
Abstract
We identified mutations in the IL7Ra gene or in genes encoding the downstream signaling molecules JAK1, JAK3, STAT5B, N-RAS, K-RAS, NF1, AKT and PTEN in 49% of patients with pediatric T-cell acute lymphoblastic leukemia (T-ALL). Strikingly, these mutations (except RAS/NF1) were mutually exclusive, suggesting that they each cause the aberrant activation of a common downstream target. Expressing these mutant signaling molecules—but not their wild-type counterparts—rendered Ba/F3 cells independent of IL3 by activating the RAS-MEK-ERK and PI3K-AKT pathways. Interestingly, cells expressing either IL7Ra or JAK mutants are sensitive to JAK inhibitors, but respond less robustly to inhibitors of the downstream RAS-MEK-ERK and PI3K-AKT-mTOR pathways, indicating that inhibiting only one downstream pathway is not sufficient. Here, we show that inhibiting both the MEK and PI3K-AKT pathways synergistically prevents the proliferation of BaF3 cells expressing mutant IL7Ra, JAK and RAS. Furthermore, combined inhibition of MEK and PI3K/AKT was cytotoxic to samples obtained from 6 out of 11 primary T-ALL patients, including 1 patient who had no mutations in the IL7R signaling pathway. Taken together, these results suggest that the potent cytotoxic effects of inhibiting both MEK and PI3K/AKT should be investigated further as a therapeutic option using leukemia xenograft models.
Similar content being viewed by others


Introduction
In the past two decades, T-cell acute lymphoblastic leukemia (T-ALL) has been investigated extensively at the genetic level, revealing several distinct T-ALL subtypes, each of which is characterized by specific oncogenic lesions.1, 2, 3, 4, 5 Because these lesions are generally considered to be the driving oncogenic event, we call these aberrations type A mutations.5, 6 Type A mutations facilitate a differentiation arrest and are accompanied by type B mutations,6, 7 which can contribute to leukemogenesis by disrupting a plethora of cellular processes (including the cell cycle, epigenetic gene regulation and apoptosis), ultimately resulting in the ectopic activation of several signaling pathways, including the NOTCH1, JAK-STAT and PI3K-AKT pathways.5, 8, 9, 10, 11, 12, 13, 14
Activating mutations in the IL7Ra gene, which encodes the interleukin-7 receptor alpha chain, have been identified in approximately 6% of pediatric ALL patients, with a slightly higher prevalence reported in pediatric T-ALL patients (9%).15, 16 The majority of mutations in IL7Ra introduce a cysteine residue in the juxta-membrane-transmembrane domain; this cysteine residue in the mutant protein facilitates the formation of intermolecular disulfide bonds, protein homodimerization and IL7-independent signaling.15, 16 The IL7Ra gene is one of many transcriptional targets of NOTCH1; specifically, NOTCH1 binds to the distal IL7Ra enhancer region.17 Under normal conditions, signaling through the heterodimeric IL7 receptor (IL7Ra-common/γ-chain) is essential for the growth and survival of developing T cells.18, 19 IL7R activation leads to the recruitment, phosphorylation and activation of the Janus kinases JAK1 and JAK3, and to the activation of the STAT5 and PI3K-AKT pathways.20 Ectopic expression of IL7 in mice is oncogenic and results in the development of gamma-delta T-cell lymphomas, which infiltrate the skin.21, 22 In mice, the development of IL7-induced T-cell lymphomas requires STAT5;23 in contrast, in human T-cell leukemias, IL7-dependent survival and cell cycle progression require PI3K-AKT signaling.24, 25 Thus, in contrast to normal T cells, the role of IL7R-driven modulation of JAK-STAT signaling in human T-ALL remains to be dissected.
Mutations in the JAK1 gene have been found in 4–27% of primary T-ALL patients,13, 14, 26, 27 as well as in acute myeloid leukemia, pre-B-ALL and solid tumors.13, 26, 27, 28, 29 Mutant JAK1 molecules transform Ba/F3 pro-B cells and activate downstream AKT and ERK signaling.13, 14, 29 Similar to the JAK2V617F mutation in myeloid disorders,30, 31, 32, 33 mutant JAK1 molecules must interact with the IL7Ra chain to drive the ligand-independent activation of STAT molecules.34, 35 The JAK3 gene can also be mutated in T-cell leukemias, as well as in acute megakaryoblastic leukemia;14, 36, 37, 38 the majority of JAK3 mutations affect the protein’s pseudokinase domain.38 JAK3 normally binds to the common γ-chain in the IL7R39 and requires JAK1 to transform Ba/F3 cells.38 Mutations in other IL7R signaling molecules have been identified in T-ALL, including PTPN2,40 N/K-RAS,10 NF1,8 PTEN, PI3K and AKT.9, 11, 41, 42
Here, we investigated the prevalence of mutations in the IL7Ra gene and its downstream signaling molecules in a pediatric T-ALL cohort. After identifying several mutations, we examined their ability to transform Ba/F3 cells and their potential to activate downstream JAK-STAT, RAS-MEK-ERK and PI3K-AKT-mTOR pathways. To find improved treatment for T-ALL patients, we tested the cytotoxic therapeutic effects of inhibiting these pathways, and we investigated the added value of using combined inhibitor therapies. Our results show that blocking two major signaling pathways downstream of the IL7R is synergistic and may be beneficial for patients with IL7R signaling mutations.
