidomide. These patients received daratumumab at 16 mg/kg at the recommended dosing schedule, in addition to pomalidomide (4 mg daily for 21 days) and dexamethasone (40 mg weekly). The primary end-point was safety, and secondary end-points were response rates and MRD negativity by NGS. Results showed that the safety profile was similar to pomalidomide/dexamethasone except the infusion reactions associated with daratumumab. Moreover, serious adverse reactions (grade 3/4) reported in⩾5% patients included pneumonia (7%). The overall response rate in the study was 60% (⩾CR/VGPR/PR were 17%/25%/18%), with ⩾VGPR in 42% of patients. The RR was 58% in those double refractory patients. The median time to response was 1 month (range=0.9–2.8 months). The median PFS and OS were 8.8 months and 17.5 months, respectively. Response rate was 60%. In patients with ⩾CR, 29% achieved MRD negativity at a threshold of 10−5. Based on this study, Dara-Pd was approved by the FDA in June 2017.
Agents with novel mechanism of action other than PIs, IMiDs or antibodies
Apart from these next-generation PIs or IMiDs, drugs with novel mechanism of action are also being studied in RRMM. Venetoclax (ABT-199) is an oral BH3 mimetic small molecule inhibiting B-cell CLL/lymphoma (BCL)-2, an anti-apoptotic protein overexpressed especially in the t(11;14) MM subtype. Based upon the activity of venetoclax (VEN) in chronic lymphocytic leukemia (CLL), single agent venetoclax has been studied in patients with advanced RRMM in a phase 1 study at dose levels ranging from 300 mg/d to 1200 mg/d (N=66)51 (Figure 2). The median number of prior lines of therapy was five, 79% of patients were refractory to the last regimen, and 61% double refractory to both bortezomib and lenalidomide. The study yielded an overall RR/⩾CR/⩾VGPR of 21%/7%/15%, with much higher responses (RR/⩾CR/⩾VGPR of 40%/14%/27%) and superior PFS in patients with t(11;14) than those without. A phase 1b study of Venetoclax in combination with bortezomib and dexamethasone (VEN-Vd) is ongoing52 (Figure 2).
Selinexor is the first drug in a new class of agents known as selective inhibitors of nuclear export.53 Selinexor is an inhibitor of exportin-1 (XPO-1), a nuclear exporter of the majority of tumor suppressor proteins, the glucocorticoid receptor, and oncoprotein mRNAs.54 As a result, selinexor induces nuclear retention and hence activation of tumor suppressors and glucocorticoid receptors, in addition to suppression of translation and expression of oncoproteins such as MYC and BCL-2. In combination with low-dose dexamethasone, selinexor (SEL) has been studied in quad-refractory (refractory to bortezomib, carfilzomib, lenalidomide and pomalidomide) or penta-refractory MM, additionally refractory to a CD38-targeting monoclonal antibody (daratumumab or isatuximab) (Figure 2). In this refractory cohort of MM, RR/⩾CR/⩾VGPR was 21%/0%/5%, with a median PFS and OS of 2.3 months and 5.5 months respectively. Interestingly, there was a higher RR in those with high-risk cytogenetics (RR: 35%) than standard-risk (18%).55
Immunotherapy in MM
In addition to these small molecules and therapeutic monoclonal antibodies, antibodies against immune check-points are undergoing clinical trials in RRMM. Normally, tumor-specific T-cell activation involves multiple steps, including priming and activation that occurs in the regional lymph node where the central check-point is localized, followed by tumor-specific cytotoxicity at the tumor site, that is, the peripheral check-point.56 Ipilimumab (a cytotoxic T-lymphocyte-associated protein (CTLA)-4 inhibitor) and pembrolizumab (anti- programmed cell death (PD) 1 inhibitor) are check-point inhibitors targeting the central and peripheral check-points respectively.57, 58 Pembrolizumab, an anti-PD1 antibody, is undergoing clinical trials in patients with RRMM in combination with either lenalidomide/dexamethasone (Pem-Rd) or pomalidomide/dexamethasone (Pem-Pd)59, 60 (Figure 2). Moreover, chimeric antigen receptor T-cells therapies targeting B-cell maturation antigen expressed mainly in plasma cells, or targeting CD19 postulated to be expressed on myeloma stem/progenitor cells, appear promising and have produced deep responses in refractory MM.61, 62 Promising data from a study of B-cell maturation antigen-targeting CAR-T cell in 19 RRMM patients has been recently presented in ASCO63 (Figure 2). The median number of infused cells was 4.7 × 106/ kg. The median follow-up times was 208 (range: 62–321) days. Seven were monitored for >6 months, of which six achieved MRD-negativity in addition to serological CR. Another 12 patients with a follow-up of <6 months achieved near CR by the modified EBMT criteria. Therefore, the overall response rate was 100% with 18 (95%) achieving ⩾near CR. Acute but manageable cytokine release syndrome was observed in 14 (74%) patients (grade 1 in 9, grade 2 in 2, and one each of grade 3 and grade 4) patient who received treatment. No relapse was observed with a median follow-up of 6 months. Therefore, B-cell maturation antigen-targeting CAR-T cells appear a promising modality of treatment.
