
Abstract
Despite the development of novel drugs, alkylating agents remain an important component of therapy in multiple myeloma (MM). DNA repair processes contribute towards sensitivity to alkylating agents and therefore we here evaluate the role of nucleotide excision repair (NER), which is involved in the removal of bulky adducts and DNA crosslinks in MM. We first evaluated NER activity using a novel functional assay and observed a heterogeneous NER efficiency in MM cell lines and patient samples. Using next-generation sequencing data, we identified that expression of the canonical NER gene, excision repair cross-complementation group 3 (ERCC3), significantly impacted the outcome in newly diagnosed MM patients treated with alkylating agents. Next, using small RNA interference, stable knockdown and overexpression, and small-molecule inhibitors targeting xeroderma pigmentosum complementation group B (XPB), the DNA helicase encoded by ERCC3, we demonstrate that NER inhibition significantly increases sensitivity and overcomes resistance to alkylating agents in MM. Moreover, inhibiting XPB leads to the dual inhibition of NER and transcription and is particularly efficient in myeloma cells. Altogether, we show that NER impacts alkylating agents sensitivity in myeloma cells and identify ERCC3 as a potential therapeutic target in MM.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) is a plasma cell malignancy with different cytogenetic subgroups and variable patient outcomes.1 Despite recent development of new drugs, including proteasome inhibitors, Imids and monoclonal antibodies, therapeutic strategies using DNA-damaging agents including melphalan, either as a consolidation regimen by autotransplantation or in combination with other drugs, remain an important component of therapy.2 Efficacy of these agents may be affected by DNA repair mechanisms, which are often dysregulated in cancer.3
Melphalan is a bifunctional alkylating agent that generates bulky monoadducts and interstrand crosslinks requiring complex and coordinated biological responses such as base excision repair, nucleotide excision repair (NER), Fanconi anemia pathway and homologous recombination (HR).4, 5, 6, 7, 8, 9 Melphalan resistance has been linked with upregulation of membrane efflux proteins, increased binding to glutathione, Fanconi anemia pathway, increased intercrosslink repair and transcriptional activity.7, 8, 10, 11, 12
The NER pathway recognizes and removes a wide range of DNA damages induced by ultraviolet (UV), tobacco, alkylating agents or DNA crosslinks. Depending on the location of the DNA damage two distinct NER pathways intervene. Global genome repair-NER recognizes and removes damages through the whole genome, whereas transcription-coupled repair-NER selectively acts from the transcribed strand of active genes. After DNA-damage recognition step, global genome repair or transcription-coupled repair converge onto the same path to perform excision of the damaged fragment and synthesis of a new DNA strand portion.13, 14
Abnormalities in NER-related genes have been described in a number of malignancies with potential impact on clinical outcome. Recurrent mutations affecting NER have been recently reported in urothelial, ovarian and breast cancer that confers higher chemosensitivity to cisplatin.15, 16, 17
Although alkylating agents remain an important component of myeloma therapy, despite the availability of novel agents, studies regarding the influence of NER on MM have not been systematically investigated. Here, we have focused on this pathway in MM and report that NER is active in MM and demonstrate that its inhibition leads to increased sensitivity to alkylating agents. We also identified that ERCC3, a major NER gene, impacts the outcome of MM patients and can be specifically targeted.
Materials and methods
Cell culture and reagents
Human MM cell lines (MMCLs) MM.1S, MM.1R, KMS11, RPMI8226, DOX40, LR5, NCIH929, IM-9, OPM2, U266, JJN3, MOLP8, KMS34, LP1, MR20, KMS18, KMS26, KMS12BM and KMS12PE, and the human stromal cell line HS5 were cultured in RPMI-1640 medium supplemented with complete medium (10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM l-glutamine) at 37 °C and 5% CO2. BJ1 cell line was cultured in supplemented DMEM medium. Xeroderma pigmentosum complementation group B (XPB) cell line corresponds to B-lymphocyte cell line featured by two xeroderma pigmentosum group B mutations obtained from the Corriel Biorepository (referenced as GM21148). Mutations are a substitution of C>T at nucleotide 1273 in exon 8 of the ERCC3 gene (c.1273C>T) [Arg425Ter (R425X)] and a T>C change at nucleotide 296 in exon 3 (c.296T>C) [Phe99Ser (F99S)]. KMS11-TKO cells that corresponds to the KMS11 cell line featured by a knock-out of MMSETII were kindly provided by Josh Lauring from the John Hopkins University, Baltimore.18
Reagents
Melphalan, bendamustine and PYR41 were purchased from Sigma (St Louis, MO, USA), spironolactone and triptolide from Selleck Chemicals LLC (Houston, TX, USA) and 4-hydroperoxycyclophosphamide from Santa Cruz Biotechnology (Dallas, TX, USA). Melflufen was obtained from Oncopeptides AB (Stockholm, Sweden).