Materials and methods
Patient samples
Written informed consent was obtained from the parents or legal guardians of each patient to use excess diagnostic material for research purposes. The study was performed in accordance with the Institutional Review Board of the Erasmus MC Rotterdam and in accordance with the Declaration of Helsinki. Leukemic cells were harvested from blood or bone marrow samples and were enriched to ⩾90% purity.
Mutation screen
We screened 146 patients for mutations in the FERM (4.1 protein, ezrin, radixin and moesin), pseudokinase and kinase domains in all four Janus kinase family members (encoded by the JAK1, JAK2, JAK3 and TYK2 genes), for STAT5BN642H, and for the mutation hotspots in the N-RAS and K-RAS genes. In this cohort, mutations in IL7Ra, NF1, PTEN and AKT had been identified previously.8, 16, 41, 42 Detailed information can be found in the Supplementary Materials and Methods.
Ba/F3 transfectants
The Gateway multi-site recombination system (Life Technologies, Carlsbad, CA, USA) was used to simultaneously clone multiple DNA fragments into our Gateway-adapted pcDNA3.1 destination vector, which contains either an SV40-driven neomycin resistance cassette or an SV40-driven puromycin resistance cassette. Ba/F3 cells were transfected by electroporation, and bulk-transfected cells were enriched to >95% purity using the CD271 (that is, the low-affinity nerve growth factor receptor or LNGFR) MicroBead kit and magnetic separation (Miltenyi Biotec, Bergisch Gladbach, Germany).
Doxycycline-inducible expression in Ba/F3 cells
We developed a doxycycline-inducible system using murine Ba/F3 cells (DSMZ, Braunschweig, Germany), a cell line that normally requires IL3 for survival and proliferation (Supplementary Figure S1). Cell line identity was confirmed by DNA fingerprinting and cells were regularly tested for mycoplasma contamination. IL3 was withdrawn from the medium at regular intervals to ensure that all selected Ba/F3 lines remained IL3-dependent. Each transfected line was exposed to doxycycline for 24 h, after which IL3-independent proliferation and activation of signaling molecules were measured. All growth curves (±s.d., n=3) and IC50 determination of the inhibitors (±s.d., n=3) have been performed in at least three independent experiments and are representative.
Cell survival assay
Patient cells were thawed and immediately cultured in duplicate in 384-well plates (10 000 cells/well) in RPMI medium containing 10% heat-inactivated fetal calf serum. Plated cells were incubated in a humidified atmosphere of 5% CO2 at 37 ºC. Inhibitors were added 2 h after plating the cells. Survival of patient cells was determined 72 h after addition of the inhibitor(s) by the intracellular ATP content as an indirect measure of the number of viable cells using ATPlite 1 Step solution (Perkin Elmer, Waltham, MA, USA). Detailed information can be found in the Supplementary Materials and Methods.
Inhibitors
The following inhibitors were obtained from Selleck Chem (Munich, Germany, unless otherwise indicated): JAK inh 1 (Merck Millipore, Billerica, MA, USA; #420099), ruxolitinib (#S1378), pimozide (Sigma-Aldrich, Zwijndrecht, The Netherlands; #P1793), Ly294002 (Cell Signaling Technology, Leiden, The Netherlands; #9901), MK-2206 (#S1078), rapamycin (#S1039), CI-1040 (Axon Medchem, Groningen, The Netherlands; #1368), AZD6244 (#S1008) and GDC-0941 (#S1065).
Antibodies
The following antibodies used for western blot analyses were obtained from Cell Signaling Technology (unless otherwise indicated): phospho-AKTS473 (#9271), phospho-ERK1/2 (#4370), phospho-JAK1 (#3331), phospho-JAK2 (#3771), phospho-MEK1/2 (#9154), phospho-mTOR (#2971), phospho-p70S6Kinase (#9204), phospho-STAT1 (#9167), phospho-STAT3 (#9145), phospho-STAT5 (#9351), phospho-TYK2 (#9321), DYKDDDDK (#2368), CD127 (anti-IL7Ra; R&D Systems, Minneapolis, MN, USA; #MAB306), RAS (Merck Millipore; #05-516) and β-actin (Sigma-Aldrich; #2547). The following antibodies used for flow cytometry were obtained from Miltenyi Biotec: CD127-FITC (#130-094-888) and CD271-APC (#130-091-884).
Statistics
Statistical analyses were performed using SPSS version 15.0. The Pearson’s chi-square test was used to test for differences in normally distributed data. If the number of patients in the individual groups was fewer than five, the Fisher’s exact test was used. Statistical significance for continuously distributed data was tested using the Mann–Whitney U test. Differences were considered to be significant at P<0.05 (two-sided).
