
Abstract
Acute promyelocytic leukemia (APL) is a subtype of myeloid leukemia characterized by differentiation block at the promyelocyte stage. Besides the presence of chromosomal rearrangement t(15;17), leading to the formation of PML-RARA (promyelocytic leukemia-retinoic acid receptor alpha) fusion, other genetic alterations have also been implicated in APL. Here, we performed comprehensive mutational analysis of primary and relapse APL to identify somatic alterations, which cooperate with PML-RARA in the pathogenesis of APL. We explored the mutational landscape using whole-exome (n=12) and subsequent targeted sequencing of 398 genes in 153 primary and 69 relapse APL. Both primary and relapse APL harbored an average of eight non-silent somatic mutations per exome. We observed recurrent alterations of FLT3, WT1, NRAS and KRAS in the newly diagnosed APL, whereas mutations in other genes commonly mutated in myeloid leukemia were rarely detected. The molecular signature of APL relapse was characterized by emergence of frequent mutations in PML and RARA genes. Our sequencing data also demonstrates incidence of loss-of-function mutations in previously unidentified genes, ARID1B and ARID1A, both of which encode for key components of the SWI/SNF complex. We show that knockdown of ARID1B in APL cell line, NB4, results in large-scale activation of gene expression and reduced in vitro differentiation potential.
Similar content being viewed by others

Introduction
Acute promyelocytic leukemia (APL) is a distinct subtype of myeloid leukemia characterized by differentiation block resulting in accumulation of leukemic promyelocytes in the bone marrow. The disease is defined by a single genetic alteration involving chromosomal translocation 15;17, leading to the formation of oncogenic fusion protein PML-RARA (promyelocytic leukemia-retinoic acid receptor alpha).1, 2, 3 A minority of APL cases (<5%) harbor fusion of RARA gene with other partners including PLZF, NPM1, NUMA1 and STAT5B.4, 5 Although the translocations involving RARA are a constant event in APL, additional genetic events have been identified, including somatic alterations of FLT3 and NRAS genes.6, 7 Moreover, expression of PML-RARA in mice is associated with long latency of onset of disease and variable penetrance.8, 9, 10, 11 This suggests that secondary genetic abnormalities contribute to the pathogenesis of APL. Sequencing of a mouse APL genome revealed recurrent mutations in Jak1 and deletions comprising Kdm6a gene.12 Previous next-generation sequencing studies, which have aimed to identify cooperating mutations in APL, were limited by either small sample size13, 14 or focus on newly diagnosed APL.7
Targeted therapy with all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) have revolutionized the treatment of APL in the past three decades.15, 16 Recent advances have included high efficacy of ATO as a frontline agent in newly diagnosed APL17, 18, 19, 20 and high rates of cure and shorter time to remission observed with the combination of ATRA and ATO.21, 22 Both ATRA and ATO target PML-RARA fusion protein and trigger its degradation by non-overlapping mechanisms, leading to restoration of normal retinoid signaling.15, 23 Despite the remarkable improvement in the treatment outcome in APL, a number of resistance-related treatment failures and relapses are observed in both ATRA- and ATO-treated APL.17, 18, 19, 20, 24, 25, 26 Somatic mutations of RARA and PML genes associated with treatment resistance have been reported in APL relapse.27, 28, 29, 30, 31, 32 However, landscape of genetic aberrations acquired at relapse following these two discrete treatment regimens has not been systematically examined.
In this study, whole-exome and targeted exome sequencing of a large APL cohort was used to uncover mutational spectrum both at initial diagnosis and relapse. Samples from 212 APL cases (comprising 30 primary/relapse pair) including relapses arising subsequent to either ATRA or ATO treatment were analyzed. A mutational signature distinctive from other myeloid leukemias was identified and recurrent mutations of SWI/SNF members ARID1A and ARID1B were discovered. Sequencing data demonstrated that APL relapse is characterized by acquisition of frequent alterations in PML and RARA genes, specifically in ATO- and ATRA-treated cases, respectively. Our experiments also demonstrate that silencing of ARID1B leads to impaired differentiation of NB4 cells in response to ATRA.