NER repair assay
We measured NER using the DDB2 proteo-probe as described previously.19 Briefly, MMCL cells and samples of primary myeloma cells were grown on polylysine-coated glass coverslips, or wall-less 24-well glass slides (Electron Microscopy Sciences, Hatfield, PA, USA). When using multiwell glass slides, cells were grown in 15 μl drops per well and evaporation was prevented by covering the slide with a sealing fluid (Curiox Biosystems Inc., San Carlos, CA, USA). After overnight incubation, cells were irradiated with 15–20 J/m2 UV-C at 254 nm using a StrataLinker 2400 (Stratagene, Agilent Technologies, Santa Clara, CA, USA). The cells were fixed intact or 5 and 120 min after irradiation. The proteo-probe treatment was performed as described by Dreze et al.19 To obtain hybridization of the DDB2 proteo-probe to the damaged DNA, we applied the proteo-probe diluted in phosphate-buffered saline-bovine serum albumin to the fixed cells for 60 min at 37 °C. After washing in phosphate-buffered saline, we labeled the hybridized proteo-probe for 1 h at 37 °C with 5 mg/ml anti-HA antibody (Cell Signaling Technology Inc., Danvers, MA, USA) diluted in phosphate-buffered saline-bovine serum albumin. The proteo-probe–HA antibody complex was labeled similarly with 6.67 mg/ml goat anti-mouse Alexa Fluor488 (Life Technologies, Carlsbad, CA, USA). After washing, the coverslips or slides were mounted in fluorogel medium containing 4′,6-diamidino-2-phenylindole (DAPI; Electron Micoscopy Science). Proteo-probe and DAPI staining were imaged using a × 20/0.45 plan-APOCHROMAT or a × 63/1.4 oil plan-APOCHROMAT objective on an upright fluorescent microscope (Imager M2, Zeiss, Germany) coupled with an AxioCam MRM camera (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA). The platform was controlled by the Axiovision 4.8 Software (Zeiss, Carl Zeiss Microscopy, LLC). We processed images using the CellProfiler Software (Broad Institute, Cambridge, MA, USA, www.cellprofiler.org).20 DAPI staining was used to define the area of the nuclei, and the fluorescence signal intensity of the DDB2 proteo-probe was quantified for each nucleus. A minimum of 100 nuclei per condition was analyzed in each experiment. Data were analyzed with GraphPad Prism 6 (GraphPad Software Inc., Jolla, CA, USA). In addition, we used anti (6-4) photoproducts antibody (Cosmo Bio USA, Inc., Carlsbad, CA, USA) to confirm our findings as a supplemental method in transient knockdown and five primary samples. Each patient sample was evaluated using a MMCL as a positive control.
Cell viability assay
Cell viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) in 96-well tissue culture plates after 24 h exposure to drugs. Cell lines were plated at 15 000 cells per well in triplicates for each dose of melphalan, melflufen, bendamustine, 4-hydroperoxycyclophosphamide or triptolide. The GraphPad Prism 6 Software was used to plot curve fits, to perform statistical analyses and to establish half-maximal inhibitory concentration (IC50) values. For combination studies, spironolactone (10 μM) or triptolide (10 nM) was added 15 to 30 min before the alkylating agent.
Cell proliferation and apoptosis assay
MM cell proliferation was measured using [3H]thymidine (Perkin-Elmer, Boston, MA, USA) incorporation assay as described previously. Apoptosis was evaluated by flow cytometric analysis following Annexin-V staining.
Western blotting
Soluble cell lysates, chromatin fractions or immunoprecipitated protein samples were heated at 70 °C for 10 min with NuPAGE sample loading buffer. Proteins separated by electrophoresis on a NuPAGE Bis-Tris 4–12% gradient gel (Novex) were transferred to a 0.45 μm pore nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). For immunoblotting, we used the following primary antibodies: rabbit anti-XPB (1:1000; Santa Cruz Biotechnology), anti-xeroderma pigmentosum complementation group C (XPC; 1/1000; Cell Signaling Technology Inc.) and anti-GAPDH (1:1000; Cell Signaling Technology Inc.). Goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase were used at 1:5000 (Cell Signaling Technology Inc.). All antibodies were diluted in phosphate-buffered saline-0.05% Tween. Visualization was performed with a LAS-4000 Luminescent Image Analyzer using SuperSignal West Pico Reagent (Thermo Fisher Scientific Waltham, MA, USA).
NER gene expression profile and OS in MM
We established a list of 70 genes involved in NER based on the KEGG, Reactome, mSigdb database and from the literature (Supplementary Table 1). We have performed differential gene expression analysis for each NER gene between normal plasma cells and newly diagnosed MM samples, and we also analyzed copy-number abnormalities by using samples from the IFM-DFCI 2009 trial. Data were generated by RNA-sequencing and CytoScan HD Array (Affymetrix, Santa Clara, CA, USA). Methods for RNA-sequencing were described elsewhere.21 We used RNA-sequencing data for MMCLs from Dr Jonathan Keats’ laboratory (http://www.keatslab.org/).
We analyzed NER gene expression profiles and correlate it with overall survival (OS) using Cox regression and log-rank test, in the IFM-DFCI 2009 (RNA-sequencing data set) data set and the IFM 2005-01 data set (Affymetrix (Sacremento, CA, USA) Exon ST 1.0 Array, GSE39754). The IFM-DFCI 2009 data set includes 16 normal patients and 292 newly diagnosed MM patients treated with RVD (lenalidomide, bortezomib, dexamethasone) with or without autologous stem cell transplant. All the samples were used for gene expression and copy number profiling while only samples from patients treated with high dose melphalan and auto-transplant were considered for OS evaluation. The IFM 2005-01 data set includes 170 newly diagnosed patients treated with VAD (vincristine, adriamycine, dexamethasone) as induction therapy followed by autologous stem cell transplantation after high-dose melphalan.
Patient samples
MM samples and healthy peripheral blood mononuclear cells were obtained after informed consent was provided, in accordance with the Declaration of Helsinki and under the auspices of a Dana-Farber Cancer Institute Institutional Review Board approved protocol. Primary CD138+ plasma cells were purified from bone marrow aspirates using anti-CD138 microbeads as described previously (Miltenyi Biotech, Auburn, CA, USA).
