
Abstract
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm characterized by hyperplastic megakaryopoiesis and myelofibrosis. We recently described the upregulation of MAF (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog) in PMF CD34+ hematopoietic progenitor cells (HPCs) compared to healthy donor. Here we demonstrated that MAF is also upregulated in PMF compared with the essential thrombocytemia (ET) and polycytemia vera (PV) HPCs. MAF overexpression and knockdown experiments shed some light into the role of MAF in PMF pathogenesis, by demonstrating that MAF favors the megakaryocyte and monocyte/macrophage commitment of HPCs and leads to the increased expression of proinflammatory and profibrotic mediators. Among them, we focused our further studies on SPP1 and LGALS3. We assessed SPP1 and LGALS3 protein levels in 115 PMF, 47 ET and 24 PV patients plasma samples and we found that SPP1 plasma levels are significantly higher in PMF compared with ET and PV patients. Furthermore, in vitro assays demonstrated that SPP1 promotes fibroblasts and mesenchymal stromal cells proliferation and collagen production. Strikingly, clinical correlation analyses uncovered that higher SPP1 plasma levels in PMF patients correlate with a more severe fibrosis degree and a shorter overall survival. Collectively our data unveil that MAF overexpression contributes to PMF pathogenesis by driving the deranged production of the profibrotic mediator SPP1.
Similar content being viewed by others


Introduction
Primary myelofibrosis (PMF) is a Philadelphia-negative (Ph−) myeloproliferative neoplasm (MPN). Ph− MPNs include polycythemia vera (PV) and essential thrombocytemia (ET) besides PMF and share a myeloproliferative phenotype with clonal hematopoietic stem/progenitor cells expansion. On the basis of the WHO classification of Ph− MPNs, the primary criteria for differential diagnosis of PMF from ET and PV are the presence of hyperplastic and dysplastic megakaryocytes with maturation defects and the bone marrow (BM) fibrosis, that results from reticulin and/or collagen fibers deposition.1 The malignant hematopoietic cells, mainly megakaryocytes and monocytes, are endowed of a proinflammatory and profibrotic phenotype in PMF patients.2 The altered interplay between these cells and the BM stromal cells in turn drives the development of myelofibrosis, osteosclerosis and extramedullary hematopoiesis, that are all features of PMF pathogenesis.3
Great efforts to unravel the genomic landscape of MPNs have provided an increasing knowledge of the genomic lesions responsible for the uncontrolled clonal proliferation, with the identification of ‘driver’ mutations (JAK2, MPL and CALR) that directly or indirectly lead to a deranged JAK/STAT signaling.4, 5, 6, 7 Nevertheless, none of the genomic lesions described to date is able to completely recapitulate all the pathological features of PMF in vivo. Therefore, further efforts should be focused on elucidating the molecular mechanisms behind the skewed crosstalk between the malignant hematopoietic cells and BM microenvironment to develop novel therapeutic options to dampen the proinflammatory and profibrotic phenotype of malignant hematopoietic cells in PMF patients.2
To shed light on the molecular mechanisms underlying PMF pathogenesis, we recently compared the gene expression profiles of CD34+ hematopoietic progenitor cells (HPCs) from PMF patients and healthy donors (HDs).8 This study provided many insights into the molecular mechanisms potentially relevant for PMF pathogenesis; notably we found the strong upregulation of the transcription factor MAF (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog) in CD34+ HPCs from PMF patients.8
MAF belongs to the large-Maf family of transcription factors that act as homo- or heterodimers by binding to the Maf-responsive elements.9, 10 c-Maf gene gives rise to two splicing isoforms, MAF variant 1 (RefSeq NM_005360.3) and MAF variant 2 (RefSeq NM_001031804.1). The proteins encoded differ for their carboxy-terminal portion. Translocations involving c-Maf locus are present in 5% of multiple myeloma patients, whereas an additional 50% of cases overexpresses MAF through an unknown mechanism.11 In both these conditions, the oncogenic activity of MAF is linked to its overexpression.
As MAF is upregulated in PMF HPCs compared with both the HD-derived and the ET/PV patients-derived counterparts, here we investigated the role of MAF in PMF pathogenesis.
Collectively, our data unveil that the upregulation of MAF in PMF patients drives the expression of proinflammatory and profibrotic genes in the HPCs progeny, therefore contributing to establish the deranged BM microenvironment, that is a hallmark of PMF.
Materials and methods
Ethic statements
Human CD34+ cells were purified upon donor’s informed written consent from umbilical cord blood (CB) samples, collected after normal deliveries, according to the institutional guidelines for discarded material. Monocytes of HDs and PMF patients, and PMF CD34+ cells were purified from peripheral blood (PB) samples. Plasma samples were collected from 186 patients with MPNs (115 PMF, 24 PV and 47 ET) and 60 HDs. BM mesenchymal stromal cells (MSC) were collected from 5 HDs and 5 PMF patients. All subjects provided informed written consent, and the study was performed under the AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative) protocol (Comitato Etico Area Vasta dell'Azienda Ospedaliero-Universitaria Careggi, approval date: 22 April, 2011, approval file number # 2011/0014777).
MAF expression analysis in PMF and HD CD34+ cells
The raw Affymetrix data previously published8 for CD34+ cells isolated from PMF patients (n=38) and HDs (n=30; n=14 from BM and n=16 from PB) were downloaded from the GEO repository (Series GSE85190). PMF patients were further characterized by the JAK2, MPL or CALR mutation carried and the BM fibrosis degree (Supplementary Table S1). The raw Affymetrix data for CD34+ cells from PV (n=24) and ET patients (n=27; RZ and RM, manuscript submitted) were also included for MAF expression analysis.
The probe level data were normalized and converted into expression values by using the robust multiarray average procedure in Partek GS. 6.6 Software Package (http://www.partek.com).
The comparative analyses of MAF expression in CD34+ cells from MPN patients and HDs were performed by using the Kruskal–Wallis H-test when comparing more than two independent groups. Next, the differences between sample pairs were assessed by the Mann–Whitney U test.