Role of salvage ASCT
In patients who relapsed after a prior ASCT, salvage ASCT has been shown to be an effective consolidation after reinduction therapy in multiple retrospective studies,64, 65 which showed that the most important factor predicting PFS after salvage ASCT was the duration of remission from the first ASCT. In particular, patients with a PFS of ⩾18 months after initial ASCT are most likely to benefit from a salvage autotransplant. However, patients with a <18-month duration of response after initial ASCT should not be considered for a second autograft in the relapsed setting because this group will probably only experience ASCT-related toxicity without substantial clinical benefit.65 In the era of novel agents, the only RCT to demonstrate the role of salvage ASCT in myeloma patients at first relapse/progression at least 12 months after prior ASCT was the UK Myeloma X study.66, 67 In this study, relapsing MM patients who achieved at least stable disease after PAD re-induction (bortezomib/ doxorubicin/dexamethasone) were randomized to receive salvage ASCT (N=89) or weekly oral cyclophosphamide (N=85) as consolidation, which showed a superior time to disease progression (19 months versus 11 months, HR: 0.45; P<0.0001), and recently OS (67 months versus 52 months, HR: 0.56; P=0.022) in those receiving salvage ASCT compared with oral cyclophosphamide consolidation.66 Given that only 174 of 297 enrolled patients were randomized, a relatively large number of patients did not get randomized. Insufficient stem cell yield was the main reason that patients were not included in the randomization, illustrating the importance of mobilizing sufficient stem cells for at least two ASCTs during the first stem cell harvest.65, 66 Therefore, salvage ASCT remains a standard consolidation after re-induction in MM patients relapsing/progressing after first ASCT.65, 66, 67 This is particularly important in many countries where patients do not have access to novel agents.
Role of salvage allogeneic HCT
Salvage allogeneic HCT in MM, in the form of reduced intensity conditioning (RIC) or non-myeloablative allogeneic SCT (NST), is performed, particularly in younger patients with either multiple relapses or short initial remission after ASCT. However, the data are conflicting. For instance, large retrospective registry analysis suggested no benefit of a salvage allogeneic HCT when compared to a second autotransplant.68 In contrast, when HLA typing was performed after relapse from a prior ASCT such that patients with donor (donor group) received RIC/NST allo-SCT while those without donors (no donor group) received a second ASCT, De Lavallade et al. reported a higher PFS but not OS in the ‘donor’ than ‘no donor’ group.69 Similarly, when MM relapses were treated with novel agent-based induction, Patriarca et al. reported a significant benefit in 2-year PFS for patient with a donor than for those without (42 versus 18%) (P<0.0001) with similar 2-year OS of 54% and 53% respectively in patients relapsing after an initial autograft.70 Therefore, RIC/NST allo-SCT may warrant further study, particularly in those relapsing within 12 months from prior ASCT, which remains an unmet medical need.64, 65
Proposed treatment algorithm for management of RRMM
Relapses may be classified into ‘initial’ (drug-sensitive), ‘intermediate’ or ‘advanced’ (drug-refractory disease) relapses by the number of prior lines of therapy or refractoriness to PI and/or IMiD, not by the time from diagnosis to relapse (Figure 3).