Results
IL7R signaling mutations in T-ALL are mutually exclusive
To measure the prevalence of mutations in IL7Ra—and/or its downstream signaling molecules—in pediatric T-ALL, we compared the mutations identified in JAK family kinase genes, STAT5B and RAS genes with mutations that we previously identified in the IL7Ra, NF1, PTEN and AKT genes8, 16, 41, 42 (Table 1 and Supplementary Figure S1a). We found no mutations in either JAK2 or TYK2. In contrast, mutations were identified in the JAK1 and JAK3 genes of 10 patients; 2 of these patients had mutations in both JAK1 and JAK3. The JAK1R724H, JAK3M511I and JAK3R657Q mutations have been reported by others;13, 14, 26, 36, 38 the remaining five JAK1 mutations were identified by us as part of this study and recently modeled in the putative JAK1 protein structure.43 Mutations in either N-RAS or K-RAS were identified in 15 patients, and inactivating deletions and mutations in NF1 had been previously detected in three additional patients.8 NF1 is a RAS-GTPase-activating protein that catalyzes the hydrolysis of active RAS-GTP into inactive RAS-GDP. Three patients with a mutation in N-RAS, K-RAS or NF1 also had a JAK1 and/or JAK3 mutation, three patients had a mutation in IL7Ra, and four patients had a PTEN-inactivating event. Aside from RAS, the overlap of mutations in IL7Ra, JAK, STAT5B, PTEN and AKT is rare in T-ALL patients. Thus, in our cohort, mutations in IL7Ra, JAK, STAT5B, PTEN and AKT were mutually exclusive from each other, and occurred rarely with mutations in either N-RAS or K-RAS. We found that patients with the TLX subtype of T-ALL had a high prevalence of IL7Ra mutations (P=0.001), whereas patients with the proliferative or ETP-ALL subtype had no mutations in IL7Ra (Table 2). Only one JAK mutation was identified in the TALLMO patient group (P=0.039), and mutations in RAS or NF1 were most prevalent among ETP-ALL14 and TLX patients (P=0.016 and P=0.044, respectively; Table 2). Overall, activating mutations in the IL7Ra, JAK-STAT, RAS-MEK-ERK or PI3K-PTEN-AKT-mTOR pathways were identified in 49% of our pediatric T-ALL patients and were nearly mutually exclusive, suggesting that these mutations are functionally redundant in T-ALL.
Mutations in IL7R signaling molecules can transform cells
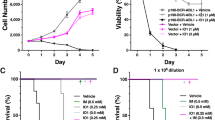
Next, we asked whether expressing the identified IL7R signaling mutations could transform Ba/F3 cells, rendering the cells IL3-independent. We generated doxycycline-inducible expression constructs for C-terminal DDK-tagged mutant JAK1, JAK3 and AKT, as well as non-tagged mutant IL7Ra and N-RAS proteins; for each mutant protein, a construct encoding the corresponding wild-type protein was also generated (Supplementary Figures S1b and S1c). Stably transfected bulk cell lines were sorted based on the constitutive expression of truncated NGFR (Supplementary Figure S1d); none of these bulk cell lines were able to proliferate in the absence of doxycycline and IL3 (data not shown). Treating the cells with doxycycline induced the expression of wild-type or mutant IL7Ra (Supplementary Figure S1d), DDK-tagged JAK (Supplementary Figure S1e), DDK-tagged AKT (Supplementary Figure S1e) and RAS (Supplementary Figure S1f). The doxycycline-induced expression of the mutant molecules IL7RaRFCPH (as a representative for all cysteine-containing IL7Ra mutants), JAK1R724H, JAK1T901G, JAK3M511I, JAK3R657Q, N-RASG12D and AKTE17K transformed Ba/F3 cells, rendering them IL3-independent. In contrast, inducing the expression of the respective wild-type counterparts failed to transform any of the bulk cell lines (Figures 1a and b). Importantly, all bulk lines grew equally well in the presence of IL3 (Supplementary Figure S1g), indicating that the observed differences in growth rates in the absence of IL3 reflect differences in the transforming potential of the mutant molecules.

Transforming potential of activating mutations in IL7R signaling molecules in Ba/F3 cells. (a, b) Growth curves of Ba/F3 cell lines expressing the indicated IL7Ra, JAK1, JAK3, N-RAS and AKT constructs (n=3 experiments per group) following induction with doxycycline. N-RASG12D ‘Dox-grown’ represent the growth curve of N-RASG12D mutant Ba/F3 cells after full adaptation to the mutant molecule. Each culture was started with 2 × 105 cells on day 0 after extensive washing to remove IL3. The inset in panel b shows an expanded view of the first 12 days of culture. (c–e) Western blot analysis of DDK-tagged and phosphorylated/total proteins from Ba/F3 cells treated in the presence or absence of IL3 or doxycycline (DOX). β-Actin was used as a loading control.
Interestingly, the cysteine-containing mutant IL7RaRFCPH was able to form homodimers (Supplementary Figure S2), which is required for interleukin-independent signaling and transformation.15, 16 In contrast, the non-cysteine-containing IL7RaV253GPSL mutant did not transform Ba/F3 cells (Figure 1a), nor did it form homodimers (Supplementary Figure S2). The cell lines expressing either wild-type IL7Ra or the IL7RaGPSL mutant grew in the presence of IL7 (data not shown), suggesting that IL7RaGPSL forms a signaling-competent receptor in the presence of IL7. The AKTE17K cell line proliferated at a slower rate than the mutant JAK and IL7Ra lines. Lastly, although expressing N-RASG12D did not provide an immediate proliferation advantage, within 7 days of doxycycline induction, these cells reached a rate of proliferation that was on par with the mutant JAK and IL7Ra cell lines (Figure 1b). The activating STAT5BN642H mutation transforms Ba/F3 cells44 but was not functionally investigated as part of this study.