Statistical analysis
The statistics used for in vitro experiments data analysis were based on two-tailed Student’s t-tests for averages comparison in paired samples. Data were analyzed by using Microsoft Excel (Microsoft Office, 2008 release) and are reported as mean±standard error of the mean (s.e.m.). P<0.05 was considered significant. Statistical analyses for clinical correlation were performed using the IBM Statistical Package for Social Sciences (SPSS) statistics v23. Comparisons between groups were performed by using the Mann–Whitney U test, and the chosen level of significance was P<0.05 with a two-tailed test. Correlation between two variables were examined by simple regression analysis. Patients with plasma levels of SPP1 and LGALS3 greater than the median value reported in normal plasma were considered high level patients. The cumulative probability of overall survival (OS) was estimated using the Kaplan–Meier method. Differences in OS among the groups were compared by using a log-rank test in univariate analysis.
Results
MAF expression levels in CD34+ cells from MPN patients
Microarray data for human primary CD34+ HPCs from PMF patients (n=38 from PB; n=20 JAK2-mutated, n=3 MPL-mutated, n=9 CALR-mutated and n=6 triple-negative) and HDs (n=14 from BM and n=16 from PB) were obtained from a data set that we previously published.8 The MAF expression data in BM-derived CD34+ cells from ET (n=27) and PV (n=24) patients were collected from an unpublished data set (RZ and RM, manuscript submitted).
As we previously reported,8 MAF was strongly upregulated in PMF patients compared with HDs (fold change (FC)=8.69, P=4.25 × 10−12; Figure 1a). Both the MAF splicing variants, namely MAF variant 1 (hereafter reported as MAFVAR1, RefSeq NM_005360.3) and MAF variant 2 (MAFVAR2, RefSeq NM_001031804.1), were upregulated in PMF versus HD CD34+ cells (Supplementary Results and Supplementary Figure S1). In addition, MAF was remarkably upregulated in PMF CD34+ cells compared with ET (FC=2.61, P=1.62 × 10−5) and PV counterparts (FC=2.99, P=1.07 × 10−6; Figure 1a). On the contrary, no significant correlation between the mutation carried (JAK2, MPL, CALR) and the expression levels of MAF in PMF patients was found (Figure 1b). Although the expression of MAF appeared to be slightly lower in triple-negative compared with JAK2-, MPL- and CALR-mutated PMF patients (Figure 1b), this difference was not statistically significant (P=0.093). Furthermore, the same analysis restricted to the JAK2V617F-positive PMF and ET patients did not highlight any correlation between the JAK2V617F allele burden and the MAF expression levels. Indeed, CD34+ cells from PMF patients with different allele burden displayed superimposable MAF expression levels (P>0.05). No differences were detected between JAK2V617 homozygous and heterozygous PMF patients (P=0.3837). Overlapping MAF expression levels were similarly detected in ET patients regardless of their JAK2V617F allele burden (P>0.05) (Figure 1c).

Expression levels of MAF in CD34+ cells from MPN patients. Expression levels of MAF in CD34+ cells from MPN patients and HDs. Gene expression levels were measured by microarray analysis performed by Affymetrix platform as detailed in Norfo et al.8(please, see the ‘Materials and Methods’ section). MAF expression levels are reported as Robust Multiarray Analysis (RMA)-normalized log2 signals, which were obtained by using the Partek GS software. Boxes represent the interquartile range that contains 50% of the subjects, the horizontal line in the box marks the median, and bars show the range of values. Data in a are representative of 38 PMF, 27 ET, 24 PV and 30 HD samples (14 from BM and 16 from PB). Only PMF (n=38) and HD (n=30) samples are shown in b, where PMF samples are classified into JAK2-mutated (n=20), MPL-mutated (n=3), CALR-mutated (n=9) and triple-negative (n=6), based on the mutational status. Only JAK2V617F-positive PMF (n=20) and ET (n=16) samples are shown in c, where samples are classified based on the JAK2V617F allele burden. BM, bone marrow; ET, essential thrombocythemia; MPN, myeloproliferative neoplasm; PMF, primary myelofibrosis; PV, polycythemia vera; HD, healthy donor; PB, peripheral blood; n, number of samples.
Effects of STAT5 activation on MAF expression
We asked whether the JAK/STAT signaling could underlie the overexpression of MAF in MPNs. As detected by Western Blot (Supplementary Figure S2A), MAF protein level resulted increased in MPL-expressing Ba/F3 cells upon treatment with TPO and, therefore, STAT5 activation. Conversely, the treatment of JAK2V617F-expressing Ba/F3 cells with increasing doses of ruxolitinib caused a reduction of STAT5 activation and MAF expression level (Supplementary Figure S2B).
Interestingly, ruxolitinib treatment lead to a reduction of MAF expression level in PMF CD34+ cells (Supplementary Figure S2C) that was even more remarkable and dose-dependent in PMF monocytes (Supplementary Figure S2D).
Overall, these data confirm the connection between the enhanced JAK/STAT signaling and the overexpression of MAF in MPNs.
Role of MAF in HPCs lineage commitment and differentiation
To understand whether MAF could be relevant for PMF pathogenesis, we studied the effects of its overexpression on the lineage commitment and differentiation of HD-derived CD34+ HPCs. Collectively, our data demonstrated that MAF overexpression enhanced the monocyte/macrophage and megakaryocyte differentiation of HD HPCs (Supplementary Results, Supplementary Table S2 and Supplementary Figure S3). In line with these observations, MAF knockdown in PMF HPCs restrained their megakaryocyte and monocyte/macrophage differentiation (Supplementary Results and Supplementary Figure S4).
Gene expression profile of MAF-overexpressing cells
To gain insights into whether and how the upregulation of MAF detected in HPCs from PMF patients (Figure 1) could be relevant for PMF pathogenesis, in a set of three independent experiments CB-derived CD34+ HPCs transduced with the LMAFVAR1IDN, LMAFVAR2IDN and LXIDN retroviral vectors were profiled by Affymetrix U219 Array (Supplementary Results; Supplementary Figures S5 and S6A–C).