Proposed algorithm for the treatment of ‘initial’, ‘intermediate’ or ‘advanced’ myeloma relapses. Those with ‘initial’ relapse may have been treated with bortezomib (V)-based, lenalidomide (R)-based, or VR-based induction regimens. In ‘initial’ relapse, in which patients had prior bortezomib-based induction regimens including VTd, VCd, VMP or VTP, if they are bortezomib-refractory, the first choice of salvage is Dara-Rd (red font), followed by KRd. In resource-restricted countries, doublets such as Rd is feasible (blue font). On the other hand, if patients are only bortezomib-exposed but not resistant, Dara-Rd and Dara-Vd (red font) are the most potent options, followed by other PI-based triplets (KRd, Ixa-Rd or VRd), or less expensive doublet (Kd, Rd) (blue font). Moreover, one may repeat the original regimen if the duration of response is ⩾2 years. In those who relapse from Rd induction, if they are R-refractory, pomalidomide combinations (PCd, VPd, Pd), second-generation PI-combinations (KPd, Kd) or V-based regimens (Dara-Vd, VCd, Vd) are viable options. By contrast, in those R-sensitive relapse, Dara-Rd and Dara-Vd are the most potent options, followed by KRd, Ixa-Rd, VRd, Elo-Rd, pomalidomide combinations (PCd, KPd, VPd), or less expensive regimens such as VCd, Vd or Kd (blue font). For patients who relapse on VRd induction, they are double V- and R-refractory, hence managed as ‘intermediate’ relapse. However, if they relapse 2 years after VRd induction, they remain V- and R-sensitive; hence, both V- and R-containing regimens will remain viable. Therefore, Dara-Rd and Dara-Vd are first choices, followed by KRd, Ixa-Rd, Elo-Rd, in addition to pomalidomide-containing regimens such as KPd, VPd, or to repeat VRd. In the ‘intermediate’ relapse category, who may be double V/R-refractory, pomalidomide, daratumumab and carfilzomib-combinations are the main options in addition to single agent daratumumab. In ‘advanced’ relapse, clinical trials of agents with novel mechanism including exportin-1 inhibitor (selinexor combinations), or BCL2 inhibitor (venetoclax combinations) in addition to immunotherapy (anti-BCMA CAR-T cells, allo-HSCT or anti-PD1 antibodies) have to be considered.
Upon relapse, one may use drugs that are naïve to the patient, drugs of a different class, a more potent version of the same class, or repeat the same regimen. For instance, in patients relapsing from Rd, one may consider PI-based regimens (including bortezomib-, carfilzomib- or ixazomib-based regimens), antibody-based or pomalidomide combinations, preferably in the form of a triplet. Finally, the original induction regimen can be repeated in case there is a sufficiently long duration of response, for example ⩾2 years.71
Despite that there is no head-to-head comparison among these novel agents, information about efficacy of the respective regimens may be inferred from the RCTs if the study design, end-points and control arms are comparable.25 For instance, multiple RCTs included patients relapsing with 1−3 prior lines of therapy, compared ‘X’-Rd with Rd as control, using PFS as primary end-point with both the intervention and control arms being continued till disease progression or intolerable side-effects. With this framework, there were four clinical trials using Rd as control including Aspire (KRd versus Rd, HR: 0.69), Tourmaline MM1 (Ixa-Rd versus Rd, HR: 0.74), Eloquent-2 (Elo-Rd versus Rd, HR: 0.70) and Pollux (Dara-Rd versus Rd, HR: 0.37), among which the HR of Pollux was lowest (Table 1). Therefore, Dara-Rd is likely the most potent regimen among these viable salvage regimens at relapse.
Based on these considerations, a treatment algorithm is proposed for management of relapses (Figure 3). The regimens are recommended based on efficacy (by HR in the respective RCT), prior regimen used, sensitivity to prior regimens (bort- or len-), and data from phase 3 studies, or at least phase 2 studies. A triplet is preferred whenever possible. Moreover, while V- or R-refractory cases mandate use of alternative agents, alternative agents are preferred but not mandatory in V-/R-exposed but sensitive patients. In V-refractory patients, KRd or Elo-Rd can be considered but not Ixa-Rd as V-refractory patients were excluded in Tourmaline MM1. Finally, while novel agent triplets are generally preferred, to accommodate affordability in less affluent countries, less expensive regimens (blue font) in the form of triplet (PCd instead of KPd) or (Kd or Vd (Aspire) instead of KRd in case of relapse from, for example, VTD) are also included.