Mutations in the IL7R pathway induce ligand-independent downstream signaling
Next, we investigated the ability of the mutant and wild-type molecules to activate downstream IL7R signaling (Figures 1c and e). First, we confirmed that the STAT5, RAS-MEK-ERK and AKT-mTOR pathways were activated in all bulk cell lines upon the addition of IL3 following overnight IL3 starvation. Upon doxycycline induction, each cell line expressed its wild-type or mutant protein within 2–4 h (Figures 1c and e, Supplementary Figure S3). In the presence of IL7, the cell lines expressing wild-type IL7Ra, IL7RaGPSL and IL7RaRFCPH activated downstream signaling pathways; in the absence of IL7, only the cysteine mutant IL7RaRFCPH activated downstream signaling. In addition to activating the JAK-STAT and PI3K-AKT-mTOR pathways,16, 45 IL7RaRFCPH can also activate MEK-ERK signaling (Supplementary Figure S3). Expressing the JAK1R724H, JAK1T901G, JAK3M511I or JAK3R657Q mutants—but not wild-type JAK1 or JAK3—activated the MEK-ERK and PI3K-AKT-mTOR pathways, as well as the downstream kinase S6K (Figures 1c and e and Supplementary Figure S3). N-RASG12D robustly activated downstream MEK-ERK signaling, as well as AKT-mTOR and the downstream target S6K (Figure 1e). Interestingly, inducing the expression of wild-type N-RAS also activated the same downstream molecules, albeit with a slower time course than the N-RASG12D mutant (Supplementary Figure S3), even though wild-type N-RAS did not transform Ba/F3 cells (Figure 1b). Similar effects were observed with respect to the AKTE17K mutant and its wild-type counterpart: induction of both proteins led to their self-activation and the downstream activation of mTOR and S6K (Supplementary Figure S3), whereas only the AKTE17K line was able to transform Ba/F3 cells (Figure 1b).
A close examination of signaling strength revealed that the JAK1 mutant molecules (that is, JAK1R724H and JAK1T901G) activated downstream signaling more robustly than the IL7RaRFCPH, JAK3M511I and JAK3R657Q mutants (Figure 1e). Indeed, the JAK3 mutants only weakly activated downstream signaling (Supplementary Figure S3), even though the transforming efficiency of the JAK3 mutants was similar to—if not higher than—the JAK1 mutants (Figure 1a). Thus, the signaling strength of these molecules does not appear to be correlated with their transforming potential.
Pharmacological inhibition of the IL7R pathway
Next, we tested a variety of pharmacological inhibitors for their ability to block signaling and cell proliferation, as well as their ability to induce cell death in cell lines expressing mutant signaling molecules (Figures 2 and 3). Importantly, in the absence of doxycycline (but in the presence of IL3), all cell lines were equally responsive to the inhibitors. Upon addition of doxycycline (and in the absence of IL3), the mutant IL7Ra, JAK1 and JAK3 lines became sensitive to the selective JAK1/2 inhibitor ruxolitinib; in contrast—and as expected—inducing the expression of the mutant N-RAS and AKT molecules induced ruxolitinib resistance (Figure 2a). Consistent with these results, ruxolitinib blocked the activation of STAT5, MEK, ERK, AKT and mTOR in the IL7RaRFCPH and JAK1T901G mutant lines, but not in the N-RASG12D or AKTE17K mutant lines (Supplementary Figure S4).

Summary of the effects of various inhibitors of IL7R signaling molecules in Ba/F3 cell lines expressing wild-type or mutant IL7Ra, JAK, AKT and RAS. In each plot, the mean ±s.d. IC50 values are shown for each Ba/F3 line (n=3) after doxycycline induction in the absence of IL3. For mutant AKT and N-RAS, two independent lines were tested. The maximum concentrations used were 5 μM for ruxolitinib (a), 125 μM for CI-1040 (b), 16.7 μM for Ly294002 (c) and 5 μM for MK-2206 (d). Cell lines that showed no effect at the maximum inhibitor concentration were considered to be completely resistant to that inhibitor, and thus no IC50 was obtained.

The effect of using combinations of inhibitors on downstream signaling in cell lines expressing mutant IL7Ra and JAK molecules. (a) Schematic representation of the IL7R signaling pathways. The molecules with activating and inactivating mutations found in T-ALL patients are indicated with stars and 'stop' signs, respectively. The inhibitors used in this study are listed in red. The green arrows indicate activation, the dashed arrows indicate putative activation, the red arrows mediate inactivating processes, and the red lines indicate inhibition. (b) Western blot analysis of phosphorylated proteins in the indicated doxycycline-treated Ba/F3 lines in the presence of the indicated inhibitors (24 h). The following concentrations were used: tipifarnib, 10 μM; CI-1040, 10 μM; Ly294002, 10 μM; MK-2206, 2 μM; rapamycin 1 μM. β-Actin was used as a loading control.