Materials and methods
Sample collection and genomic DNA extraction
Genomic DNA or frozen cell pellets from disease/remission stages were obtained from various institutes and hospitals. Informed consent was obtained in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of respective institutes. Genomic DNA was extracted from cell pellets using DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA). DNA was quantified using Qubit Fluorometer (Life Technologies, Carlsbad, CA, USA) and DNA integrity was assessed by agarose gel electrophoresis. DNA was amplified using REPLI-g UltraFast Mini Kit (Qiagen) for samples with insufficient quantity. DNA was sheared using Covaris instrument (Woburn, MA, USA) and assessed on 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) before library preparation.
Whole-exome and targeted capture sequencing
For whole-exome sequencing, DNA libraries were prepared using SureSelect Human All Exon (50 Mb) Kit (Agilent), as per the manufacturer’s instructions. For targeted capture library, customized RNA baits were designed to capture exons of 398 genes. Libraries were prepared using SureSelect XT2 Target Enrichment System (Agilent) for Illumina Multiplexed Sequencing and sequenced on HiSeq 2000 platform (Illumina, San Diego, CA, USA).
Somatic variant discovery and validation
Paired-end reads of 100 bp were aligned to hg19 reference genome using bwa mem (http://arxiv.org/abs/1303.3997v2) in default mode. Duplicates were marked with Samblaster.33 Resulting bam files were further processed according to Genome Analysis Toolkit (GATK) best practices (https://www.broadinstitute.org/gatk/guide/bp_step.php?p=1), by performing Indel Realignment and Base Quality Recalibration. Minimum mapping quality of 15 was considered for variant calling. Somatic variants were called using two methods: ebCall34 and VarScan2 (version 2.3.8).35 Mutations supported by either <5 reads or those supported by reads in only one direction were excluded. Also, variants with variant allele frequencies <0.05 were disregarded. Processed somatic variants were later filtered for false positives using varscan2 fpfilter command. Final confident variants were annotated using annovar and oncotator. Commonly found SNPs in general population were filtered out using dbSNP135, 1000 Genomes and our in-house SNP databases, while retaining the clinically associated variants. For samples without germline samples, we retained variants that passed the criteria outlined in Supplementary Figure S1. Oncoplots were drawn using mafTools (https://github.com/PoisonAlien/mafTools) and ComplexHeatmap bioconductor package. Clonality analysis was carried out using sciClone.36 Mutual exclusion relationships between frequently mutated genes were assessed by Mutation-Relation Test.37 All somatic mutations reported in this study were validated by Sanger sequencing. The sequencing data have been deposited in the NCBI Sequence Read Archive under accession code SRP070515.
Generation of ARID1B knockdown cell lines
Short hairpin (sh) RNA sequences were cloned into AgeI/EcoRI sites of pLKO.1 lentiviral vector (Sigma, St Louis, MO, USA) and viral particles were generated by transfection in 293T cells, as described previously.38 Viral supernatant was collected 48 and 72 h after transfection, and used to transduce NB4 cells (provided by Dr Wee Joo Chng, Singapore) two times, 24 h apart. Cells were selected in puromycin to generate stably knockdown cell lines. The target sequence for human ARID1B sh2 and sh3 are 5′-GCAGGACATGTACAACCAAAG-3′ and 5′-GCCGAATTACAAACGCCATAT-3′, respectively.
Results
Analysis of APL exomes at diagnosis and relapse
Whole-exome sequencing was used to characterize the genetic alterations in 12 primary diagnosis APL with eight matched relapse samples (Table 1 and Supplementary Table S1). Complete remission samples that were negative for PML-RARA fusion allele were used as germline control. We achieved a mean depth of 117x (range 79–159x) and an average 81% of nucleotides were covered by at least 20 reads (Supplementary Figure S2). We validated a total of 115 somatic non-silent mutations in 109 genes using Sanger sequencing (Supplementary Table S2). We observed that the average number of nonsynonymous and synonymous mutations at relapse remained unchanged compared with the initial diagnosis (two-tailed Student's t-test) (Figure 1a). Recurrent mutations were observed in FLT3, WT1 and PML genes, with mutations of PML detected only in relapse samples. Mutations in other novel genes such as ARID1A, CEBPE, AKT1, KAT6A and HK3 were also detected in one sample each (Supplementary Table S2 and Supplementary Figure S3). Among the eight diagnosis–relapse pairs, six samples had mutations that were shared between the two disease stages, indicating re-emergence of disease subclone following therapy (Supplementary Figure S3). Although, no non-silent mutation common to both diagnosis and relapse was detected in DS1 and DS6, PML-RARA fusion was the common genetic lesion, indicating that the relapse evolved from an ancestral clone (Figure 1b and Supplementary Figure S3). Although sequencing depth was inadequate to discover minor subclones, clustering analysis outlined an evolutionary pattern where the founding clone or a subclone at primary disease acquired additional lesions at relapse (number of variants in DS11 were insufficient for clustering) (Figure 1b), similar to as previously described for acute myeloid leukemia (AML).39, 40, 41

Characteristics of somatic mutations detected at primary and relapse APL. (a) Comparison of mutational load between primary and relapse APL. Boxplots depict median and range for numbers of nonsynonymous and synonymous variants identified per exome. Difference between average number of variants between two groups was estimated using two-tailed Student's t-test. (b) Clustering analysis using variant allele frequencies (VAFs) of all somatic variants (VAF ⩾0.08) observed at primary and relapse APL. DS2 and DS6 are shown as two representative cases, which harbor common or exclusive somatic mutations, respectively, at initial diagnosis and relapse.
