
Abstract
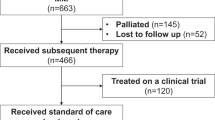
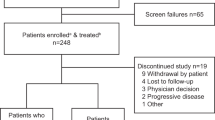
Introduction of new myeloma therapies offers new options for patients refractory to immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs). In this multicenter study, patients with relapsed multiple myeloma, who have received at least three prior lines of therapy, are refractory to both an IMiD (lenalidomide or pomalidomide) and a PI (bortezomib or carfilzomib), and have been exposed to an alkylating agent were identified. The time patients met the above criteria was defined as time zero (T0). Five hundred and forty-three patients diagnosed between 2006 and 2014 were enrolled in this study. Median age at T0 was 62 years (range 31–87); 61% were males. The median duration between diagnosis and T0 was 3.1 years. The median number of lines of therapy before T0 was 4 (range 3–13). The median overall survival (OS) from T0 for the entire cohort was 13 (95% confidence interval (CI) 11, 15) months. At least one regimen recorded after T0 in 462 (85%) patients, with a median (95% CI) progression-free survival and OS from T0 of 5 (4, 6), and 15.2 (13, 17) months, respectively. The study provides the expected outcome of relapsed multiple myeloma that is refractory to a PI and an IMiD, a benchmark for comparison of new therapies being evaluated.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014; 28: 1122–1128.
Pulte D, Jansen L, Castro FA, Emrich K, Katalinic A, Holleczek B et al. Trends in survival of multiple myeloma patients in Germany and the United States in the first decade of the 21st century. Br J Haematol 2015; 171: 189–196.
Ozaki S, Handa H, Saitoh T, Murakami H, Itagaki M, Asaoku H et al. Trends of survival in patients with multiple myeloma in Japan: a multicenter retrospective collaborative study of the Japanese Society of Myeloma. Blood Cancer J 2015; 5: e349.
Binder M, Rajkumar SV, Ketterling RP, Dispenzieri A, Lacy MQ, Gertz MA et al. Occurrence and prognostic significance of cytogenetic evolution in patients with multiple myeloma. Blood Cancer J 2016; 6: e401.
Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471: 467–472.
Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012; 120: 1067–1076.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014; 25: 91–101.
Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hajek R et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016; 17: 27–38.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015; 372: 142–152.
Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med 2016; 374: 1621–1634.
Usmani SZ, Weiss BM, Plesner T, Bahlis NJ, Belch A, Lonial S et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016; 128: 37–44.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016; 387: 1551–1560.
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015; 373: 621–631.
Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015; 373: 1207–1219.
Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 2012; 26: 149–157.
Kumar SK, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc 2004; 79: 867–874.
Rajkumar SV, Harousseau JL, Durie B, Anderson KC, Dimopoulos M, Kyle R et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 2011; 117: 4691–4695.
Kumar SK, LaPlant B, Roy V, Reeder CB, Lacy MQ, Gertz MA et al. Phase 2 trial of ixazomib in patients with relapsed multiple myeloma not refractory to bortezomib. Blood Cancer J 2015; 5: e338.
Jakubowiak A, Offidani M, Pegourie B, De La Rubia J, Garderet L, Laribi K et al. Randomized phase 2 study: elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016; 127: 2833–2840.
San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014; 15: 1195–1206.
Acknowledgements
Janssen Pharmaceuticals provided funding for the study.
Author contributions
All authors (except BGMD) provided patient data and were involved in manuscript preparation. SKK was involved in the statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
SKK: research funding (Abbvie, Celgene, Janssen, Merck, Novartis, Roche, Sanofi and Takeda) and honoraria (Skyline Diagnostics); MAD: honoraria (Celgene, Janssen, Amgen and Takeda); ET: research funding and honoraria (Amgen, Celgene and Janssen), and honoraria (Novartis, GSK, Bristol Myers Squibb and Takeda); HG: research funding, advisory board and honoraria (Janssen, Celgene, Novartis and BMS), research funding and honoraria (Chugai), and advisory boards (Amgen and Takeda); JH: consultancy, honoraria and advisory board (Amgen), honoraria and advisory board (Janssen, Celgene and Novartis), honoraria (Bristol Myers Squibb) and research funding (Sanofi); MB: speakers bureau and advisory board (Celgene, Janssen-Cilag, Amgen, Novartis, Takeda and Bristol Myers Squibb); MA: speakers bureau (Janssen); AO: advisory board and consultancy (Amgen and Janssen), and consultancy (Takeda); MVM: honoraria and advisory board (Janssen, Celgene, Takeda and Amgen); RV: research support and consultancy/honoraria (Amgen, Celgene and Takeda), and consultancy/honoraria (Bristol Myers Squibb, Janssen, Karyopham and Abbvie); H Lokhorst: research funding and advisory board (Janssen), and research funding (Genmab); NvdD: research funding (Janssen, Celgene, Amgen and BMS) and advisory board (Janssen, Celgene, Amgen, BMS and Novartis); TM: research funding (Amgen and Celgene); MH: advisory board (Celgene, Janssen, Amgen and Takeda); H Ludwig: speakers bureau (Celgene, Takeda, Amgen and Janssen-Cilag), advisory board (Celgene, Amgen, Janssen-Cilag, AbbVie and Bristol Myers Squibb) and research funding (Takeda and Amgen); MD: consultancy (Amgen, Bristol Myers Squibb, Celgene, Janssen and Takeda) and research funding (Celgene and Janssen); U-HM: advisory board (Amgen and Takeda) and honoraria (Amgen, Celgene, Takeda, Mundipharma, Janssen and Novartis); SZU: advisory board (Amgen, Celgene and Skyline Diagnostics), speakers bureau (Amgen, Celgene and Takeda) and research funding (Amgen, Celgene, Janssen, Sanofi, Pharmacyclics, Array Biopharma and Takeda); PM: advisory board (Celgene, Takeda, Janssen, Novartis and Amgen); LR: honoraria (Janssen and Celgene); all the other authors declare no conflicts of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

Cite this article
Kumar, S., Dimopoulos, M., Kastritis, E. et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia 31, 2443–2448 (2017). https://doi.org/10.1038/leu.2017.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.138
This article is cited by
-
CTPS1 is a novel therapeutic target in multiple myeloma which synergizes with inhibition of CHEK1, ATR or WEE1
Leukemia (2024)
-
A Phase I Trial Evaluating the Addition of Lenalidomide to Patients with Relapsed/Refractory Multiple Myeloma Progressing on Ruxolitinib and Methylprednisolone
Targeted Oncology (2024)
-
ML-based sequential analysis to assist selection between VMP and RD for newly diagnosed multiple myeloma
npj Precision Oncology (2023)
-
ASH highlights 2022—multiple myeloma
memo - Magazine of European Medical Oncology (2023)
-
Post-infusion Costs Associated with Idecabtagene Vicleucel Treatment for Patients with Relapsed/Refractory Multiple Myeloma in the KarMMa Trial
Advances in Therapy (2023)