In ‘Initial’ relapse (Figure 3), in which patients had prior bortezomib-based induction regimens including VTd, VCd, VMP or VTP, if they are bortezomib-refractory, the first choice of salvage is Dara-Rd (red font, Figure 3) because of highest efficacy based on HR. KRd and Elo-Rd but not Ixa-Rd can be considered viable triplets as 15−22% V-refractory patients had been enrolled in the respective RCTs. However, Kd cannot be recommended for V-refractory relapses, as V-refractory patients had been excluded in the Endeavor study. In resource-restricted countries, doublet such as Rd is feasible (blue font, Figure 3). On the other hand, if patients are only bortezomib-exposed but not refractory, Dara-Rd and Dara-Vd (red font, Figure 3) are preferred based on their low HRs (Table 1), followed by other PI-based triplets (KRd, Ixa-Rd, Elo-Rd or even VRd), or less expensive doublet (Kd, Rd) (blue font, Figure 3). Moreover, one may consider to repeat the original regimen if the duration of response is ⩾2 years.71 In those who relapse from Rd induction (Figure 3), if they are R-refractory (for example, relapse during Rd treatment), antibody-based regimen (Dara-Vd), pomalidomide combinations (PCd, VPd, Pd), second-generation PI-combinations (KPd, Kd) or V-based regimens (VCd, Vd) are viable options. Moreover, less expensive options include PCd, VCd, Pd, Vd or Kd (blue font, Figure 3) By contrast, in those R-sensitive relapse, Dara-Rd and Dara-Vd are the most potent options, followed by KRd, Ixa-Rd, VRd, Elo-Rd, or pomalidomide combinations (PCd, KPd, VPd), or less expensive regimens such as VCd, Vd or Kd (blue font, Figure 3). For patients who relapse while on VRd (Figure 3), they are double V- and R-refractory, hence managed as ‘intermediate’ relapse. However, if they relapse 2 years after VRd induction, they remain V- and R-sensitive; hence both V- and R-containing regimens will remain viable options. Therefore, Dara-Rd and Dara-Vd are first choices, followed by KRd, Ixa-Rd, Elo-Rd, in addition to pomalidomide-containing regimens such as PCd, KPd, VPd, or Pd, or to repeat VRd (Figures 2 and 3). In the ‘intermediate’ relapse category, in which patients may be double V/R-refractory, pomalidomide, daratumumab and carfilzomib-combinations are the main options in addition to single agent daratumumab. In ‘advanced’ relapse, clinical trials of agents with novel mechanism including exportin-1 inhibitor (selinexor combinations), or BCL2 inhibitor (venetoclax combinations) in addition to immunotherapy (anti-B-cell maturation antigen CAR-T cells, allo-HSCT or anti-PD1 antibodies) have to be considered (Figures 2 and 3).
Discussion
A few points are noteworthy. First, there is no head-to-head comparison between next-generation novel agents. For instance, there is no study to compare carfilzomib with ixazomib. Moreover, while carfilzomib appears more potent than bortezomib in the ENDEAVOR study in relapsing MM,19 the dose of carfilzomib was 56 mg/m2, instead of 27 mg/m2 used in the ASPIRE study for RRMM.15
Secondly, among the novel agents discussed above, two agents lack single agent activity, elotuzumab and panobinostat.72 As a result, while daratumumab can be used as a single agent in RRMM, elotuzumab has to be used in combination with lenalidomie/dexamethasone. Moreover, while the clinical efficacy of bortezomib and panobinostat is a successful clinical translation from the bench to bedside by which the aggresome pathway is targeted, the role of panobinostat in RRMM is unclear because of the availability of salvage agents that carry single agent activity in MM including carfilzomib, ixazomib, pomalidomide, daratumumab, venetoclax and selinexor, efficacy of which was further enhanced when combined with additional agents.
Thirdly, in this era of next-generation novel agents, the role of salvage ASCT as a consolidation to prolong PFS and OS remains important,66, 67 hence still a standard in RRMM patients achieving a response to next-generation novel agent combinations.