Inhibiting STAT5 with pimozide had no effect on any of the mutant lines, with the exception of a moderate effect on the JAK1 mutant lines (data not shown); thus, signaling molecules other than STAT5 are important for maintaining cell viability and proliferation. Although the N-RASG12D mutant line was resistant to the PI3K inhibitor Ly294002 and the AKT inhibitor MK-2206 (Figures 2c and d), this line was sensitive to the RAS inhibitor tipifarnib (data not shown) and the MEK inhibitor CI-1040 (Figure 2b). In this cell line, tipifarnib reduced the levels of phosphorylated MEK and phosphorylated ERK, whereas CI-1040 increased the levels of phosphorylated MEK but decreased the levels of phosphorylated ERK (Supplementary Figure S4). Both JAK mutant lines were also sensitive (to varying degrees) to the MEK inhibitor CI-1040, whereas the IL7RaRFCPH and AKTE17K lines were completely resistant to CI-1040 (Figure 2b). Moreover, inhibiting MEK increased AKT phosphorylation in the IL7RaRFCPH and JAK1T901G lines (Supplementary Figure S4); this effect is likely a cellular escape mechanism used to activate an alternative survival pathway. The JAK1 and JAK3 mutant lines were also sensitive to inhibitors of PI3K and AKT (Ly294002 and MK-2206, respectively); in contrast, the IL7RaRFCPH line was completely resistant to these inhibitors (Figures 2c and d). Because most JAK mutants are very sensitive to PI3K inhibition and respond to a lesser degree to MEK inhibition, JAK mutants may preferentially signal via PI3K-AKT. As expected, the AKTE17K line was highly sensitive to both Ly294002 and MK-2206 (Figures 2c and d, Supplementary Figure S4). Finally, the N-RASG12D line retained S6K activity in the presence of PI3K, AKT and mTOR inhibitors (Supplementary Figure S4), providing further evidence that S6K is a common target downstream of PI3K-AKT-mTOR and RAS-MEK-ERK pathways and can be used as a measure of activity for both pathways (Figure 3a).
As shown above, the IL7Ra and JAK mutant proteins activated both the RAS-MEK-ERK and the PI3K-AKT-mTOR pathways; moreover, inhibiting MEK led to the activation of AKT. Therefore, we tested the effect of treating cells with various combinations of MEK, PI3K and AKT inhibitors, using phosphorylated S6K levels as a measure of signaling activity (Figure 3b). Individually, none of these inhibitors completely silenced downstream signaling in either the IL7RaRFCPH or the JAK1T901G cell line. In contrast, combining CI-1040 with Ly294002 or MK-2206 completely blocked the activation of ERK, AKT, mTOR and S6K (Figure 3b). Thus, we hypothesized that these combinations of inhibitors may exert synergistic cytotoxic effects in cells carrying mutations in IL7 signaling molecules.
Synergistic inhibition using combinations of MEK and PI3K/AKT inhibitors
To test the hypothesis that applying combinations of inhibitors has a synergistic effect on cytotoxicity, we first exposed each mutant cell line to serial dilutions of the MEK inhibitor AZD6244, the PI3K inhibitor GDC-0941 and the AKT inhibitor MK-2206, each of which is used clinically. After we obtained IC50 values for each inhibitor, cytotoxicity was then measured using serial dilutions of inhibitor combinations that were prepared at three different fixed ratios (1:1, 4:1 and 1:4); for an example of this approach, see Supplementary Figure S5. Synergy was then tested by calculating the combination index for each inhibitor combination’s dose–response curve relative to the respective single inhibitors’ dose–response curves. For each mutant line (with the exception of AKTE17K), the MEK+PI3K and/or MEK+AKT inhibitor combinations were synergistic (Table 3). Interestingly, these two combinations had a synergistic effect in the N-RASG12D line, in which AKT is activated (Figure 1e, Supplementary Figure S3).
Lastly, to investigate the potential clinical relevance of these findings, we tested the synergistic effects of MEK+PI3K and MEK+AKT inhibitor combinations using primary leukemic cells obtained from 11 T-ALL patients. Specifically, we tested the efficacy of the MEK inhibitors AZD6244 and trametinib, the PI3K inhibitor GDC-0941, the PI3K/mTOR inhibitor GDC-0980 and the AKT inhibitor MK-2206, as well as various combinations of these inhibitors (Table 4). Six of the 11 patient samples had a measurable synergistic response to the inhibitor combinations tested. Five cases were relatively resistant, because the IC50 of one or both of the inhibitors could not be determined and/or the maximum efficacy was lower than is necessary to determine the effective dose for 50 or 75% of responding cells. Therefore, no synergy could be determined in these five cases. Interestingly, one (patient #3821) of the five samples in which we could not measure a synergistic response had no mutations in NOTCH1, IL7Ra, JAK1, JAK3 or RAS and was resistant to all inhibitors tested, suggesting that the survival and proliferation of these leukemic cells do not require these signaling pathways.
Discussion
Here, we report that activating mutations in the signaling molecules IL7Ra, JAK1/3,STAT5B, PTEN and AKT were mutually exclusive in a cohort of 146T-ALL patients, indicating that these mutations have shared mechanisms for cell survival and/or proliferation. Ten out of 24 N-RAS/K-RAS/NF1 mutations occurred in combination with other IL7R pathway mutations. In Down Syndrome ALL, RAS and JAK2 mutations are mutually exclusive.46 Some of our mutations—for example, mutations in the IL7Ra, JAK and RAS genes—were more prevalent in patients with the TLX and ETP-ALL subtypes of T-ALL. These cases frequently also carry mutations in NOTCH1 and/or FBXW7; in particular, most patients with the TLX subtype carry strongly activating mutations in NOTCH1.47 A similar association with Notch1 mutations was also reported in mouse models of T-ALL induced by mutations in K-Ras and RasGRP1.48, 49, 50
The majority of mutations in the IL7Ra gene introduce a cysteine residue, which facilitates receptor homodimerization and IL7-independent signaling.15, 16 Although the cysteine mutant IL7RaRFCPH confers IL7-independent growth and signaling, the function of non-cysteine mutations (for example, IL7RaGPSL) is not currently understood. We found that the non-cysteine IL7RaGPSL mutant supports the growth of Ba/F3 cells better than the wild-type IL7Ra; thus, non-cysteine IL7Ra mutations may promote leukemogenesis by increasing the IL7 response.