A list of 729 differentially expressed genes (DEGs) in both LMAFVAR1IDN- and LMAFVAR2IDN-transduced samples compared to the controls was identified (Supplementary Tables S3 and S4).
Microarray data unveiled that several genes known to promote fibrosis (for example, ENPP2,12 and ADAM9,13 Supplementary Figure S6A) and inflammation (for example, TNF, CCL2,14 and CCL3,15 Supplementary Figure S6B) were remarkably upregulated upon MAF overexpression. Noteworthy, many of them encoded for secreted proteins (Supplementary Figure S6C).
Owing to their putative role in inflammation and fibrosis, which are key mechanisms in PMF pathogenesis, a subset of transcripts coding for inflammation and fibrosis mediators was selected for the qRT-PCR validation (Supplementary Table S5). TaqMan qRT-PCR was performed in LMAFVAR1IDN-, LMAFVAR2IDN- and LXIDN-transduced cells from an independent set of five experiments and allowed us to validate the expression of 7 out of 8 genes (87.5%) (Supplementary Figures S6D and E).
Expression of IL-8, THBS1, CCL2, PLAUR, MMP9, LGALS3 and SPP1 in culture supernatants from MAF-overexpressing cells and in plasma from MPN patients
The secreted proteins coded by the 7 qRT-PCR-validated genes were therefore assessed by means of an enzyme-linked immunosorbent assay (ELISA) (a) in the culture supernatants from LMAFVAR1IDN-, LMAFVAR2IDN- and LXIDN-transduced cells, collected at 3 days post-purification and (b) in plasma samples of PMF patients (n=30) and HDs (n=10) (Supplementary Table S6). As shown in Figure 2, subpanels i, all the proteins selected displayed significantly higher expression levels in both the MAF-overexpressing samples compared with the controls. In addition, IL-8, CCL2, PLAUR, MMP9, LGALS3 and SPP1 displayed significantly higher plasma levels in PMF patients than in HDs (P<0.05; Figures 2a–f, subpanels ii), whereas no difference was detected for THBS1 (Figure 2gii).

Detection of a subset of secreted molecules in LMAFVAR1IDN-, LMAFVAR2IDN- and LXIDN-transduced cells and in PMF patients plasma by ELISA. Protein levels of seven secreted molecules (IL-8, a; CCL2, b; PLAUR, c; MMP9, d; LGALS3, e; SPP1, f; THBS1, g) were measured by ELISA in culture supernatants from LMAFVAR1IDN, LMAFVAR2IDN and LXIDN-transduced cells (subpanels i) and plasma from PMF patients and HDs (subpanels ii). Protein levels are expressed as ng/ml. Protein levels in culture supernatants from LMAFVAR1IDN-, LMAFVAR2IDN- and LXIDN-transduced cells (subpanels i) are reported as mean±standard error of the mean (s.e.m.). *P<0.05; **P<0.01 versus LXIDN. The results come from three independent experiments. Plasma levels in PMF patients and HDs (subpanels ii) are shown as dot plots. Boxes represent the interquartile range that contains 50% of the subjects, the horizontal line in the box marks the median, and the bars show the range of values. Data are representative of 30 PMF and 10 HD samples. Comparisons between HDs and PMF patients were performed by using the Mann–Whitney U test, and the chosen level of significance was P<0.05 with a two-tailed test. ELISA, enzyme-linked immunosorbent assay; HD, healthy donor; PMF, primary myelofibrosis.
As the increase of SPP1 and LGALS3 in the plasma of PMF patients had never been reported before, we further analyzed SPP1 and LGALS3 plasma levels in 85 additional PMF patients and 60 HDs, together with 47 ET and 24 PV patients (Supplementary Table S6). As shown in Figure 3a, even though increased also in ET and PV patients versus HDs, SPP1 plasma levels were significantly higher in PMF versus ET (P=0.0139) and PV patients (P=0.0453). On the contrary, LGALS3 plasma levels were increased in all the MPN patients versus HDs (P<0.00001), with no remarkable differences among PMF, PV and ET (Figure 3b).

Detection of SPP1 and LGALS3 in the plasma of MPN patients by ELISA. Protein levels of SPP1 (a) and LGALS3 (b) were measured by ELISA in plasma samples derived from PMF (n=115), ET (n=47) and PV (n=24) patients and HDs (n=60). Protein levels are expressed as ng/ml. Boxes represent the interquartile range that contains 50% of the subjects, the horizontal line in the box marks the median, and the bars show the range of values. Comparisons between two groups were performed by using the Mann–Whitney U test, and the chosen level of significance was P<0.05 with a two-tailed test. ELISA, enzyme-linked immunosorbent assay; ET, essential thrombocythemia; HDs, healthy donors; n, number of samples; PMF, primary myelofibrosis; PV, polycythemia vera.
Assessment of the mechanistic relation between MAF and SPP1/LGALS3 expression
To shed some light on the mechanistic connection between the increased MAF expression and the overproduction of SPP1 and LGALS3, we assessed by Luciferase reporter assay the ability of MAF to transactivate SPP1 and LGALS3 expression. The proximal promoter regions of SPP1 and LGALS3 genes were cloned upstream of the Firefly Luciferase gene. Luciferase assay demonstrated that MAF is able to transactivate SPP1 promoter, as increasing doses of MAF-coding vector caused a dose-dependent induction in the luciferase activity (Figure 4ai). Conversely, MAF did not significantly affect LGALS3 promoter-driven luciferase expression (Figure 4aii).