Finally, we need to address the enormous costs of the combination of next-generation novel agents with immune antibody-based therapies in order to assure access to these therapies for patients worldwide.73
In summary, the efficacy of next-generation PIs, IMiDs, and monoclonal antibodies in patients with RRMM has been demonstrated in RCTs. Optimal combination of these agents remains to be defined. Evaluation of these agents in frontline induction is underway, which will hopefully result in deeper responses, as well as superior PFS and OS of MM patients. With the advent of monoclonal antibodies, a major breakthrough has been established in the treatment of MM. The future of MM is promising as we use combinations of novel and immune therapies earlier in the disease course.
References
Palumbo A, Anderson K . Multiple myeloma. N Engl J Med 2011; 364: 1046–1060.
Egan JB, Shi C-X, Tembe W, Christoforides A, Kurdoglu A, Sinari S et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012; 120: 1060–1066.
Harousseau J-L, Attal M, Avet-Loiseau H . The role of complete response in multiple myeloma. Blood 2009; 114: 3139–3146.
Zent C, Wilson C, Tricot G, Jagannath S, Siegel D, Desikan K et al. Oligoclonal protein bands and Ig isotype switching in multiple myeloma treated with high-dose therapy and hematopoietic cell transplantation. Blood 1998; 91: 3518–3523.
Chim C, Lie A, Chan E, Leung Y, Cheung S, Chan S et al. A staged approach with vincristine, adriamycin, and dexamethasone followed by bortezomib, thalidomide, and dexamethasone before autologous hematopoietic stem cell transplantation in the treatment of newly diagnosed multiple myeloma. Ann Hematol 2010; 89: 1019–1027.
Chim C, Chan EY . Oligoclonal reconstitution masquerading as myeloma relapse. Ann Hematol 2013, 1–2.
Tovar N, de Larrea CF, Aróstegui JI, Cibeira MT, Rosiñol L, Rovira M et al. Natural history and prognostic impact of oligoclonal humoral response in patients with multiple myeloma after autologous stem cell transplantation: long-term results from a single institution. Haematologica 2013; 98: 1142–1146.
Alejandre ME, Madalena LB, Pavlovsky MA, Facio ML, Corrado C, Milone G et al. Oligoclonal bands and immunoglobulin isotype switch during monitoring of patients with multiple myeloma and autologous hematopoietic cell transplantation: a 16-year experience. Clin Chem Lab Med 2010; 48: 727–731.
Zamarin D, Giralt S, Landau H, Lendvai N, Lesokhin A, Chung D et al. Patterns of relapse and progression in multiple myeloma patients after auto-SCT: implications for patients’ monitoring after transplantation. Bone Marrow Transplant 2013; 48: 419–424.
Blade J, Samson D, Reece D, Apperley J, Bjorkstrand B, Gahrton G et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Br J Haematol 1998; 102: 1115–1123.
Sonneveld P, Broijl A . Treatment of relapsed and refractory multiple myeloma. Haematologica 2016; 101: 396–406.
Rajkumar SV, Harousseau J-L, Durie B, Anderson KC, Dimopoulos M, Kyle R et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011; 117: 4691–4695.
Harousseau J-L, Avet-Loiseau H, Attal M, Charbonnel C, Garban F, Hulin C et al. Achievement of at least very good partial response is a simple and robust prognostic factor in patients with multiple myeloma treated with high-dose therapy: long-term analysis of the IFM 99-02 and 99-04 trials. J Clin Oncol 2009; 27: 5720–5726.
Gaballa S, Saliba RM, Srour S, Lu G, Brammer JE, Shah N et al. Outcomes in patients with multiple myeloma with TP53 deletion after autologous hematopoietic stem cell transplant. Am J Hematol 2016; 91: 10.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015; 372: 142–152.
Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016; 374: 1621–1634.
San Miguel J, Weisel K, Moreau P, Lacy M, Song K, Delforge M et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol 2013; 14: 1055–1066.
Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med 2016; 375: 754–766.
Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016; 17: 27–38.
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015; 373: 621–631.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016; 387: 1551–1560.
Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015; 2015: 1207–1219.
Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016; 375: 1319–1331.