Mutational topography of primary APL
Next, to uncover the repertoire of somatic mutations in APL, targeted sequencing was performed on exons of 398 genes, including 109 genes identified in our discovery cohort. The additional genes (listed in Supplementary Table S3) comprised those either previously implicated in hematological malignancies or identified in earlier leukemia sequencing studies. In this frequency cohort, we sequenced 22 paired diagnosis and relapse samples. In addition, 131 unpaired newly diagnosed and 47 unpaired relapse samples, making a total of 200 individual APL samples, were also sequenced (Supplementary Table S4). The number of samples analyzed in this study is summarized in Table 1. The mean sequencing depth was 272x (range 40–865x), with 90% of target bases covered >20x (Supplementary Figure S4). C–T transition was the most frequent nucleotide substitution at both primary and relapse APL (Supplementary Figure S5).
To identify significantly mutated genes in APL, we excluded from further analysis several genes (listed in Supplementary Table S5) that frequently exhibit high rate of variants because of large size and constitute potential false positives in cancer sequencing studies.42, 43 Somatic variants identified in bioinformatics analysis were verified using Sanger sequencing. Overall, 210 non-silent point mutations and small insertions/deletions were validated in our frequency cohort. In addition, the presence of FLT3-ITD (fms-like tyrosine kinase 3-internal tandem duplication) was examined using the Genomon ITDetector44 and verified using PCR analysis.
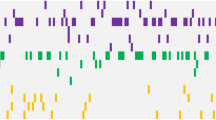
Our collective analysis of discovery and frequency cohorts revealed FLT3 as most frequently altered gene in primary APL (Figure 2), similar to observation in an earlier study.7 This included 27% cases with FLT3-ITD (45 out of 165 initial diagnosis samples) and 16% cases (26 mutations in 25 samples) with point mutations of FLT3 (Figure 2 and Supplementary Tables S1). In fact, alteration of FLT3 gene was a sole genetic aberration present in 25% of newly diagnosed APL cases (39/153 in frequency cohort), signifying their cooperativity with PML-RARA fusion in driving the disease. The somatic mutations of FLT3 predominantly occurred in the tyrosine kinase domains 1 and 2 and in the juxtamembrane domain, with D835 residue most frequently mutated (18/27 cases) (Supplementary Figure S6 and Supplementary Tables S2 and S6). Other known AML genes that were recurrently mutated in our cohort included WT1 (14%), NRAS (10%), KRAS (4%), ETV6 (1%) and EZH2 (1%) (Figure 2). Mutations of NRAS gene clustered at amino-acid residues G12, G13 and V14, whereas KRAS mutations occurred at G12 and Q61 residues (Supplementary Figure S6 and Supplementary Table S6). In addition, missense mutations at K117 and A146 were also detected in KRAS. Majority of WT1 mutations resulted in truncated protein (seven nonsense, seven frameshift insertions/deletions and one splice site mutation out of 23 mutations). In addition, eight missense substitutions were clustered in the C-terminus region encoding Zn-finger domains (Supplementary Figure S6). Our sequencing analysis also revealed the absence of FLT3-ITD or non-silent mutation in any known driver genes in a quarter of primary APL samples (27%; 43/153 frequency cohort), suggesting that a single detectable genetic lesion (PML-RARA fusion) was sufficient to drive the disease.