Role of MAF in the regulation of SPP1 and LGALS3 expression. (a) MAFvar1 and MAFvar2-dependent transactivation of human SPP1 promoter-driven (i) and LGALS3 promoter-driven (ii) luciferase expression. The amounts (in ng) of co-transfected plasmids are reported. Renilla-normalized Firefly luciferase levels (mean±s.e.m.;n=5 for SPP1 promoter and n=5 for LGALS3 promoter, respectively) are normalized by setting as =1 the pXP1-SPP1(−853/+22)/LXIDN empty vector-transfected and the pXP1-LGALS3(−1720/+35)/LXIDN empty vector-transfected samples, respectively. Error bars represent s.e.m. *P<0.05 compared to pXP1-SPP1(−853/+22)/LXIDN empty vector-transfected (i) or pXP1- pXP1-LGALS3(−1720/+35)/LXIDN empty vector-transfected (ii) samples, respectively. (b) Expression levels of MAFvar1, MAFvar2, SPP1 and LGALS3 upon MAF knockdown in HD-derived (i) megakaryocytes (n=6) and (ii) monocytes (n=6) as well as (iii) in PMF patients-derived CD34+ cells (n=5) were measured by qRT-PCR 24 h post-nucleofection. Expression levels are reported as mean RQ±s.e.m. respect to the NegCTRsiRNA sample, set as calibrator. *P<0.05; **P<0.01; ***P<0.001 versus NegCTRsiRNA. (c) Expression levels of proinflammatory cytokines IL-8, IL-6, TNF-α and IL-1β in HDs monocytes are measured by qRT-PCR after 24 h of culture in the presence or absence of rhSPP1 or rhLGALS3 and are reported as RQ (mean±s.e.m.; n=6) respect to the untreated control samples, set as calibrator. *P<0.05 versus untreated control. IL-1β, interleukin-1β; IL-6, interleukin-6; IL-8, interleukin-8; n, number of experiments; rh, recombinant human; RQ, relative quantity; s.e.m., standard error of the mean; TNF-α, tumor necrosis factor-alpha.
Overall, these data indicated that MAF is able to directly transactivate the expression of SPP1, but not of LGALS3.
Next, we wondered which cell types could be responsible for SPP1 and LGALS3 secretion. Our data strongly suggested that monocytes and megakaryocytes mainly account for SPP1 and LGALS3 production (Supplementary Results and Supplementary Figures S7A and B). Indeed, SPP1 and LGALS3 expression levels were downregulated upon MAF knockdown in monocytes, whereas only SPP1 was significantly downregulated in megakaryocytes (Figures 4bi and ii). Noteworthy, SPP1 expression was remarkably reduced upon MAF knockdown in PMF CD34+ cells (Figure 4biii).
Overall, these data reveal that MAF is able to transactivate SPP1 expression in monocytes and megakaryocytes. Of note, the downregulation of SPP1 was also observed in PMF-derived HPCs and monocytes after ruxolitinib treatment (Supplementary Results and Supplementary Figures S7C and D).
Effects of recombinant human SPP1 and LGALS3 on inflammation
To investigate the role of MAF-induced SPP1 and LGALS3 in inflammation, the expression of proinflammatory mediators (namely, IL-8, IL-6, IL-1β and TNF-α) in HDs-derived monocytes treated with recombinant human (rh)SPP1 or rhLGALS3 was analyzed. qRT-PCR data showed that both SPP1 and LGALS3 enhanced the expression of proinflammatory mediators in monocytes (Figure 4c).
Effects of rhSPP1 and rhLGALS3 on human fibroblasts and MSCs
Owing to the key role of BM fibrosis in PMF pathogenesis, we next moved to unravel the effects of SPP1 and LGALS3, whose increased plasma levels in PMF patients had never been reported before, on fibroblasts proliferation and collagen production.
First, we treated normal human dermal fibroblasts with either rhSPP1 or rhLGALS3. A sample treated with rhTGF-β1 was included as positive control.
The treatment with rhSPP1 or rhLGALS3 in culture promoted fibroblasts growth, as demonstrated by the increased cell counts (P<0.05; Figure 5a) and cell density (Supplementary Figure S8A) in rhSPP1- and rhLGALS3-treated fibroblasts at 24 and 48 h of culture. In addition, the increase in cell count detected upon treatment with rhSPP1 or rhLGALS3 was reversed by the simultaneous treatment with SPP1 (Figure 5bi) or LGALS3 (Figure 5bii) neutralizing antibody, respectively.

Effects of SPP1 and LGALS3 on normal human fibroblasts. Effects of rhSPP1 and rhLGALS3 on proliferation of NHDFs. NHDFs were cultured in the absence or presence of 500 ng/ml rhSPP1, 1000 ng/ml rhLGAL3 or 5 ng/ml rhTGF-β1 (as positive control) for 24 and 48 h. (a) Evaluation of cell growth by Trypan Blue exclusion assay after 24 and 48 h of treatment. Results are shown as number of cells, starting from 5000 cells per sample, as reported in ‘Materials and Methods’ section. Values are reported as mean±standard error of the mean (s.e.m.); n=6. *P<0.05 versus untreated control. (b) Effect of an anti-SPP1 (i) and anti-LGALS3 (ii) neutralizing antibody on SPP1-induced NHDFs cell growth. NHDFs were treated with rhSPP1 (i) or rhLGALS3 (ii) in the absence or presence of anti-SPP1 neutralizing antibody (2.5 μg/ml, bi) or anti-LGALS3 neutralizing antibody (10 μg/ml, bii) or an isotype-matched antibody for 24 h. Cell growth was evaluated by Trypan Blue exclusion assay. For each sample the fold of increase in cell counts after 24 h of culture is shown as normalized to the number of cells plated (t0). Values are reported as mean±s.e.m. (n=6). (c) Expression levels of COL1A1, an extracellular matrix-related gene involved in fibrosis, measured by qRT-PCR after 24 h of culture in the presence or absence of rhSPP1, rhLGALS3 or rhTGF-β1 and are reported as RQ (mean±s.e.m.; n=3) respect to the untreated control samples, set as calibrator. COL1A1, collagen type 1 alpha 1; NHDFs, normal human dermal fibroblasts; n=number of experiments; rh, recombinant human.