Kumar SK, Callander NS, Alsina M, Atanackovic D, Biermann JS, Chandler JC et al. Multiple myeloma, version 3.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw 2017; 15: 230–269.
Rajkumar SV, Kyle RA . Progress in myeloma—a monoclonal breakthrough. N Engl J Med 2016; 375: 1390–1392.
Dimopoulos M, Goldschmidt H, Niesvizky R, Joshua D, Chng W-J, Oriol A et alOverall survival of patients with replased or refractory multiple myeloma treated with carfilzomib and dexamethasone versus bortezomib and dexamethasone in the randomized phase 3 endeavor trial EHA 2017. EHA Learning Center: Madrid, 2017 Haematologica 2017; 102 (S2): 168.
Moreau P, Joshua D, Chng W, Palumbo A, Goldschmidt H, Hájek R et al. Impact of prior treatment on patients with relapsed multiple myeloma treated with carfilzomib and dexamethasone vs bortezomib and dexamethasone in the phase 3 ENDEAVOR study. Leukemia 2017; 31: 115–122.
Richardson PG, Avet-Loiseau H, Palumbo A, Viterbo L, Nagler A, Ganly P et al. Efficacy and safety of ixazomib plus lenalidomide-dexamethasone (IRd) vs placebo-rd in patients (pts) with relapsed/refractory multiple myeloma (RRMM) by cytogenetic risk status in the global phase III Tourmaline-MM1 study. J Clin Oncol 2016; 34 (15_suppl): 8018.
Baz RC, Martin TG, Lin H-Y, Zhao X, Shain KH, Cho HJ et al. Randomized multicenter phase 2 study of pomalidomide, cyclophosphamide, and dexamethasone in relapsed refractory myeloma. Blood 2016; 127: 2561–2568.
Richardson PG, Bensmaine A, Doerr T, Wang J, Zaki MH . MM-007: a phase 3 trial comparing the efficacy and safety of pomalidomide (POM), bortezomib (BORT), and low-dose dexamethasone (LoDEX [PVD]) versus BORT and LoDEX (VD) in subjects with relapsed or refractory multiple myeloma (RRMM). J Clin Oncol 2015; 33 (15_suppl): TPS8610.
Catley L, Weisberg E, Kiziltepe T, Tai Y-T, Hideshima T, Neri P et al. Aggresome induction by proteasome inhibitor bortezomib and α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006; 108: 3441–3449.
Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA 2005; 102: 8567–8572.
Carew JS, Medina EC, Esquivel I, Juan A, Mahalingam D, Swords R et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J cell Mol Med 2010; 14: 2448–2459.
San-Miguel JF, Hungria VT, Yoon S-S, Beksac M, Dimopoulos MA, Elghandour A et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014; 15: 1195–1206.
Vogl DT, Raje NS, Jagannath S, Richardson PG, Hari P, Orlowski RZ et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res 2017; 23: 3307–3315, clincanres2526.2016.
Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016; 128: 384–394.
Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016; 128: 37–44.
San-Miguel J, Weisel K, Cook G, Leiba M, Suzuki K, Kumar S et alEfficacy by cytogenetic risk status for daratumumab in combination with lenalidomide and dexamethason or bortezomib and dexamethason in replapsed or refractory multiple myeloma EHA 2017. EHA Learning Center: Madrid, 2017 Haematologica 2017; 102 (S2): 1.
Joshua Ryan Richter TGM, Vij Ravi, Cole Craig, Atanackovic Djordje, Zonder Jeffrey A., Kaufman Jonathan L. et al. J Clin Oncol 2016; 34.
Raab MS, Chatterjee M, Goldschmidt H, Agis H, Blau I, Einsele H et al. A phase I/IIa study of the CD38 antibody MOR202 alone and in combination with pomalidomide or lenalidomide in patients with relapsed or refractory multiple myeloma. Blood 2016; 128: 1152.
van de Donk NW, Moreau P, Plesner T, Palumbo A, Gay F, Laubach JP et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 2015; 127: 681–695.
Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother 2013; 62: 1841–1849.
Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008; 112: 1329–1337.