Activating mutations in the IL7R signaling pathway can act at different levels and to varying degrees. For example, the JAK3 mutant Ba/F3 lines grew robustly in the absence of IL3, but their potential to activate downstream signaling molecules was relatively weak compared with the JAK1 and IL7Ra mutants. The strongest level of activation was conferred by the JAK1 mutations, conferring even stronger activation than the IL7RaRFCPH mutant. These differences in activation strength may be due to different properties of the mutant molecules. JAK3 mutant Ba/F3 cells signal to STAT5 and ERK, but this signaling was generally weaker than that in the JAK1 mutant.38 Furthermore, IL7Ra mutations that facilitate the formation of IL7Ra homodimers primarily recruit JAK1 molecules rather than JAK3 molecules, activate JAK1 but not JAK3 and also require JAK1 (but not JAK3) to activate STAT5.16, 51 Conversely, mutant JAK3 molecules require a functional cytokine receptor complex, likely via binding to the common γ-chain;39, 52 however, they also require JAK1 for ligand-independent signaling.38 This may explain why two of our patients with JAK3 mutations also have a JAK1 mutation. Weak signaling by JAK3 mutants cannot be explained by low endogenous expression of the IL7Ra/IL2cγ heterodimeric receptor in Ba/F3 cells, as these JAK3 mutations also result in weaker downstream signaling compared with JAK1 and IL7Ra mutations when measured in SUPT1 and P12 Ichikawa T-ALL cell lines (both of which express IL7Ra/IL2cγ receptors; data not shown).
Mutations in the IL7R signaling pathway may provide a therapeutic window of opportunity. We used the Ba/F3 model system to measure cellular responses to a variety of signaling inhibitors in the context of specific individual mutations. Strikingly, the IL7Ra, JAK and RAS mutant lines had different responses to MEK, PI3K and AKT inhibitors. For example, JAK mutants seem to be more sensitive to PI3K inhibition and less to MEK inhibition, suggesting JAK mutants preferentially signal through PI3K-AKT. Moreover, inhibiting MEK increased the activation of AKT in some cell lines, possibly because of a cellular escape mechanism. Combining a MEK inhibitor with either a PI3K inhibitor or an AKT inhibitor robustly blocked downstream signaling and had a synergistic cytotoxic effect in nearly all Ba/F3 lines tested. This synergy underscores the importance of both MEK-ERK and PI3K-AKT-mTOR downstream pathways, as well as the need for combined inhibition of these pathways. Interestingly, none of the lines responded to the STAT5 inhibitor pimozide, suggesting that activation of STAT5 may not be a common survival pathway downstream of mutant IL7Ra or JAK molecules. Ba/F3 cells are transformed by the activating STAT5BN642H mutation,44 but this mutation was not functionally investigated as part of this study; nonetheless, it would be interesting to assess the sensitivity of STAT5BN642H mutant cells to the inhibitors tested here.
Combined therapy using MEK and PI3K inhibitors has been suggested as a viable treatment option for several solid tumors.53, 54, 55, 56 With respect to acute myeloid leukemia, MEK and AKT inhibitors have been combined in a current phase II study (trial NCT01907815). This combination of inhibitors may prevent the cross-activation of one pathway upon inhibition of the other,57, 58 for example, as suggested for the activation of AKT by RAS (this study) and for the activation of ERK by the PI3K-dependent feedback loop involving mTORC1.59 More than half of the primary T-ALL patient samples that we tested had a synergistic response to inhibitors of the RAS-MEK and PI3K-AKT pathways, suggesting that these patients will benefit from compounds that inhibit downstream IL7R signaling. Historically, our laboratory and others have correlated in vitro cytotoxicity to in vivo response for many chemotherapeutics and many patients, so it seems that the in vitro data are a good indicator of in vivo response,60, 61, 62, 63, 64 especially in T-ALL.65 Patient cells only survive in culture for several days, but do not proliferate. The in vitro cytotoxicity assay measures cell survival over the course of 3 days in the absence of cytokines and stromal cell support. This assay was chosen because cytokines and stromal support conditions may induce cell growth, which would lead to results in which the potential effects of inhibitors on cell survival versus cell growth could not be separated. The efficacy of inhibitors should not reflect the efficacy of cell cycle inhibition but should reflect the potential to kill leukemia cells. Moreover, cytokine addition or stromal support does not consistently support growth of all patient samples, adding an extra variable in the results. Interestingly, one primary T-ALL patient (case #3976) lacks mutations in IL7R signaling, but leukemic cells from this patient had a synergistic response to MEK+PI3K and MEK+AKT inhibitor combinations, suggesting the presence of additional mutations and/or oncogenic mechanisms that apparently depend on the MEK-ERK and PI3K-AKT-mTOR signaling pathways. This finding also underscores the importance of performing in vitro inhibitor testing in addition to screening for mutations; this dual diagnostic strategy can be used to stratify patients in specific treatment groups.