At last, we tried to elucidate whether rhSPP1 and rhLGALS3 could stimulate the fibroblasts to express collagen type III (COL3A1) and collagen type I (COL1A1), the main components of reticulin and collagen fibers, respectively. qRT-PCR data showed that treatment with rhSPP1 and rhLGALS3 induced the expression of COL1A1 (Figure 5c), but not of COL3A1 (data not shown).
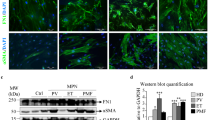
Given the role of MSCs in BM remodeling, we also investigated the effects of rhSPP1 and rhLGALS3 on HD (Supplementary Figures S8B–D) and PMF BM MSCs (Figure 6). The treatment with rhSPP1 and rhLGALS3 increased both HD (Supplementary Figure S8B) and PMF MSCs proliferation (Figure 6a). This effect was reverted by treatment with SPP1 (Supplementary Figure S8Ci and Figure 6bi) or LGALS3 neutralizing antibodies (Supplementary Figure S8Cii and Figure 6bii).

Effects of SPP1 and LGALS3 on PMF mesenchymal stromal cells. PMF-derived MSCs were cultured in the absence or presence of 500 ng/ml rhSPP1, 1000 ng/ml rhLGAL3 or 5 ng/ml rhTGF-β1 (used as positive control) for 48 h. (a) Cell proliferation was monitored by Trypan Blue exclusion assay after 48 h of treatment. Results are shown as number of cells, starting from 1.6 × 104 cells per sample. Values are reported as mean±standard error of the mean (s.e.m.). The results come from three independent experiments. *P<0.05; **P<0.01 versus untreated control. (b) Effect of an anti-SPP1 (i) and anti-LGALS3 (ii) neutralizing antibody on MSCs cell proliferation. PMF MSCs were treated with rhSPP1 (i) or rhLGALS3 (ii) in the absence or presence of anti-SPP1 neutralizing antibody (2.5 μg/ml, bi) or anti-LGALS3 neutralizing antibody (10 μg/ml, bii) or an isotype-matched antibody for 48 h. Cell proliferation was evaluated by Trypan Blue exclusion assay. For each sample the fold of increase in cell counts after 48 h of culture was normalized to the number of cells plated at t0. Values are reported as mean±s.e.m. (n=5). *P<0.05. (c) Expression levels of the fibrotic marker COL1A1 in PMF MSCs were measured by qRT-PCR after 48 h of culture with rhSPP1, rhLGALS3 or rhTGF-β1 and are reported as RQ (mean±s.e.m.; n=5) respect to the untreated control sample, set as calibrator. *P<0.05; **P<0.01. COL1A1, collagen type 1 alpha 1; MSCs, mesenchymal stromal cells; rh, recombinant human; n, number of experiments.
In addition, rhSPP1 and rhLGALS3 induced COL1A1 expression in both HDs (Supplementary Figure S8D) and PMF MSCs (Figure 6c), whereas no modulation of COL3A1 mRNA level was observed (data not shown).
Collectively, these results suggest that SPP1 and LGALS3 could promote fibroblast and MSC growth and collagen I production and therefore could be involved in BM fibrosis in PMF patients.
Noteworthy, we also unveiled that the production of SPP1 was higher in PMF versus HD MSCs. Strikingly, the treatment with PMF monocytes-conditioned medium strongly increased the SPP1 protein release by PMF MSCs (Supplementary Results and Supplementary Figures S8E and F).
Correlations between MAF, SPP1 and LGALS3 expression and clinical features of PMF patients
As our data collectively supported the idea of a profibrotic role for MAF/SPP1 axis, we further analyzed the expression levels of MAF in CD34+ cells from PMF patients according to their BM fibrosis grade. Strikingly, we uncovered that MAF expression levels in CD34+ cells were significantly higher in overt fibrotic (MF-2/MF-3) compared with prefibrotic (MF-0/MF-1) PMF (Figure 7a).

Clinical features of PMF patients according to MAF expression levels in CD34+ cells and SPP1 plasma levels. (a) MAF expression levels in CD34+ cells isolated from PMF patients at diagnosis according to the BM fibrosis stage they presented. Patients were divided in prefibrotic (MF-0/1, n=7) and overt fibrotic (MF-2, n=9 and MF-3, n=14) based on the BM fibrosis grading. Gene expression levels were measured by microarray analysis performed by Affymetrix platform as described in Norfo et al.8 MAF expression levels are reported as Robust Multiarray Analysis (RMA)-normalized log2 signals, which were obtained by using the Partek GS software. Boxes represent the interquartile range that contains 50% of the subjects, the horizontal line in the box marks the median, and bars show the range of values. The comparison between MF-0/1 and MF-2/3 groups was performed by using the Mann–Whitney U test. (b) Protein levels of SPP1 in the plasma of PMF patients (n=115) according to BM fibrosis grade. PMF patients were classified based on grade of fibrosis in prefibrotic (MF-0/1, n=54) and overt fibrotic (MF-2, n=43 and MF-3, n=18). Protein levels were measured by ELISA and are expressed as ng/ml. Plasma levels of SPP1 are shown as dot plots. Boxes represent the interquartile range that contains 50% of the subjects, the horizontal line in the box marks the median, and the bars show the range of values. Comparisons between two groups were performed by using the Mann–Whitney U test. (c) Kaplan–Meier estimates of overall survival in 115 patients with PMF, stratified by the plasma levels of SPP1. Patients with SPP1 plasma levels greater than the median value observed in the plasma of HDs were considered patients with high SPP1 levels. BM, bone marrow; ELISA, enzyme-linked immunosorbent assay; HDs, healthy donors; MF, myelofibrosis; PMF, primary myelofibrosis; n, number of samples.