Dimopoulos MA, Lonial S, White D, Moreau P, Mateos M-V, Miguel JS et alPhase 3 eloquent-2 study: extended 4-year follow-up of elotuzumab plus lenalidomide/dexamethasone vs lenalidomide/dexamethasone in replapsed/refractory multiple myeloma EHA 2017. EHA Learning Center: Madrid, 2017 Haematologica 2017; 102 (S2): 167.
Lacy MQ, LaPlant BR, Laumann KM, Kumar S, Gertz MA, Hayman SR et al. Pomalidomide, bortezomib and dexamethasone (PVD) for patients with relapsed lenalidomide refractory multiple myeloma (MM). Blood 2014; 124: 304–304.
Shah JJ, Stadtmauer EA, Abonour R, Cohen AD, Bensinger WI, Gasparetto C et al. Carfilzomib, pomalidomide, and dexamethasone for relapsed or refractory myeloma. Blood 2015; 126: 2284–2290.
Jakubowiak AJ, Rosenbaum CA, Stephens L, Kukreti V, Cole C, Zimmerman T et al. Final Results of Phase (PH) 1/2 Study of Carfilzomib, Pomalidomide, and Dexamethasone (KPD) in Patients (PTS) with Relapsed/Refractory Multiple Myeloma (RRMM): A Multi-Center MMRC Study. European Hematology Association: Madrid, Spain, 2017; p 680.
Nooka AK, Joseph N, Boise LH, Gleason C, Kaufman JL, Lonial S . Clinical Efficacy of daratumumab, pomalidomide and dexamethasone in relapsed, refractory myeloma patients: utility of retreatment with daratumumab among refractory patients. Blood 2016; 128: 492.
Branca A, Buros A, Yoon D, Suva LJ, Weinhold N, Rasche L et al. Daratumumab single agent and daratumumab plus pomalidomide and dexametasone in relapsed/refractory multiple myeloma: a real life retrospective evaluation. Blood 2016; 128: 4516.
Chari A, Suvannasankha A, Fay JW, Arnulf B, Kaufman JL, Ifthikharuddin JJ et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017; 130: 974–981.
Kumar S, Vij R, Kaufman JL, Mikhael J, Facon T, Pegourie B et al. Venetoclax monotherapy for relapsed/refractory multiple myeloma: safety and efficacy results from a phase I study. Blood 2016; 128: 488.
Moreau P, Chanan-Khan AA, Roberts AW, Agarwal AB, Facon T, Kumar S et al. Venetoclax combined with bortezomib and dexamethasone for patients with relapsed/refractory multiple myeloma. Blood 2016; 128: 975.
Conforti F, Wang Y, Rodriguez JA, Alberobello AT, Zhang Y-W, Giaccone G . Molecular pathways: anticancer activity by inhibition of nucleocytoplasmic shuttling. Clin Cancer Res 2015; 21: 4508–4513.
Tai Y, Landesman Y, Acharya C, Calle Y, Zhong M, Cea M et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia 2014; 28: 155–165.
Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D et al. Selinexor and low dose dexamethasone (Sd) in patients with lenalidomide, pomalidomide, bortezomib, carfilzomib and anti-CD38 Ab refractory multiple myeloma (MM): STORM study. Blood 2016; 128: 491.
Chen DS, Mellman I . Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39: 1–10.
Pardoll DM . The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264.
Rosenblatt J, Avigan D . Targeting the PD-1/PD-L1 axis in multiple myeloma: a dream or a reality. Blood 2016; 129: 275–279.
San Miguel J, Mateos M-V, Shah JJ, Ocio EM, Rodriguez-Otero P, Reece D et al. Pembrolizumab in combination with lenalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma (RRMM): keynote-023. Blood 2015; 126: 505–505.
Badros AZ, Kocoglu MH, Ma N, Rapoport AP, Lederer E, Philip S et al. A phase II study of anti PD-1 antibody pembrolizumab, pomalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma (RRMM). Blood 2015; 126: 506–506.
Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016; 128: 1688–1700.
Garfall AL, Maus MV, Hwang W-T, Lacey SF, Mahnke YD, Melenhorst JJ et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med 2015; 373: 1040–1047.
Fan F, Zhao W, Liu J, He A, Chen Y, Cao X et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol 2017; 35 (18_suppl): LBA3001.