Finally, our results may also be relevant to precursor B-ALL patients with mutations in IL7R signaling molecules.14, 15 Furthermore, the incidence of RAS mutations is higher in relapsed patients with ALL, and this is likely due to selection of RAS mutant subclones during therapy.66
In conclusion, we report that combining MEK inhibitors with PI3K or AKT inhibitors has synergistic cytotoxic effects in leukemic cells carrying mutations at various levels in the IL7R signaling pathway. In addition, this combination of inhibitors may also be toxic to cells without apparent IL7R pathway mutations. Therefore, the cytotoxic effects of combining MEK inhibitors with PI3K and/or AKT inhibitors warrant further study of in vitro and in vivo models using leukemic cells from primary and relapsed ALL patients.
References
Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1: 75–87.
Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood 2005; 106: 274–286.
Van Vlierberghe P, van Grotel M, Tchinda J, Lee C, Beverloo HB, van der Spek PJ et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood 2008; 111: 4668–4680.
Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 2011; 19: 484–497.
Meijerink JP . Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2010; 23: 307–318.
Van Vlierberghe P, Pieters R, Beverloo HB, Meijerink JP . Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br J Haematol 2008; 143: 153–168.
Meijerink JP, den Boer ML, Pieters R . New genetic abnormalities and treatment response in acute lymphoblastic leukemia. Semin Hematol 2009; 46: 16–23.
Balgobind BV, Van Vlierberghe P, van den Ouweland AM, Beverloo HB, Terlouw-Kromosoeto JN, van Wering ER et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood 2008; 111: 4322–4328.
Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009; 114: 647–650.
Kawamura M, Ohnishi H, Guo SX, Sheng XM, Minegishi M, Hanada R et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leuk Res 1999; 23: 115–126.
Palomero T, Dominguez M, Ferrando AA . The role of the PTEN/AKT pathway in NOTCH1-induced leukemia. Cell Cycle 2008; 7: 965–970.
Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306: 269–271.
Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med 2008; 205: 751–758.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012; 481: 157–163.
Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med 2011; 208: 901–908.
Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet 2011; 43: 932–939.
Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci USA 2014; 111: 705–710.
Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med 1994; 180: 1955–1960.
Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity 1995; 2: 223–238.
Foxwell BM, Beadling C, Guschin D, Kerr I, Cantrell D . Interleukin-7 can induce the activation of Jak 1, Jak 3 and STAT 5 proteins in murine T cells. Eur J Immunol 1995; 25: 3041–3046.
Rich BE, Campos-Torres J, Tepper RI, Moreadith RW, Leder P . Cutaneous lymphoproliferation and lymphomas in interleukin 7 transgenic mice. J Exp Med 1993; 177: 305–316.
Uehira M, Matsuda H, Hikita I, Sakata T, Fujiwara H, Nishimoto H . The development of dermatitis infiltrated by gamma delta T cells in IL-7 transgenic mice. Int Immunol 1993; 5: 1619–1627.
Abraham N, Ma MC, Snow JW, Miners MJ, Herndier BG, Goldsmith MA . Haploinsufficiency identifies STAT5 as a modifier of IL-7-induced lymphomas. Oncogene 2005; 24: 5252–5257.
Barata JT, Cardoso AA, Nadler LM, Boussiotis VA . Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood 2001; 98: 1524–1531.
Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA . Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med 2004; 200: 659–669.
Asnafi V, Le Noir S, Lhermitte L, Gardin C, Legrand F, Vallantin X et al. JAK1 mutations are not frequent events in adult T-ALL: a GRAALL study. Br J Haematol 2010; 148: 178–179.
Jeong EG, Kim MS, Nam HK, Min CK, Lee S, Chung YJ et al. Somatic mutations of JAK1 and JAK3 in acute leukemias and solid cancers. Clin Cancer Res 2008; 14: 3716–3721.
Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA 2009; 106: 9414–9418.
Xiang Z, Zhao Y, Mitaksov V, Fremont DH, Kasai Y, Molitoris A et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood 2008; 111: 4809–4812.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
Gordon GM, Lambert QT, Daniel KG, Reuther GW . Transforming JAK1 mutations exhibit differential signalling, FERM domain requirements and growth responses to interferon-gamma. Biochem J 2010; 432: 255–265.
Hornakova T, Staerk J, Royer Y, Flex E, Tartaglia M, Constantinescu SN et al. Acute lymphoblastic leukemia-associated JAK1 mutants activate the Janus kinase/STAT pathway via interleukin-9 receptor alpha homodimers. J Biol Chem 2009; 284: 6773–6781.
Bains T, Heinrich MC, Loriaux MM, Beadling C, Nelson D, Warrick A et al. Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia 2012; 26: 2144–2146.
Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L et al. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer Discov 2012; 2: 591–597.
Degryse S, de Bock CE, Cox L, Demeyer S, Gielen O, Mentens N et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014; 124: 3092–3100.
Witthuhn BA, Silvennoinen O, Miura O, Lai KS, Cwik C, Liu ET et al. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 1994; 370: 153–157.
Kleppe M, Lahortiga I, El Chaar T, De Keersmaecker K, Mentens N, Graux C et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat Genet 2010; 42: 530–535.
Zuurbier L, Petricoin EF 3rd, Vuerhard MJ, Calvert V, Kooi C, Buijs-Gladdines JG et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 2012; 97: 1405–1413.
Mendes RD, Sarmento LM, Cante-Barrett K, Zuurbier L, Buijs-Gladdines JG, Povoa V et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 2014; 124: 567–578.