We next unraveled the correlations between SPP1 and LGALS3 plasma levels and clinical features of PMF patients. We first compared hematological, clinical and molecular characteristics of PMF patients according to the SPP1/LGALS3 expression levels in the plasma in comparison with HDs. SPP1 and LGALS3 plasma levels were significantly greater in PMF patients than in HDs (median=70.3 ng/ml, range=10.3–392.4 ng/ml in PMF patients versus median=42.2, range=4.4–87.4 ng/ml in HDs for SPP1, P<0.00001 and median=8.8 ng/ml, range=1.6–36.2 ng/ml in PMF patients versus median 5.4 ng/ml, range=1.5–12.8 ng/ml in HDs for LGALS3, P<0.00001).
On the basis of SPP1 and LGALS3 plasma levels in HDs, we defined high SPP1 (n=88 patients), low SPP1 (n=27), high LGALS3 (n=96) and low LGALS3 (n=19) levels those patients with SPP1/LGALS3 levels higher or lower, respectively, than the median value in HDs (42.2 ng/ml for SPP1 and 5.4 ng/ml for LGALS3).
Patients with higher SPP1 plasma levels suffered more frequently of constitutional symptoms (94.9%) compared with patients with normal SPP1 levels (67.1%; P=0.001).
Further clinical correlations results are available in the Supplementary Results section. The levels of SPP1 were progressively higher depending on fibrosis grade, increasing from fibrosis grade 0–1 (MF-0/1) (median=51.8 ng/ml, range=12.1–220.5 ng/ml, n=54) to grade 2 (MF-2) (median=96.0 ng/ml, range=10.3–392.4 ng/ml, n=43) and grade 3 (MF-3) (median=103.5 ng/ml, range=18.6–365.9 ng/ml, n=18) (P=0.0016 in MF-2 versus MF-0/1 and P=0.0021 in MF-3 versus MF-0/1, Figure 7b). Furthermore, higher plasma levels of SPP1 were more frequently detected in the high molecular risk (HMR) category (median=113.8 ng/ml, range=22.2–392.4 ng/ml) in comparison with the low molecular risk group (median 57.2=ng/ml, range=10.3–364.7 ng/ml) (P=0.014); no difference was observed for LGALS3 levels according to HMR status. We then evaluated the impact of SSP1 and LGAL3 plasma level on OS. In univariate analysis, patients with higher SPP1 plasma levels displayed significantly shorter OS compared to those with SSP1 level in the normal range (HR: 31, 95% CI: 1.0–183.7, P=0.006; Figure 7c) together with the IPSS variables evaluated at the time of plasma collection (P<0.001). In multivariable Cox proportional hazard regression analysis, higher SPP1 plasma levels remained significant for survival (HR: 12.7, 95% CI:1.36–119.2; P = 0.026) together with all the IPSS categories (HR: 3.7, 95% CI: 2.36–5.72; P<0.0001), therefore representing an independent prognostically adverse variable for survival in PMF.
Discussion
As we have recently unveiled the upregulation of MAF in CD34+ cells from PMF patients compared with their normal counterparts,8 in this study we further unraveled the role of MAF in PMF pathogenesis.
We uncovered that MAF is also remarkably upregulated in PMF CD34+ cells in comparison with ET and PV CD34+ cells. Notably, MAF expression levels in CD34+ cells from PMF patients are independent of the driver mutation they carry. We demonstrated that MAF promotes the HPCs commitment toward the monocyte/macrophage and megakaryocyte lineages, in line with previous reports.16, 17 Of note, both megakaryocytes and monocytes are key players in PMF pathogenesis.18, 19, 20, 21 Gene expression profile of MAF-overexpressing cells provided further insights into the role of MAF in PMF pathogenesis, as they unveiled the enhanced expression of genes related to inflammation and fibrosis.
PMF, as well as ET and PV, is associated with a chronic nonresolving inflammation.22 This long-lasting inflammatory state is fueled by the continuous release of proinflammatory and microenvironment-regulating cytokines and chemokines, which are mainly produced by megakaryocytes, monocytes and other hematopoietic cells.23 Notably, elevated levels of circulating IL-8, IL2R, IL12 and IL15 have been introduced as independent prognostic criteria for PMF risk stratification according to the Dynamic International Prognostic Scoring System (DIPSS plus).24 Similarly, increased plasma levels of proinflammatory cytokines and chemokines, such as TNFα, IL-6 and CCL4, have been detected in PMF patients and their reduction has been related to the clinical benefits (that is, the resolution of splenomegaly and constitutional symptoms) of ruxolitinib treatment.23, 25
Our study unveils that the upregulation of MAF in CD34+ cells from PMF patients is at least in part responsible for the induction of a proinflammatory state and for the increased expression of proinflammatory mediators in their progeny. Some of these molecules (for example, IL-8, CCL2, PLAUR, SPP1 and LGALS3) showed higher plasma levels in PMF patients compared to HDs. Although CCL2,26 IL-824 and PLAUR27 have been already reported to be increased in PMF patients in comparison with HDs, no data were available for SPP1 and LGALS3,28, 29 respectively. However, both these molecules have been associated with other inflammatory disorders. The upregulation of SPP1 is a hallmark of inflammatory neuropathologies such as multiple sclerosis,30 Parkinson’s and Alzheimer’s disease.31 Similarly, LGALS3 promotes inflammation by stimulating macrophage activation and migration and LGALS3 levels are associated with neutrophils accumulation in patients with chronic obstructive pulmonary disease.32
As SPP1 and LGALS3 increased plasma levels in PMF patients had never been reported before, we further unraveled their role in PMF-related inflammation by demonstrating that SPP1 and LGALS3 enhance the expression of proinflammatory mediators (namely, IL-8, IL-6, IL-1β and TNF-α) in monocytes.
No correlation between SPP1/LGALS3 plasma levels and LDH levels, neutrophil or monocyte counts was found in PMF patients, suggesting that the increased SPP1/LGALS3 plasma levels did not exclusively reflect the ongoing chronic inflammation in PMF.