Atanackovic D, Schilling G . Second autologous transplant as salvage therapy in multiple myeloma. Br J Haematol 2013; 163: 565–572.
Giralt S, Garderet L, Durie B, Cook G, Gahrton G, Bruno B et al. American Society of Blood and Marrow Transplantation, European Society of Blood and Marrow Transplantation, Blood and Marrow Transplant Clinical Trials Network, and International Myeloma Working Group Consensus Conference on salvage hematopoietic cell transplantation in patients with relapsed multiple myeloma. Biol Blood Marrow Transplant 2015; 21: 2039–2051.
Cook G, Ashcroft AJ, Cairns DA, Williams CD, Brown JM, Cavenagh JD et al. The effect of salvage autologous stem-cell transplantation on overall survival in patients with relapsed multiple myeloma (final results from BSBMT/UKMF Myeloma X Relapse [Intensive]): a randomised, open-label, phase 3 trial. Lancet Haematol 2016; 3: e340–e351.
Cook G, Williams C, Brown JM, Cairns DA, Cavenagh J, Snowden JA et al. High-dose chemotherapy plus autologous stem-cell transplantation as consolidation therapy in patients with relapsed multiple myeloma after previous autologous stem-cell transplantation (NCRI Myeloma X Relapse [Intensive trial]): a randomised, open-label, phase 3 trial. Lancet Oncol 2014; 15: 874–885.
Freytes CO, Vesole DH, LeRademacher J, Zhong X, Gale RP, Kyle RA et al. Second transplants for multiple myeloma relapsing after a previous autotransplant-reduced-intensity allogeneic vs autologous transplantation. Bone Marrow Transplant 2014; 49: 416–421.
De Lavallade H, El-Cheikh J, Faucher C, Fürst S, Stoppa A, Coso D et al. Reduced-intensity conditioning allogeneic SCT as salvage treatment for relapsed multiple myeloma. Bone Marrow Transplant 2008; 41: 953–960.
Patriarca F, Einsele H, Spina F, Bruno B, Isola M, Nozzoli C et al. Allogeneic stem cell transplantation in multiple myeloma relapsed after autograft: a multicenter retrospective study based on donor availability. Biol Blood Marrow Transplant 2012; 18: 617–626.
Palumbo A, Rajkumar SV, San Miguel JF, Larocca A, Niesvizky R, Morgan G et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem-cell transplantation. J Clin Oncol 2014; 32: 587–600.
Gyawali B, Prasad V . Drugs that lack single-agent activity: are they worth pursuing in combination? Nat Rev Clin Oncol 2017; 14: 193–194.
Rajkumar SV, Harousseau JL . Next-generation multiple myeloma treatment: a pharmacoeconomic perspective. Blood 2016; 128: 2757–2764.
Acknowledgements
We thank Dr Bart Barlogie, Hematology and Medical Oncology, Mount Sinai Hospital, New York, USA, & Professor Jean Luc Harousseau, University of Nantes, France for critical review of the manuscript. We also thank Dr Elaine Au, Division of Immunology, Department of Pathology, Queen Mary Hospital, for providing the immunofixation gel picture in Figure 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chim, C., Kumar, S., Orlowski, R. et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia 32, 252–262 (2018). https://doi.org/10.1038/leu.2017.329
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.329
This article is cited by
-
Comparative performance of scFv-based anti-BCMA CAR formats for improved T cell therapy in multiple myeloma
Cancer Immunology, Immunotherapy (2024)
-
A prospective, multicenter, observational study of ixazomib plus lenalidomide-dexamethasone in patients with relapsed/refractory multiple myeloma in Japan
Annals of Hematology (2024)
-
A phase I first-in-man study to investigate the pharmacokinetics and safety of liposomal dexamethasone in patients with progressive multiple myeloma
Drug Delivery and Translational Research (2023)
-
Real-World Outcomes of Belantamab Mafodotin for Relapsed/Refractory Multiple Myeloma (RRMM): Preliminary Results of a Spanish Expanded Access Program (EAP)
Oncology and Therapy (2023)
-
PD-1 and LAG-3-positive T cells are associated with clinical outcomes of relapsed/refractory multiple myeloma patients
European Journal of Medical Research (2022)