Small interfering RNA interference
We used ERCC3 and XPC small interfering RNA (siRNA) (s4796, s14929; Invitrogen, Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions. Non-targeting scrambled negative control siRNA (AM4611; Invitrogen, Life Technologies) was used as a negative control. RPMI8226 and LR5 cells were transiently transfected with 2 μM of ERCC3, XPC and scrambled siRNA by electroporation using the AMAXA Technology (Lonza Cologne AG, Cologne, Germany).
Lentiviral-mediated stable gene knockdown
Hairpin-containing PLKO.1 plasmids were obtained from Sigma Mission (Sigma-Aldrich Corp. St Louis, MO, USA). Packaged viral particles were used to infect MM cells using polybrene media (final concentration 8 μg/ml). Infected MM cells were selected by puromycin (0.5 μg/ml) (Sigma) for 72 h, and then left to recover. Knockdown efficacy was determined by western blotting and cells were used for functional studies as described above.
Lentiviral-mediated gene overexpression
LentiORF clone of human ERCC3-GFP-tagged (clone ID: PLOHS_100006334, cat. no. OHS5899) and lentiORF turbo RFP control (cat. no. OHS5833) were purchased from GE Healthcare Dharmacon (Lafayette, CO, USA). MM cells were transduced in polybrene media (final concentration 8 μg/ml) for 8 h. Infected cells were selected by puromycin for 72 h and left to recover before the functional studies.
Transcription assay
RNA labeling by 5-ethynyl uridine (5-EU) incorporation was evaluated using the Click-iT RNA Imaging Kit (Invitrogen) following the manufacturer's protocol. 5-EU signal and DAPI staining were imaged using the imaging platform described above. We processed images using the CellProfiler Software.20 DAPI staining was used to define the area of the nuclei, and the fluorescence signal intensity of 5-EU was quantified for each nucleus.
HR assay
We used fluorescence-based HR assay substrate (pDRGFP; Addgene, Cambridge, MA, USA).22 Briefly, FLO-1 cells stably transfected with HR substrate (Addgene) were transfected with a plasmid expressing I-Sce I enzyme. The cells were then treated with scrambled siRNA and with SiERCC3 for 72 h, and subsequently evaluated for confocal microscopy. HR was assessed from the fluorescence intensity of each microscopic field divided by the total number of cells in the corresponding field. Average background fluorescence per cells, determined from untransfected cells, were subtracted from values of transfected cells.
Results
NER activity is heterogeneous in MM
We evaluated NER activity in 20 MM cell lines with different cytogenetic and p53 background using the novel in vitro assay developed and previously validated by us.19 We evaluated the NER efficiency by measuring the extent of unrepaired (6-4) photoproducts after UV exposure in MM cell lines. Although all cell lines eventually repaired >95% of UV-induced DNA damage over time (between 2 and 4 h after UV exposure: Supplementary Figure 1), we observed heterogeneity in the ability of myeloma cell lines to repair UV-induced DNA damage after 2 h as seen in Figure 1a and b. The difference in ability to repair was not influenced by p53 deletion or other cytogenetic characteristics, except for the t(4;14) translocation, which was consistently and significantly associated with a more rapid NER phenotype (Figure 1c). To get further insight and to explain these observed differences in NER activity, we evaluated the mutational profile and NER-related gene expression profile of 15 MMCLs included in our study. We observed 9 different missense mutations in NER genes in 8 out of 15 cell lines (Supplementary Table 1); 5 mutations were associated with a slower NER phenotype but no NER deficiency (Supplementary Figure 1). Both MM1S and MM1R cells presented mutations in the xeroderma pigmentosum complementation group A (XPA) gene (mutation D70H), and MM1R was also mutated in the ERCC6 gene (mutation L682I). No NER gene was significantly differentially expressed between t(4;14) and non t(4;14) MMCL (Supplementary Figure 3).

NER proficiency is heterogeneous in MM cell lines. (a) NER proficiency of 20 MM cell lines. We used an NER functional assay to evaluate NER proficiency. Each grey bar corresponds to a different MMCLs and represents the DDB2 proteo-probe signal per cell 120 min after exposure to UV-C. The intensity of the signal corresponding to the DNA damage was evaluated through fluorescence microscopy. Pictures were taken with the Axiovision 4.8 Software (Zeiss, Carl Zeiss Microscopy, LLC) × 63 magnification, and the images were processed using the CellProfiler Software.20 Data in this graph are normalized to the average value of the maximum intensity of DDB2 proteo-probe signal that was observed 5 min after UV. A minimum of 100 nuclei per condition was evaluated for each cell line. The results are representative of at least two independent experiments. At least 108 nuclei were analyzed in each experiment (range from 108 to 2815). p53 status refers to either homozygous deletion or homozygous mutation (*). (b) Pictures of NER functional assay. This figure shows pictures of NER evaluation in two myeloma cell lines (NCIH929 and KMS12PE). Cells were cultured on polylysine-coated coverslips overnight and then half of the cells were irradiated with UV-C. Cells were next fixed 5 and 120 min after UV exposure and without UV. The pictures illustrate NER heterogeneity, showing that NCIH929 completely repairs after 2 h, whereas KMS12PE does not. DAPI staining was used to define the area of the nuclei, and the fluorescence signal intensity of the DDB2 proteo-probe was quantified for each nucleus. (c) t(4;14) MM cell lines are featured by rapid NER phenotype. The graph represents the normalized level of UV-induced damages 120 min after exposure to UV in t(4;14) and non-t(4;14) MMCLs. t(4;14) cell lines repaired significantly faster UV-induced DNA damages. Statistical analysis was performed with a Student’s t-test.