Interestingly, even though several data demonstrate that MPN-associated oncogenes can induce a proinflammatory response by both malignant and non-malignant cells, emerging evidence suggests that chronic inflammation itself may foster clonal evolution in MPNs33, 34 by increasing the risk of oxidative DNA damage and consequently promoting the emergence of novel genomic lesions. Therefore, the question of whether the MPN-associated genomic lesions boost inflammation or vice versa still remains unanswered.22 As JAK/STAT signaling is involved in both malignant hematopoiesis and inflammation, we cannot exclude that chronic inflammation in the BM rather than JAK/MPL/CALR-driven oncogenic signaling could underlie the upregulation of MAF in CD34+ cells. However, our data demonstrate that MAF contributes to enhance the proinflammatory microenvironment that is a major feature of PMF and, to a lesser extent, of the other MPNs.
Concerning the development of BM fibrosis and stromal changes characterizing PMF,18, 19, 20, 21 the most acknowledged theory is that they result from an overproduction of growth factors by malignant hematopoietic cells, mainly monocytes and megakaryocytes. The proinflammatory/profibrotic cytokines and growth factors released (for example, TGFbeta, PDGF, bFGF and VEGF) in turn act on stromal cells leading to the development of BM fibrosis, osteosclerosis and other pathological features of PMF.18, 19, 20, 21
Here we demonstrate that the upregulation of MAF in malignant HPCs from PMF patients contributes to enhance the expression of several profibrotic mediators, such as CCL2,35 IL-8,36 MMP9,37 LGALS3 and SPP1 in their progeny. Indeed, all the latter molecules show significantly increased plasma levels in PMF patients in comparison with HDs. We showed that SPP1 is a direct transcriptional target of MAF, whereas the mechanism of LGALS3 overproduction upon MAF overexpression still remains unclear. Interestingly, both LGALS3 and SPP1 have been previously linked to the development of liver fibrosis.38, 39 In line with these data, we demonstrated that SPP1 promotes fibroblasts and MSCs proliferation and collagen production. In addition, in line with the increasing evidences suggesting that stromal cells may foster key pathophysiological features of PMF,3 we showed that the production of SPP1 is significantly higher in PMF MSCs compared with their normal counterparts and, interestingly, their crosstalk with PMF monocytes strongly enhances the SPP1 production by MSCs.
Our study showed that SPP1 plasma levels in PMF patients have a prognostic value as they were able to distinguish prefibrotic (MF-0/MF-1) and overt fibrotic (MF-2/MF-3) PMF patients.40, 41 Indeed, higher SPP1 plasma levels correlate with a more severe fibrosis degree in PMF patients. Strikingly, we also uncovered a significant correlation between the higher SPP1 plasma levels and a shorter OS in PMF patients.
As a whole, in this study we demonstrate that the upregulation of MAF in malignant CD34+ cells is a key mechanism for the induction of a proinflammatory and profibrotic phenotype in their differentiated progeny (Supplementary Figure S9). Both JAK2, MPL and CALR mutations converge toward the STAT5 activation,4, 42, 43 and, consequently, the STAT5-driven transactivation of MAF expression.44 Indeed, JAK inhibition by ruxolitinib treatment reduced MAF expression in PMF HPCs and monocytes.
Nonetheless, the upregulation of MAF is also detected in triple-negative PMF patients; moreover, JAK2-mutated PMF patients with different allele burden- and therefore different STAT5 activation- display superimposable MAF expression levels. In addition, MAF expression is further enhanced in PMF versus ET and PV CD34+ cells, in spite of the same driver mutations. Therefore, we speculate that other molecular mechanisms should be involved in the control of MAF expression in PMF.
In the present study, we showed for the first time an increased expression of LGALS3 and SPP1 in MPN patients’ plasma compared to HD; however, while LGALS3 could be considered a putative serum marker of MPN, SPP1 increased expression is more remarkable in PMF patients, in which it is related to a greater degree of BM fibrosis and shorter OS.
As a whole, in the present study we demonstrated that MAF–SPP1 axis is involved in the development of BM fibrosis with potential prognostic value in PMF patients.
References
Barbui T, Thiele J, Gisslinger H, Finazzi G, Vannucchi AM, Tefferi A . The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev 2016; 30: 453–459.
Desterke C, Martinaud C, Ruzehaji N, Le Bousse-Kerdiles MC . Inflammation as a keystone of bone marrow stroma alterations in primary myelofibrosis. Mediat Inflamm 2015; 2015: 415024.
Le Bousse-Kerdiles MC . Primary myelofibrosis and the “bad seeds in bad soil” concept. Fibrogenesis Tissue Repair 2012; 5: S20.
Bianchi E, Norfo R, Pennucci V, Zini R, Manfredini R . Genomic landscape of megakaryopoiesis and platelet function defects. Blood 2016; 127: 1249–1259.
Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 2015; 372: 601–612.
Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014; 123: 2220–2228.
Tenedini E, Bernardis I, Artusi V, Artuso L, Roncaglia E, Guglielmelli P et al. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. Leukemia 2014; 28: 1052–1059.
Norfo R, Zini R, Pennucci V, Bianchi E, Salati S, Guglielmelli P et al. miRNA-mRNA integrative analysis in primary myelofibrosis CD34+ cells: role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood 2014; 124: e21–e32.
Blank V . Small Maf proteins in mammalian gene control: mere dimerization partners or dynamic transcriptional regulators? J Mol Biol 2008; 376: 913–925.
Eychene A, Rocques N, Pouponnot C . A new MAFia in cancer. Nat Rev Cancer 2008; 8: 683–693.
Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004; 5: 191–199.
Oikonomou N, Mouratis MA, Tzouvelekis A, Kaffe E, Valavanis C, Vilaras G et al. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol 47: 566–574.
Schwettmann L, Wehmeier M, Jokovic D, Aleksandrova K, Brand K, Manns MP et al. Hepatic expression of A disintegrin and metalloproteinase (ADAM) and ADAMs with thrombospondin motives (ADAM-TS) enzymes in patients with chronic liver diseases. J Hepatol 2008; 49: 243–250.
Kolattukudy PE, Niu J . Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ Res 2012; 110: 174–189.
Menten P, Wuyts A, Van Damme J . Macrophage inflammatory protein-1. Cytokine Growth Factor Rev 2002; 13: 455–481.