Recurrently mutated genes in primary APL. Mutational topography of newly diagnosed APL is depicted. Top 20 mutated genes validated by Sanger sequencing are shown and are color-coded for various types of mutation. Genes are displayed according to decreasing mutational frequency from top to bottom. Samples are displayed as columns and arranged to emphasize mutual exclusivity. Samples in which either no driver mutation was detected in our analysis pipeline or do not harbor mutations of top 20 genes are not included in the matrix. Bars on the right side depict absolute number of mutations in each gene.
Mutation-relation test37 was used to examine the association between mutations identified in primary APL. FLT3-ITD rarely co-occurred with mutations in FLT3 or WT1 genes (P<0.01). Similarly, mutations of Ras pathway genes (NRAS and KRAS) were predominantly detected in cases with wild-type FLT3 (FLT3-ITD vs NRAS mutation, P=0.022; FLT3-ITD vs KRAS mutation, P=0.024; FLT3 mutation vs NRAS mutation, P=0.027).
APL exhibits distinct mutational signature among myeloid leukemia
Sequencing of a large cohort enabled us to contrast the mutational pattern of APL to other AML subtypes. Mutational frequencies of recurrently mutated genes in newly diagnosed APL were compared with non-M3 AML samples in TCGA data set. Somatic mutations in known leukemia-related genes (e.g. DNMT3A, NPM1, TET2, IDH1, IDH2 and ASXL1) were absent or rarely observed in APL (Figure 3a). This was evident not only in the primary disease, as previously noted by Welch et al.,7 but also at relapse (two cases with mutation of TET2; one case with mutations of DNMT3A; no mutation of NPM1, ASXL1, IDH1 and IDH2 genes were observed in our study) (Supplementary Table S6). An increased incidence of FLT3 alterations occurred in APL compared with other AML subtypes, although the difference did not reach statistical significance (FLT3-ITD: 27.3% in APL vs 18.9% in other AML, P=0.191; mutant FLT3: 15.8% in APL vs 9.7% in other AML, P=0.078) (Supplementary Figure S7). On the other hand, mutational frequency of other known drivers such as NRAS, KRAS, EZH2 and ETV6 were comparable between primary APL and other AML (Supplementary Figure S7). Our sequencing data also revealed recurrent mutations in several genes either previously unidentified in APL or mutated at higher frequency compared with other AML subtypes. These included two members of the SWI/SNF complex, ARID1B and ARID1A (Fisher's exact test, P<0.05), both of which exhibited high proportion of loss-of-function alterations including frameshift insertions/deletions or nonsense mutations (Figures 3a–c). We also noted recurrent mutations of other lesser recognized genes such as CEBPE, NUMA1, RUNX1T1, HUWE1, HK3 and USP9X. These observations highlight a distinctive mutational profile for APL, distinguishing it from other AML subtypes.

Distinct mutational profile of newly diagnosed APL. (a) Bars represent mutational rates of genes mutated at significantly different frequencies (Fisher’s exact test; P<0.05) between APL and other AML subtypes at initial diagnosis. Frequency of validated non-silent mutations in 165 newly diagnosed APL samples analyzed in this study was compared with 176 non-APL AML samples from the TCGA database. TTN, which is most frequently mutated in public exomes,42 and MT-CYB, which was not included in our targeted sequencing, were excluded from this representation. (b and c) Schematic representation of protein domains of ARID1A (b) and ARID1B (c) was drawn with DOG 2.0 software (http://dog.biocuckoo.org) using domain information from NCBI Protein database (http://www.ncbi.nlm.nih.gov/protein). Location and type of all somatic mutations verified at primary and relapse APL are shown. Circles depicting individual mutations are color-coded for different types of mutations.