Identification of NER genes as potential therapeutic targets
To find potential new therapeutic targets related to the NER pathway, we analyzed our RNA-sequencing and SNP array profiling data from primary myeloma samples from 292 MM patients and 16 healthy donors and identified differentially expressed NER genes and NER genes impacting OS. We observed that 34 NER-related genes are differentially expressed between MM cells and normal plasma cells and 23 genes are affected by copy-number alterations (amplification in 19 and deletion in 4) (Figure 2 and Supplementary Table 2). For OS analysis, we used two independent data sets (IFM 2005-01 and IFM-DFCI 2009). We identified in both data sets that high ERCC3 expression significantly impacts OS (Figure 2), suggesting its potential as a therapeutic target. ERCC3 expression was the only NER gene that negatively impacts OS in both data sets. In addition, in a multivariate analysis, we found that ERCC3 expression is an independent prognosis factor (Supplementary Table 3).

NER pathway is dysregulated in MM. (a) Multiple NER-related genes are dysregulated in MM. The Circos plot represents the variation of copy number and the expression level of 70 NER-related genes in a cohort of 292 newly diagnosed patients in comparison with 16 samples from normal plasma cells, based on RNA-sequencing and CytoScan Array data. Each dot corresponds to one NER gene. The outer circle represents gene expression level, green dots represent significantly higher expressed genes, purple dots to significantly lower expressed genes and gray dots to genes with no differential expression. The inner circle represents copy-number abnormalities. Red dots correspond to NER genes with amplifications, green dots are related to genes featured by deletions and gray dots relate to NER genes with no copy-number abnormalities. (b) XPC is overexpressed in context of amplification in MM. The right panel graph represents the correlation between XPC expression and XPC amplification in the same cohort. The left panel figure represents the level of expression of XPC in 292 myeloma patients (MM) in comparison with 16 normal plasma cells from healthy donors (N). MM patients significantly overexpress XPC (adj. P-value=1 × 10^5). (c) ERCC3 expression significantly impact OS in MM treated with alkylating agents. We evaluated the impact of ERCC3 expression level in MM patients in two different data sets (IFM-DFCI 2009 and IFM 2005-01). We observed that high ERCC3 expression is significantly associated with poor outcome and shorter OS in MM. The figure represents Kaplan–Meier curbs based on ERCC3 median expression level.
We also investigated public whole-exome sequencing data from a large cohort of MM patients to identify a recurrent mutation in specific NER-related genes. Although no recurrent mutation was identified, sporadic mutations in different NER genes were found raising the possibility of involvement of the common pathway (Supplementary Table 4).23, 24
NER contributes to alkylating agent sensitivity in MM cells
As NER is involved in alkylating agent-induced DNA-damage repair, we evaluated the role of NER on alkylating agent sensitivity in MM. We evaluated the sensitivity to melphalan in our panel of MM cell lines and we observed that five cell lines with the highest IC50 for melphalan were also featured by a rapid NER activity (Supplementary Figure 2).
To confirm the specific impact of NER inhibition on alkylating agents’ sensitivity, we next evaluated the impact of knockdown of two NER-related genes, ERCC3 (gene coding for XPB), which we identified as an independent prognosis marker in MM, and xeroderma pigmentosum, complementation group C (XPC), which is significantly overexpressed in myeloma in context of amplification (Figure 2 and Supplementary Table 2). As seen in Figure 3, ERCC3 and XPC knockdown inhibits NER and significantly increases sensitivity to melphalan in both cell lines RPMI8226 and LR5. Similarly, significant increase in melphalan sensitivity was observed using two shRNA-mediated knockdown targeting ERCC3 in the MM1S cell line (Figures 3e and f). Conversely, ERCC3-stable overexpression in MM1S and RPMI8226 decreased sensitivity to melphalan (Figures 3g–i).

Specific knockdown of NER-related genes increases sensitivity to melphalan in MMCLs. (a and b) ERCC3 and XPC knockdowns impair NER and increase sensitivity to melphalan. The figure represents the impact of XPC and ERCC3 knockdown on NER (left graph) and on melphalan sensitivity (right graph) in RPMI8226 and LR5 cell lines. Three independent experiments were processed 72 h after transfection with scrambled siRNA or siRNA targeting ERCC3 or XPC. The impact of knockdown on NER was evaluated with the measurement of remaining (6-4) photoproducts signal 120 min after UV-C exposure, using anti-(6-4) photoproducts antibody. Cell viability was evaluated by CellTiter-Glo (Promega Corporation, Madison, WI, USA). (c and d) Knockdown efficiency. Western blot evaluation of XPB and XPC levels 72 h after transfection with scrambled or specific siRNA. (e and f) Stable knockdown of ERCC3 increases sensitivity to melphalan. The figure represents the impact of ERCC3-stable knockdown in MM1S cell line on melphalan sensitivity. Cell viability was assessed by CellTiter-Glo 1 month after knockdown and puromycin selection. Confirmation of XPB knockdown was assessed by western blot. (g–i) Stable overexpression of ERCC3 increases resistance to melphalan. Apoptotic and cell death were assessed in MM1S and RPMI8226 cells transduced with either control (RFP-GFP-positive cells) or ERCC3-GFP particles (GFP-positive cells) by flow cytometric analysis following Annexin-V and propidium iodide (PI) staining after exposure to melphalan at several doses. The percentage of live cells corresponds to the proportion of lives cells on the total of GFP- or RFP-positive cells. Confirmation of XPB overexpression was assessed by western blot.