Cante-Barrett K, Uitdehaag JC, Meijerink JP . Structural modeling of JAK1 mutations in T-ALL reveals a second contact site between pseudokinase and kinase domains. Haematologica e-pub ahead of print 27 January 2016.
Bandapalli OR, Schuessele S, Kunz JB, Rausch T, Stutz AM, Tal N et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 2014; 99: e188–e192.
Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest 2008; 118: 3762–3774.
Nikolaev SI, Garieri M, Santoni F, Falconnet E, Ribaux P, Guipponi M et al. Frequent cases of RAS-mutated Down syndrome acute lymphoblastic leukaemia lack JAK2 mutations. Nat Commun 2014; 5: 4654.
Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia 2010; 24: 2014–2022.
Chiang MY, Xu L, Shestova O, Histen G, L'Heureux S, Romany C et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest 2008; 118: 3181–3194.
Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood 2008; 112: 3373–3382.
Oki T, Kitaura J, Watanabe-Okochi N, Nishimura K, Maehara A, Uchida T et al. Aberrant expression of RasGRP1 cooperates with gain-of-function NOTCH1 mutations in T-cell leukemogenesis. Leukemia 2012; 26: 1038–1045.
Haan C, Rolvering C, Raulf F, Kapp M, Druckes P, Thoma G et al. Jak1 has a dominant role over Jak3 in signal transduction through gammac-containing cytokine receptors. Chem Biol 2011; 18: 314–323.
Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM et al. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science 1994; 266: 1039–1042.
Guenther MK, Graab U, Fulda S . Synthetic lethal interaction between PI3K/Akt/mTOR and Ras/MEK/ERK pathway inhibition in rhabdomyosarcoma. Cancer Lett 2013; 337: 200–209.
Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2011; 2: 135–164.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008; 14: 1351–1356.
Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA 2013; 110: 4015–4020.
Aksamitiene E, Kiyatkin A, Kholodenko BN . Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem Soc Trans 2012; 40: 139–146.
Castellano E, Downward J . RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011; 2: 261–274.
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118: 3065–3074.
Den Boer ML, Harms DO, Pieters R, Kazemier KM, Gobel U, Korholz D et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol 2003; 21: 3262–3268.
Friedman HS, Schold SC Jr., Muhlbaier LH, Bjornsson TD, Bigner DD . In vitro versus in vivo correlations of chemosensitivity of human medulloblastoma. Cancer Res 1984; 44: 5145–5149.
Kaspers GJ, Pieters R, Van Zantwijk CH, Van Wering ER, Van Der Does-Van Den Berg A, Veerman AJ . Prednisolone resistance in childhood acute lymphoblastic leukemia: vitro-vivo correlations and cross-resistance to other drugs. Blood 1998; 92: 259–266.
Klumper E, Pieters R, Veerman AJ, Huismans DR, Loonen AH, Hahlen K et al. In vitro cellular drug resistance in children with relapsed/refractory acute lymphoblastic leukemia. Blood 1995; 86: 3861–3868.
Pieters R, Huismans DR, Loonen AH, Hahlen K, Van Der Does-Van Den Berg A, van Wering ER et al. Relation of cellular drug resistance to long-term clinical outcome in childhood acute lymphoblastic leukaemia. Lancet 1991; 338: 399–403.
Escherich G, Troger A, Gobel U, Graubner U, Pekrun A, Jorch N et al. The long-term impact of in vitro drug sensitivity on risk stratification and treatment outcome in acute lymphoblastic leukemia of childhood (CoALL 06-97). Haematologica 2011; 96: 854–862.
Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 2014; 124: 3420–3430.
Acknowledgements
This study was supported by the Cancer-Free Children Foundation (Stichting Kinderen Kankervrij, KiKa), grants KiKa2008-29 (KC-B and WS), KiKa2013-116 (KC-B). This study was also supported by the European Commission Horizon 2020 SME 662461 project (GJRZ and RCB). We thank Judith de Vetter, Martine Prinsen, Jelle Dylus and Jeroen de Roos of the Netherlands Translational Research Center for performing the synergy experiments.
Author contributions
KC-B, JAPS-H, JGCAMB-G, JCMU, WKS, JvdZ, RCB and GJRZ performed the experiments and/or analyzed the data; KC-B and JPPM wrote the manuscript; RP and JPPM designed and supervised the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
GJRZ and RCB are founders and shareholders of The Netherlands Translational Research Center B.V. The remaining authors declare no conflict of interests.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Canté-Barrett, K., Spijkers-Hagelstein, J., Buijs-Gladdines, J. et al. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T-cell acute lymphoblastic leukemia. Leukemia 30, 1832–1843 (2016). https://doi.org/10.1038/leu.2016.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.83
This article is cited by
-
Pediatric T-cell acute lymphoblastic leukemia blast signature and MRD associated immune environment changes defined by single cell transcriptomics analysis
Scientific Reports (2023)
-
Integrated multi-omics analyses reveal homology-directed repair pathway as a unique dependency in near-haploid leukemia
Blood Cancer Journal (2023)
-
Interleukin-7 regulates CD127 expression and promotes CD8+ T cell activity in patients with primary cutaneous melanoma
BMC Immunology (2022)
-
JAK3 mutations and mitochondrial apoptosis resistance in T-cell acute lymphoblastic leukemia
Leukemia (2022)
-
Mutant IL7R collaborates with MYC to induce T-cell acute lymphoblastic leukemia
Leukemia (2022)