Landscape of somatic mutations at APL relapse
To obtain insights into mutations either specifically present or enriched in APL relapse, mutational frequencies at initial diagnosis and relapse (morphological relapses) were compared. Significantly, higher frequency of mutations in four genes were detected in relapse samples, including PML, RARA, RUNX1 and ARID1B (Fisher’s exact test; P<0.05) (Figure 4a). FLT3-ITD was the most frequent lesion at relapse, at a frequency similar to that observed at primary APL. On the other hand, mutations of FLT3 were enriched in the primary APL samples and their incidence in relapse samples was significantly reduced (15.8% in diagnosis vs 5.2% in relapse; Fisher’s exact test; P<0.05) (Figures 2 and 4a). Similar to the pattern observed in FLT3, mutations of both NRAS and KRAS occurred at a higher frequency in newly diagnosed samples compared with the relapse, albeit the difference in their mutational frequencies between the two stages did not reach statistical significance (NRAS: 9.7% in newly diagnosed and 5.2% in relapse; KRAS: 3.6% in primary APL and 1.3% in relapse). On the other hand, mutational frequency of WT1 did not vary significantly between diagnosis and relapse stages (13.9% in primary APL and 18.2% in relapse).

Spectrum of somatic mutations at APL relapse. (a) Matrix displays top 15 genes recurrently mutated at relapsed APL. Each column represents a relapse sample. Genes are arranged according to decreasing mutational frequencies from top to bottom. Right panel illustrates the number of mutations for all genes. Only those relapse samples that harbor mutations of top 15 genes are included in the matrix. (b–d) Protein domains for RARA (b), PML (c) and RUNX1 (d) are drawn using the DOG 2.0 software (http://dog.biocuckoo.org), and type and location of validated mutations are illustrated. Information about protein domains was obtained from Human Protein Reference Database (http://www.hprd.org). All except one missense mutation (E699K) of PML were found in relapse samples. One frameshift deletion in PML transcript NM_033249 (NP_15025; S482fs) is not shown. All mutations of RARA were detected in relapse. Only one of the six mutations of RARA occurred in newly diagnosed samples.
Missense mutations of RARA were detected in seven relapse cases (eight mutations), all of which harbored mutations in the ligand-binding domain. One sample (222 R) had a second mutation of RARA (R217P) present in the vicinity of ligand-binding domain (Figure 4b). Three samples (four mutations), where RNA was available, showed that all four mutations of RARA were present in the PML-RARA fusion allele (Supplementary Figure S8). Similar mutations affecting the ligand-binding domain of RARA in the PML-RARA allele have also been reported previously.28, 45 Thirteen mutations of PML (in 11 cases) were observed in relapse APL, of which 12 were clustered in the B2 domain, and A216V was the most frequent alteration (Figure 4c). Although mutations in close proximity of C212-C213 dicysteine residues, which are involved in binding to PML, have been described, a missense mutation at N191 in B2 domain was also identified. A frameshift deletion in a shorter PML isoform (NM_033249; S482) was also detected in relapse APL. In addition, a newly diagnosed case harbored a missense mutation at E699 (its matched relapse sample was not available). One relapse case (249 R) harbored two mutations (a missense and a nonsense point mutation) at the C213 residue, indicating that both PML alleles were affected. RNA was available for three cases, of which two cases harbored mutations in the PML-RARA allele (Supplementary Figure S9). However, one case had mutations of D219-S220 dinucleotide in the wild-type PML allele (Supplementary Figure S9), indicating that disruption of normal PML function may have implications in APL.
We categorized our relapse samples into two groups, based on the treatment approach used at the primary disease, which was either ATRA (with or without chemotherapy) or arsenic trioxide (Table 1). Using this classification, a divergence was observed in the mutational pattern of PML and RARA genes in relapse following ATO and ATRA therapy, respectively. While 12 of 13 PML mutations (10/11 cases) occurred in post-ATO relapse, RARA mutations were more frequent post-ATRA treatment (6/8 mutations of RARA were detected post-ATRA treatment) (in 5 of 7 cases). Interestingly, one relapse sample (245 R; post-ATO relapse) had mutations of both PML and RARA genes (Supplementary Tables S4 and S6).
Although RUNX1 mutations were rare in newly diagnosed APL, they occurred at a higher frequency at relapse (0.6% at primary APL and 6.5% at relapse) (Figure 4d). Interestingly, five of the six mutations of RUNX1 were either frameshift insertion/deletions or nonsense mutations (Supplementary Table S6). Notably, we observed that mutations in ARID1B were also significantly enriched at relapse (3.0% at initial diagnosis and 11.7% at relapse), whereas ARID1A was mutated at a similar frequency at both stages of disease (4.8% at initial diagnosis and 5.2% at relapse). Both genes predominantly exhibited loss-of-function mutations, and two cases (159D and 210 R) harbored frameshift insertions of both ARID1A and ARID1B (Supplementary Table S6). Moreover, three cases with ARID1B mutations also harbored PML mutations, indicating cooperativity between the two genes (Figure 4a).