Small-molecule NER inhibitors increase sensitivity to melphalan
We next evaluated the feasibility of targeting NER using small molecules in myeloma cells. In particular, we evaluated two drugs that target XPB. We evaluated the effect of spironolactone, a recently described NER inhibitor,25 and triptolide26 on NER and sensitivity to alkylating agents in MM cell lines (Figure 4 and Supplementary Figure 4). We confirmed that both spironolactone and triptolide completely inhibit NER in MM cell lines. Importantly, while the two drugs are targeting XPB, a key component of NER and of the transcription initiation factor (TFIIH) complex, only triptolide inhibits transcription (Figures 4h and i). This difference is related to the distinct mechanisms of action of the two drugs. Spironolactone acts through XPB degradation mediated by the E1 ubiquitin ligase in a time-dependent manner, whereas triptolide inhibits the ATPase activity of XPB (Figures 4f and g and Supplementary Figure 4D).25, 27 Next, we treated MM cells with spironolactone or triptolide in combination with alkylating agents. We first evaluated the combination of the two drugs in the model of sensitive and resistant MM cell lines that has been established with the RPMI8226 cell lines. In these MMCLs, the combination significantly increases sensitivity (RPMI8226) and overcomes resistance (LR5) to melphalan and increases sensitivity to melflufen (Figure 4d), bendamustine and cyclophosphamide (Supplementary Figure 4). We confirmed this result in our panel of 20 MM cell lines. All cell lines were featured by a significant increased sensitivity to melphalan in combination with spironolactone with a mean decrease in IC50 of 50.4% (range 14–79%). Importantly, spironolactone does not impact melphalan sensitivity in the XPB-GM-21148 cell line, which harbors ERCC3 mutations and is NER deficient, strongly suggesting that the impact of spironolactone on alkylating agent sensitivity is mediated through NER (Table 1 and Figures 1a and 4e).

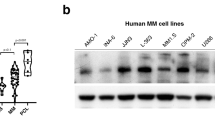
NER inhibition with spironolactone increases melphalan sensitivity in MM cell lines. (a and b) Spironolactone (sp.) and triptolide inhibit NER in myeloma cells. RPMI8226 cells were incubated with dimethyl sulfoxide (DMSO), spironolactone (10 μM) or triptolide (1 μM) for 6 h before NER evaluation. The figure represents the persistence of DNA-damage signal 150 min after exposure to UV (AFU: arbitrary fluorescent unit). Figure 4a shows representative merged pictures of DAPI and DDB2 proteo-probe signal (b). (c and d) Spironolactone (sp.) and triptolide increase sensitivity to alkylating agents. (c) The left figure represents the impact of spironolactone (10 μM) or triptolide (10 nm) on melphalan (left graph) and melflufen IC50 (right side graph) in RPMI8226 and LR5 MMCL. (d) Sensitivity to melphalan was evaluated in a panel of 20 MM cell lines in context of DMSO or spironolactone. Each dot represents the corresponding melphalan IC50 for a distinct MMCL, in combination with DMSO or spironolactone. IC50 was evaluated by CellTiter-Glo. (e) Spironolactone (sp.) increases sensitivity to melphalan through NER inhibition and ERCC3 downregulation. The figure represents the impact of spironolactone on melphalan sensitivity in GM21148-XPB cell line (green dots), which harbors two mutations in ERCC3, as compared with 20 myeloma cell lines (gray dots). Cell viability was evaluated with CellTiter-Glo. GM21148-XPB cells are not sensitized to melphalan combined with spironolactone. These results were confirmed in three independent experiments. (f and g) Spironolactone (sp.) exposure enhances time-dependant XPB degradation. This western blot shows the impact of 10 μM spironolactone on XPB expression (f) in three MMCLs (JJN3, LP1, NCIH929) after overnight exposure, and over time in the NCIH929 (g). (h and i) Triptolide inhibits transcription. After exposure to triptolide (1 μM), spironolactone (10 μM), actinomycin D (2 μM, transcription inhibitor used as a positive control) or DMSO for 6 h, the global RNA synthesis was evaluated by the measurement of 5-EU incorporation. The figure shows representative merged pictures of DAPI and 5-EU signal. The intensity of the signal was evaluated through fluorescence microscopy (h) and quantified with the CellProfiler Software. Triptolide but not spironolactone inhibits global transcription.
In addition, we evaluated the efficacy of spironolactone in the presence of stromal cells, using either the HS5 cell line or human stromal cells derived from a newly diagnosed myeloma patient. In both conditions, we observed an increased sensitivity to melphalan in the two MMCL RPMI8226 and MM1S cells, reinforcing the potential therapeutic role of NER inhibition in myeloma (Supplementary Figure 5).
NER can be monitored and targeted in MM patients
To confirm our findings in primary myeloma cells, we evaluated 12 patient MM samples (eight relapsed/refractory and four newly diagnosed patients). We found that NER was similarly active and heterogeneous (Figure 5a), confirming variability in NER proficiency in primary MM cells. We also confirmed that spironolactone inhibits NER in primary samples (Figures 5b and c). We observed a significant increased sensitivity to melphalan when used in combination with either spironolactone (n=7) or triptolide (n=4). Spironolactone increases sensitivity to melphalan in all seven primary MM samples (Figures 5d and e) but not in healthy peripheral blood mononuclear circulating cells (PBMCs). Similarly, triptolide significantly increased the sensitivity to melphalan in myeloma cells but not in healthy PBMCs. In addition, triptolide as a single agent showed a strong activity in primary myeloma cells as compared with normal PBMCs (Figure 5f), probably due to the dual inhibition of both NER and transcription.