Bianchi E, Bulgarelli J, Ruberti S, Rontauroli S, Sacchi G, Norfo R et al. MYB controls erythroid versus megakaryocyte lineage fate decision through the miR-486-3p-mediated downregulation of MAF. Cell Death Differ 2015; 22: 1906–1921.
Hegde SP, Zhao J, Ashmun RA, Shapiro LH . c-Maf induces monocytic differentiation and apoptosis in bipotent myeloid progenitors. Blood 1999; 94: 1578–1589.
Wen QJ, Yang Q, Goldenson B, Malinge S, Lasho T, Schneider RK et al. Targeting megakaryocytic-induced fibrosis in myeloproliferative neoplasms by AURKA inhibition. Nat Med 2015; 21: 1473–1480.
Rameshwar P, Denny TN, Stein D, Gascon P . Monocyte adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-beta. J Immunol 1994; 153: 2819–2830.
Rameshwar P, Narayanan R, Qian J, Denny TN, Colon C, Gascon P . NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J Immunol 2000; 165: 2271–2277.
Wagner-Ballon O, Pisani DF, Gastinne T, Tulliez M, Chaligne R, Lacout C et al. Proteasome inhibitor bortezomib impairs both myelofibrosis and osteosclerosis induced by high thrombopoietin levels in mice. Blood 2007; 110: 345–353.
Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ et al. Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia 2016; 30: 1018–1024.
Hasselbalch HC . The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev 2013; 24: 133–145.
Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A . Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol 2011; 29: 1356–1363.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 363: 1117–1127.
Pardanani A, Begna K, Finke C, Lasho T, Tefferi A . Circulating levels of MCP-1, sIL-2R, IL-15, and IL-8 predict anemia response to pomalidomide therapy in myelofibrosis. Am J Hematol 2011; 86: 343–345.
Jensen MK, Riisbro R, de Nully Brown P, Brunner N, Hasselbalch HC . Elevated soluble urokinase plasminogen activator receptor in plasma from patients with idiopathic myelofibrosis or polycythaemia vera. Eur J Haematol 2002; 69: 43–49.
Koopmans SM, Bot FJ, Schouten HC, Janssen J, van Marion AM . The involvement of Galectins in the modulation of the JAK/STAT pathway in myeloproliferative neoplasia. Am J Blood Res 2012; 2: 119–127.
Moura LG, Tognon R, Nunes NS, Rodrigues LC, Ferreira AF, Kashima S et al. Different expression patterns of LGALS1 and LGALS3 in polycythemia vera, essential thrombocythemia and primary myelofibrosis. J Clin Pathol 2016; 69: 926–929.
Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 2001; 294: 1731–1735.
Brown A . Osteopontin: a key link between immunity, inflammation and the central nervous system. Transl Neurosci 2012; 3: 288–293.
Li LC, Li J, Gao J . Functions of galectin-3 and its role in fibrotic diseases. J Pharmacol Exp Ther 2014; 351: 336–343.
Hasselbalch HC . Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res 2013; 37: 214–220.
Hasselbalch HC . Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012; 119: 3219–3225.
Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One 2016; 11: e0158156.
Zimmermann HW, Seidler S, Gassler N, Nattermann J, Luedde T, Trautwein C et al. Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS One 2011; 6: e21381.
Tan TK, Zheng G, Hsu TT, Lee SR, Zhang J, Zhao Y et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab Invest 2013; 93: 434–449.
Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, Lu Y et al. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 2016.;66: 1123–1137.
Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci USA 2006; 103: 5060–5065.
Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A . European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005; 90: 1128–1132.
Guglielmelli P, Pacilli A, Rotunno G, Rumi E, Rosti V, Delaini F et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017; 129: 3227–3236.
Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016; 127: 1325–1335.
Marty C, Pecquet C, Nivarthi H, El-Khoury M, Chachoua I, Tulliez M et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 2016; 127: 1317–1324.
Rani A, Afzali B, Kelly A, Tewolde-Berhan L, Hackett M, Kanhere AS et al. IL-2 regulates expression of C-MAF in human CD4 T cells. J Immunol 2011; 187: 3721–3729.
Acknowledgements
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC), Project Number #10005 ‘Special Program Molecular Clinical Oncology 5 × 1000’ to AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative, http://www.progettoagimm.it); AIRC Project Number #15337. Italian Ministry of Health (Progetti di ricerca giovani ricercatori, Ricerca Finalizzata 2011–2012) Project Number #GR-2011-02352109).
Author contributions
AMV and RM designed the study; PG enrolled patients and analyzed clinical correlations; GCF, CM, TF, NB, BM, LE, SiSa, contributed patients cell samples and performed molecular characterization; VR, FP, AR enrolled patients; MAA performed MSC experiments; SeRo and SS performed microarray and WB experiments; CR and ZP performed real time-PCR assays; SaRu and EB performed ELISA assays; GB and LT performed luciferase reporter assays; RZ, RN, VP and ET performed data analysis; SaRu and EB performed gene overexpression experiments; EB, SF, AMV and RM wrote the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ruberti, S., Bianchi, E., Guglielmelli, P. et al. Involvement of MAF/SPP1 axis in the development of bone marrow fibrosis in PMF patients. Leukemia 32, 438–449 (2018). https://doi.org/10.1038/leu.2017.220
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.220
This article is cited by
-
Inhibition of ERK1/2 signaling prevents bone marrow fibrosis by reducing osteopontin plasma levels in a myelofibrosis mouse model
Leukemia (2023)
-
Macrophage frequency in the bone marrow correlates with morphologic subtype of myeloproliferative neoplasm
Annals of Hematology (2021)
-
Identification of key genes in atrial fibrillation using bioinformatics analysis
BMC Cardiovascular Disorders (2020)
-
Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants
Scientific Reports (2019)
-
SPP1 inhibition improves the cisplatin chemo-sensitivity of cervical cancer cell lines
Cancer Chemotherapy and Pharmacology (2019)