Knockdown of ARID1B leads to impaired differentiation of NB4 cells
Next, we investigated the functional consequences of the loss of ARID1B, one of the novel mutated gene in APL, on growth and differentiation of APL cells. To replicate the deficiency of ARID1B caused by truncating lesions, we used shRNA-mediated gene knockdown approach to stably downregulate expression of ARID1B in NB4 cells. Two shRNA sequences (sh2 and sh3) significantly silenced the expression of ARID1B at both the transcript and protein levels (Figures 5a and b), without affecting the expression of ARID1A, a closely related member of ARID1 family (Supplementary Figure S10). Suppression of ARID1B did not significantly alter the growth of NB4 cells in liquid culture over 4 days (Supplementary Figure S11). In the colony-forming assay, ARID1B knockdown cells exhibited a moderate increase in the number of colonies observed after 9 days (Figure 5c). NB4 cells can undergo granulocytic differentiation upon ATRA treatment, and we used this in vitro model to assess the differentiation of ARID1B-deficient cells. We noticed that knockdown of ARID1B resulted in a small increase in CD11b expression compared with control shRNA-transduced cells without stimulation with ATRA (Supplementary Figure S12). Treatment of cells with different concentrations of ATRA-induced expression of CD11b in a dose- and time-dependent manner. However, increase in the proportion of CD11b-expressing cells in ATRA- vs vehicle-treated cells was lower in the ARID1B knockdown cells compared with the control cells. This observation suggests a role of ARID1B in in vitro differentiation towards the granulocytic lineage.

Silencing of ARID1B impairs ATRA-induced neutrophil differentiation of NB4 cells. (a and b) Generation of NB4 cells with stable knockdown of ARID1B. Cells transduced with ARID1B shRNA (sh2 or sh3) display significant suppression of ARID1B transcript (two primer pairs) (a) and protein levels (b) compared with the control transduced (con) cells. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript levels were used to normalize for RNA in quantitative real time-PCR (qRT-PCR) analysis in (a) and bars represent mean±s.d. (c) Clonogenic ability of ARID1B knockdown NB4 cells compared with control cells. Cells were plated in methylcellulose media, and colonies were enumerated after 9 days. Bars represent mean±s.e.m. (d) ATRA-induced differentiation of control and ARID1B knockdown NB4 cells was determined by surface expression of CD11b. Cells were treated with 0.1, 1 or 10 μm ATRA, and the proportion of CD11b-expressing cells was determined by flow cytometry at indicated times. The difference in the percentage of CD11b+ cells obtained in ATRA- vs DMSO-treated cells at each time point is depicted. Data represent mean±s.e.m. of four to five experiments (*P<0.05; Student’s t-test).
Gene expression changes induced by silencing of ARID1B
We performed RNA sequencing of control and ARID1B knockdown NB4 cells to examine transcriptomic changes caused by suppression of ARID1B. Gene expression analysis revealed differential expression of 1028 genes (false discovery rate <0.05). Significantly, expression of a majority of genes (740/1028) was elevated in the ARID1B knockdown cells compared with the control cells, whereas 288 genes were downregulated (Figure 6a and Supplementary Figure S13). Gene ontology analysis of upregulated genes showed a marked enrichment for several pathways related to immune response, signal transduction and hematopoietic differentiation, whereas downregulated genes were over-represented for cell adhesion and WNT signaling (Figure 6b). Using quantitative real time-PCR, we validated elevated expression of several genes involved in hematopoietic development and immune regulation in ARID1B knockdown cells. These included GATA1, GATA2, IKZF2, NFE2, IRF5, BPI, PRG2 and PRTN3, whereas the expression of WNT7B and HOXC4 was downregulated (Figure 6c). In accordance with increased expression of GATA1 and GATA2, gene set enrichment analysis verified upregulation of their target genes in ARID1B knockdown NB4 cells (Supplementary Figure S14).