ERCC3 is a therapeutic target in MM. (a) NER proficiency in primary MM samples. CD138-positive cells from MM patients’ bone marrow were purified with anti-CD138 microbeads, cultured overnight and exposed to UV-C. Each bar represents the normalized remaining DNA-damage percentage 2 h after UV-C exposure for each sample. A control MMCL was processed as a positive control for each experiment with primary samples. A minimum of 107 nuclei was analyzed in each experiment (range from 107 to 7390 nuclei). Similar to MM cell lines, primary samples are featured by a heterogeneous NER efficiency. Eight samples from newly diagnosed (white bars) and four samples from relapse/refractory myeloma patients (gray bars) were evaluated. Cytogenetic information was available for all except one sample. Sample MM6 is featured by the t(4;14) translocation, whereas other samples have a standard cytogenetic risk. Eight samples were evaluated with the DDB2 proteo-probe and four samples with the (6-4) photoproducts antibody. (b and c) Spironolactone inhibits NER in primary myeloma cells. Primary myeloma cells were incubated with dimethyl sulfoxide (DMSO), or spironolactone (10 μM) overnight before NER evaluation. The figure represents the persistence of DNA-damage signal 120 min after exposure to UV (AFU: arbitrary fluorescent unit). The pictures show the persistence of high DNA-damage signal in the presence of spironolactone confirming NER inhibition. (d and e) Spironolactone and triptolide increase sensitivity to alkylating agents in primary MM cells but not in healthy PBMCs. The figure represents the impact of spironolactone (10 μM) or triptolide (10 nm) on melphalan in 7 (3 newly diagnosed and 4 relapse/refractory) and 4 primary MM samples (3 newly diagnosed and 1 relapse/refractory) and in 5 and 4 healthy PBMCs, respectively. Each dot represents a distinct MM or PBMC sample. Viability was evaluated by CellTiter-Glo. The figure shows the significant increased sensitivity to melphalan (25 μM) in primary MM cells (d), whereas no significant increase was observed in PBMCs. Spironolactone itself impacts the viability of both MM cells and PBMCs but does not significantly increase melphalan sensitivity in healthy PBMCs. (Sp.=spironolactone; mel=melphalan). (f) Triptolide has significant antimyeloma activity as a single agent. Triptolide antimyeloma activity was evaluated in three PBMCs (blue line), four primary myeloma samples (red line) and three MM cell lines (green line). Cells were cultured with different doses of triptolide (0–250 nm) for 24 h and cell viability was measured with CellTiter-Glo. The figure shows that myeloma cells are more sensitive than PBMCs to triptolide.
Discussion
The advent of new therapies has provided major improvement in OS in MM. However, alkylating agent-based therapies, especially high-dose melphalan with stem cell transplant, remain an important component of MM therapeutics.28, 29 DNA damage response (DDR) pathways have an important role in modulating chemosensitivity to DNA-damaging agents in MM.30, 31, 32, 33 NER, one of the important DDR pathway, has been shown to remove monoadducts generated by melphalan,5, 34 but paradoxically, its role in melphalan sensitivity has not been evaluated in contrast with other mechanisms in MM cells.7, 8, 10, 11
Removal of adducts generated by alkylating agents requires NER. In this study, we evaluate the capacity of myeloma cells to perform NER by measuring the removal of UV-induced (6-4) photoproducts DNA lesions. We show an important role of NER in MM and have observed heterogeneity in NER completion in a panel of MMCL and a subset of primary samples (Figures 1 and 5). In this study, we observed that all t(4;14) cell lines tested (eight) share a rapid NER as compared with other MMCLs (Figure 1), suggesting that in t(4;14) cells the ability to repair may contribute to relative resistance to alkylating agents and the reported poor prognosis in these patients.35, 36 However, these findings do not seem to be specific to the presence of t(4;14). Indeed, non-t(4;14) cell lines and non-t(4;14) primary MM samples harbored a rapid NER phenotype while one primary sample harboring this translocation did not complete NER at 2 h (sample MM6 in Figure 5a). The TKO cells (featured by MMSETII KO) and the native KMS11 cell line harbor a similar NER phenotype (Figure 1a), and TKO and KO cells have no significant variation in NER gene expression profile (data not shown from GSE57863).37 Furthermore, no significant difference in NER-related gene expression between the t(4;14) and non-t(4;14) cell lines tested was observed (Supplementary Figure 3B).
These results may be explained by the important heterogeneity existing within t(4;14) MM38 and by other mechanisms influencing NER. Notably, 5 min after exposure to UV-C, the t(4;14) MM MMCL showed lower levels of (6-4) photoproducts than the non-t(4;14) MMCL (Supplementary Figure 2A). Moreover, less (6-4) photoproducts were detected in the t(4;14) MM cells fixed in methanol before irradiation. This suggests that NER phenotype is influenced by epigenomic differences affecting chromatin conformation and consequently UV-C’s accessibility to DNA (Supplementary Figure 6).