Gene expression changes in ARID1B knockdown NB4 cells. (a) Volcano plot depicts differentially expressed genes between ARID1B knockdown (sh2 and sh3) and control transduced NB4 cells. Horizontal line denotes false discovery rate (FDR)=0.05. Blue dots represent genes involved in ‘immune system process’ GO:0002376. (b) Functional enrichment analysis (gene ontology) of genes either upregulated or downregulated in ARID1B knockdown NB4 cells (FDR<0.05). (c) qRT-PCR analyses of selected genes identified as differentially expressed in RNA sequencing. GAPDH was used as an endogenous control to normalize for RNA quantity. Bars represent mean±s.e.m.
Discussion
This study provides the most comprehensive profiling of somatic mutations in APL to date, at both initial diagnosis and relapse. We demonstrate that the mutational spectrum of APL is notably different from other AML subtypes. Overall, the topography of somatic mutations in APL is defined by recurrent alterations of FLT3, WT1, NRAS, KRAS, ARID1B and ARID1A genes, and the absence of mutations in other common AML genes incluing DNMT3A, NPM1, TET2, ASXL1 and IDH1/2. Moreover, the repertoire of mutations at relapsed APL differs significantly from newly diagnosed cases and is characterized by alterations in PML and RARA genes, which are rarely affected by somatic mutations in primary APL. In our cohort, matched diagnosis samples were available for three relapse cases (DS4R, DS6R, 243 R) harboring four PML mutations and one case (29 R) harboring RARA mutation, all of which were negative for the corresponding mutations of PML and RARA genes. We also uncovered increased frequency of ARID1B and RUNX1 mutations in APL relapse. In addition, the mutational frequency of FLT3 was considerably lower at relapse compared with primary disease, although the proportion of FLT3-ITD was similar at the two stages of disease.
APL is distinctive from other AML subclasses in its sensitivity to ATRA- and ATO-based therapies. In this study, we also examined relapse-specific mutations in the context of therapeutic intervention used at the initial diagnosis. Notably, our relapse cohort comprised of sizable number of ATRA-free ATO-treated samples. We found that the incidence of mutations in RARA and PML genes largely segregated into post-ATRA and post-ATO relapses, respectively. This indicates distinct mechanisms are involved in acquisition of resistance following two different therapeutic regimens.
Our RT-PCR experiments for allele-specific analyses show that mutations of RARA invariably occurred on the PML-RARA fusion allele, similar to previous studies.28, 45 Mutations of PML occurred in proximity to the C212-C213 dicysteine residues, which is associated with binding of ATO to PML moiety.46, 47 These mutations of PML in the B2 domain are associated with resistance to arsenic therapy.30, 32 Although an initial report showed that mutations of PML in two ATO-resistant cases occurred on the PML-RARA allele and accounted for an impaired response to therapy,30 we detected mutations of PML either on the rearranged (two cases) or the wild-type PML allele (one case). A recent report noted a mutation in the non-rearranged allele of PML gene in a case which presented with multiple relapses.31 Also, truncating mutations of wild-type PML allele have been observed in ATRA-resistant APL,29 indicating that mutations affecting either allele of PML may have pathological consequences.
Our sequencing data uncover recurrent mutations in two novel genes, ARID1B and ARID1A, in APL. Somatic mutations in these two members of SWI/SNF chromatin remodeling complex are very frequent in gynecologic and several other solid tumors.48, 49, 50 However, incidence of somatic alterations in genes of ARID1 family is rare in myeloid leukemia,7, 51 although, recently, mutations of ARID1A have been detected in other hematological malignancies including chronic lymphocytic leukemia, natural killer/T-cell lymphoma and hairy cell leukemia.52, 53, 54 Notably, large majority of mutations in both ARID1B and ARID1A in APL were loss-of-function alterations, similar to the truncating mutations observed in solid tumors.
Germline mutations and chromosomal aberrations affecting ARID1B are associated with disorders characterized by developmental delay and intellectual disability including Coffin-Siris syndrome.55, 56 Although mutations of ARID1B have been reported in solid tumors, to our knowledge, this is the first report of ARID1B alterations in myeloid leukemia. Interestingly, mutations of ARID1B were more frequent at relapse APL compared with the newly diagnosed cases in our cohort. Our experiments show that the knockdown of ARID1B does not significantly modulate either the cell growth or colony-forming ability in vitro; however, it alters the differentiation of NB4 cells in response to ATRA. Transcriptome analysis showed that silencing of ARID1B resulted more prominently in activation of gene expression, suggesting it potentially functions as a repressor of gene expression.