The subtle regulation of NER activity may be clinically relevant as a surrogate prognosis marker or to predict sensitivity to alkylating agents and further prospective evaluation of NER should be performed to address this question. A previous study from our group, in a small cohort of myeloma patients, reported that slower capability to repair DNA damage is associated with higher sensitivity to melphalan.39
We next evaluated the role of NER on melphalan sensitivity in MM cells. We observed that melphalan-sensitive and -resistant MMCLs have a variable NER phenotype (Table 1), and cell lines with high rate of NER tends to be resistant to melphalan, as illustrated in Supplementary Figure 2. In particular, the LR5 melphalan-resistant cell line harbored a rapid repair phenotype as compared with the parental RPMI8226 cell line, which exhibited a slower repair (Figure 1 and Supplementary Figure 2). To confirm the role of NER in resistance to alkylating agent sensitivity, we knocked down XPC and ERCC3. In both cases, we observed NER deficiency and a significant increase in sensitivity to melphalan (Figure 3). We showed that ERCC3 overexpression increases resistance to melphalan and also ruled out a potential impact of ERCC3 knockdown on HR (Supplementary Figure 7).
Identification of ERCC3/XPB expression as an independent prognosis marker in myeloma in context of autotransplant (Supplementary Table 3) led us to evaluate XPB as a therapeutic target. Spironolactone, a recently described potent inhibitor of NER,25 increased the capacity of melphalan, melflufen,40 bendamustine and cyclophosphamide in killing MM cells (Figures 4 and 5 and Supplementary Figure 4). Similar results were obtained in the presence of stroma demonstrating that NER is involved in resistance to alkylating agents and confirming that its inhibition can reverse acquired resistance in MM cells (Supplementary Figure 5). Interestingly, alkylating agents in MM are already combined with other therapies such as proteasome inhibitors. Our functional NER assay revealed that bortezomib, which is known to inhibit certain DNA-damage repair pathways41 and widely used in MM, does not affect NER in MM cells (Supplementary Figure 8). Hence, NER inhibition may be another adjunct target to be considered in combination therapies.
Triptolide, which leads to the dual inhibition of NER and transcription by targeting XPB, further points toward targeting XPB in MM. As illustrated in Figure 5f, triptolide has significant efficacy in primary myeloma samples as a single agent as well as in combination with an alkylating agent. Previous studies have reported triptolide as a potential therapeutic drug in myeloma,42, 43, 44 but its specific mechanisms of action as reported here were not investigated to our knowledge.
Altogether, our data show that the NER pathway components, in particular XPB, are potential therapeutic targets in MM.
References
Morgan GJ, Walker BA, Davies FE . The genetic architecture of multiple myeloma. Nat Rev Cancer 2012; 12: 335–348.
Ludwig H, Miguel JS, Dimopoulos MA, Palumbo A, Garcia Sanz R, Powles R et al. International Myeloma Working Group recommendations for global myeloma care. Leukemia 2014; 28: 981–992.
Curtin NJ . DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 2012; 12: 801–817.
Fu D, Calvo JA, Samson LD . Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012; 12: 104–120.
Grant DF, Bessho T, Reardon JT . Nucleotide excision repair of melphalan monoadducts. Cancer Res 1998; 58: 5196–5200.
Spanswick VJ, Lowe HL, Newton C, Bingham JP, Bagnobianchi A, Kiakos K et al. Evidence for different mechanisms of 'unhooking' for melphalan and cisplatin-induced DNA interstrand cross-links in vitro and in clinical acquired resistant tumour samples. BMC Cancer 2012; 12: 436.
Spanswick VJ, Craddock C, Sekhar M, Mahendra P, Shankaranarayana P, Hughes RG et al. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood 2002; 100: 224–229.
Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS . The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005; 106: 698–705.
Alagpulinsa DA, Yaccoby S, Ayyadevara S, Shmookler Reis RJ . A peptide nucleic acid targeting nuclear RAD51 sensitizes multiple myeloma cells to melphalan treatment. Cancer Biol Ther 2015; 16: 976–986.
Gkotzamanidou M, Sfikakis PP, Kyrtopoulos SA, Bamia C, Dimopoulos MA, Souliotis VL . Chromatin structure, transcriptional activity and DNA repair efficiency affect the outcome of chemotherapy in multiple myeloma. Br J Cancer 2014; 111: 1293–1304.
Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res 2009; 69: 9367–9375.
Tagde A, Singh H, Kang MH, Reynolds CP . The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J 2014; 4: e229.
Alekseev S, Coin F . Orchestral maneuvers at the damaged sites in nucleotide excision repair. Cell Mol Life Sci 2015; 72: 2177–2186.
Kamileri I, Karakasilioti I, Garinis GA . Nucleotide excision repair: new tricks with old bricks. Trends Genet 2012; 28: 566–573.
Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Tiao G et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 2016; 48: 600–606.
Ceccaldi R, O'Connor KW, Mouw KW, Li AY, Matulonis UA, D'Andrea AD et al. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res 2015; 75: 628–634.
Vijai J, Topka S, Villano D, Ravichandran V, Maxwell KN, Maria A et al. A recurrent ERCC3 truncating mutation confers moderate risk for breast cancer. Cancer Discov 2016; 6: 1267–1275.
Lauring J, Abukhdeir AM, Konishi H, Garay JP, Gustin JP, Wang Q et al. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 2008; 111: 856–864.
Dreze M, Calkins AS, Galicza J, Echelman DJ, Schnorenberg MR, Fell GL et al. Monitoring repair of UV-induced 6-4-photoproducts with a purified DDB2 protein complex. PLoS One 2014; 9: e85896.
Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006; 7: R100.
Rashid NU, Sperling AS, Bolli N, Wedge DC, Van Loo P, Tai YT et al. Differential and limited expression of mutant alleles in multiple myeloma. Blood 2014; 124: 3110–3117.
Pierce AJ, Johnson RD, Thompson LH, Jasin M . XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13: 2633–2638.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014; 25: 91–101.
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014; 5: 2997.
Alekseev S, Ayadi M, Brino L, Egly JM, Larsen AK, Coin F . A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol 2014; 21: 398–407.
Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol 2011; 7: 182–188.
Alekseev S, Nagy Z, Sandoz J, Weiss A, Egly JM, Le May N et al. Transcription without XPB establishes a unified helicase-independent mechanism of promoter opening in eukaryotic gene expression. Mol Cell 2017; 65: 504–514. e504.
Varga C, Laubach J, Hideshima T, Chauhan D, Anderson KC, Richardson PG . Novel targeted agents in the treatment of multiple myeloma. Hematol Oncol Clin N Am 2014; 28: 903–925.
Attal M, Lauwers-Cances V, Hulin C, Facon T, Caillot D, Escoffre M et al. Autologous transplantation for multiple myeloma in the era of new drugs: a phase III study of the Intergroupe Francophone Du Myelome (IFM/DFCI 2009 Trial). Blood 2015; 126: 391–391.
Gourzones-Dmitriev C, Kassambara A, Sahota S, Reme T, Moreaux J, Bourquard P et al. DNA repair pathways in human multiple myeloma: role in oncogenesis and potential targets for treatment. Cell Cycle 2013; 12: 2760–2773.
Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC . Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009; 113: 2290–2297.
Herrero AB, San Miguel J, Gutierrez NC . Deregulation of DNA double-strand break repair in multiple myeloma: implications for genome stability. PLoS One 2015; 10: e0121581.
Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med 2014; 20: 599–606.
Souliotis VL, Dimopoulos MA, Episkopou HG, Kyrtopoulos SA, Sfikakis PP . Preferential in vivo DNA repair of melphalan-induced damage in human genes is greatly affected by the local chromatin structure. DNA Rep 2006; 5: 972–985.
Chng WJ, Dispenzieri A, Chim CS, Fonseca R, Goldschmidt H, Lentzsch S et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014; 28: 269–277.
Shah MY, Martinez-Garcia E, Phillip JM, Chambliss AB, Popovic R, Ezponda T et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016; 35: 5905–5915.
Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Ezponda T, Shah MY et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet 2014; 10: e1004566.
Moreau P, Attal M, Garban F, Hulin C, Facon T, Marit G et al. Heterogeneity of t(4;14) in multiple myeloma. Long-term follow-up of 100 cases treated with tandem transplantation in IFM99 trials. Leukemia 2007; 21: 2020–2024.
Gkotzamanidou M, Terpos E, Bamia C, Munshi NC, Dimopoulos MA, Souliotis VL . DNA repair of myeloma plasma cells correlates with clinical outcome: the effect of the nonhomologous end-joining inhibitor SCR7. Blood 2016; 128: 1214–1225.
Chauhan D, Ray A, Viktorsson K, Spira J, Paba-Prada C, Munshi N et al. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin Cancer Res 2013; 19: 3019–3031.
Neri P, Ren L, Gratton K, Stebner E, Johnson J, Klimowicz A et al. Bortezomib-induced 'BRCAness' sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011; 118: 6368–6379.
Nakazato T, Sagawa M, Kizaki M . Triptolide induces apoptotic cell death of multiple myeloma cells via transcriptional repression of Mcl-1. Int J Oncol 2014; 44: 1131–1138.
Heimberger T, Andrulis M, Riedel S, Stuhmer T, Schraud H, Beilhack A et al. The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br J Haematol 2013; 160: 465–476.
Huang X, Yang M, Jin J . Triptolide enhances the sensitivity of multiple myeloma cells to dexamethasone via microRNAs. Leuk Lymphoma 2012; 53: 1188–1195.
Acknowledgements
This work was supported by NIH grants PO1-155258 (NCM) and P50-100707 (NCM, KCA) Department of Veterans Affairs Merit Review Award 1 I01BX001584-01 (NCM) and Leukemia and Lymphoma Society Translational Research Program Award, French Foundation for the Research on Myeloma and Gammopathies (RS) and NIH grant 4P50CA100707-14 (DF/HCC SPORE in Multiple Myeloma) (RS), and The Pallotta Investigator Fund; the Marjorie Powell Allen Memorial Fund and Barbara and Paul Ferry (JBL).
Author contributions
RS, J-BL and NCM designed the research, analyzed the data and wrote the manuscript. MKS and AC performed research and analyzed the data. ASC, MF, MS, JPF, HA-L, BA, Y-TT, ER, TP, PN, SK, ML, KW, KCA and DC analyzed the data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Szalat, R., Samur, M., Fulciniti, M. et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 32, 111–119 (2018). https://doi.org/10.1038/leu.2017.182
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.182
This article is cited by
-
Crosstalk between glucocorticoid and mineralocorticoid receptors boosts glucocorticoid-induced killing of multiple myeloma cells
Cellular and Molecular Life Sciences (2023)
-
Deciphering genes associated with diffuse large B-cell lymphoma with lymphomatous effusions: A mutational accumulation scoring approach
Biomarker Research (2021)
-
Targeting of the E3 ubiquitin-protein ligase HUWE1 impairs DNA repair capacity and tumor growth in preclinical multiple myeloma models
Scientific Reports (2020)
-
A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma
Leukemia (2020)