Several other potentially novel drivers of leukemia were also identified at low mutational frequencies in this study. Among them, mutations in the transcription factor CEBPE were observed in three cases (two primary and one relapse APL), all of which were potentially deleterious alterations (frameshift insertion/deletion and splice site mutation). Loss-of-function germline alteration of CEBPE causes specific granule deficiency, a rare congenital disorder characterized by neutrophils with abnormal nuclear morphology, impaired function and lacking secondary and tertiary granule proteins.57 However, somatic mutations of CEBPE in myeloid leukemia have not been previously identified. CEBPE is a critical regulator of granulocytic differentiation and truncating mutations in this gene may have implications on differentiation of promyelocytes. Another interesting candidate is a hexokinase gene, HK3, which was mutated in three APL cases. HK3 is specifically expressed during granulocytic differentiation and was previously shown to be downregulated in APL.58 Silencing of HK3 also resulted in impaired ATRA-induced granulocytic differentiation of NB4 cells.58 Four somatic mutations (in three cases) in NUMA1 gene, located on chromosome 11 and involved in t(11;17)(q13;q21) translocation to form NUMA1-RARA fusion in variant cases of APL,59 were also observed. Two primary APL cases harbored mutations of RUNX1T1, a gene fused with RUNX1 in AML characterized by t(8;21) translocation. USP9X, an X-linked intellectual disability gene,60 which encodes for a deubiquitinase enzyme, was mutated in four APL cases. Although the function of USP9X in hematopoietic development has not been defined, all four mutations of USP9X were potentially damaging alterations (three frameshift deletion and one splice site substitution). The pathogenic implications of these novel mutations in the development of APL remain to be investigated.
References
de The H, Chomienne C, Lanotte M, Degos L, Dejean A . The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990; 347: 558–561.
Borrow J, Goddard AD, Sheer D, Solomon E . Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 1990; 249: 1577–1580.
Longo L, Pandolfi PP, Biondi A, Rambaldi A, Mencarelli A, Lo Coco F et al. Rearrangements and aberrant expression of the retinoic acid receptor alpha gene in acute promyelocytic leukemias. J Exp Med 1990; 172: 1571–1575.
Mistry AR, Pedersen EW, Solomon E, Grimwade D . The molecular pathogenesis of acute promyelocytic leukaemia: implications for the clinical management of the disease. Blood Rev 2003; 17: 71–97.
Redner RL . Variations on a theme: the alternate translocations in APL. Leukemia 2002; 16: 1927–1932.
Callens C, Chevret S, Cayuela JM, Cassinat B, Raffoux E, de Botton S et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 2005; 19: 1153–1160.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150: 264–278.
Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, Pelicci PG et al. A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci USA 1997; 94: 2551–2556.
Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ . Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood 1997; 89: 376–387.
He LZ, Tribioli C, Rivi R, Peruzzi D, Pelicci PG, Soares V et al. Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci USA 1997; 94: 5302–5307.
Westervelt P, Lane AA, Pollock JL, Oldfather K, Holt MS, Zimonjic DB et al. High-penetrance mouse model of acute promyelocytic leukemia with very low levels of PML-RARalpha expression. Blood 2003; 102: 1857–1865.
Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J Clin Invest 2011; 121: 1445–1455.
Greif PA, Yaghmaie M, Konstandin NP, Ksienzyk B, Alimoghaddam K, Ghavamzadeh A et al. Somatic mutations in acute promyelocytic leukemia (APL) identified by exome sequencing. Leukemia 2011; 25: 1519–1522.
Riva L, Ronchini C, Bodini M, Lo-Coco F, Lavorgna S, Ottone T et al. Acute promyelocytic leukemias share cooperative mutations with other myeloid-leukemia subgroups. Blood Cancer J 2013; 3: e147.
de The H, Chen Z . Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer 2010; 10: 775–783.
Park J, Jurcic JG, Rosenblat T, Tallman MS . Emerging new approaches for the treatment of acute promyelocytic leukemia. Ther Adv Hematol 2011; 2: 335–352.
Mathews V, George B, Lakshmi KM, Viswabandya A, Bajel A, Balasubramanian P et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood 2006; 107: 2627–2632.
Mathews V, George B, Chendamarai E, Lakshmi KM, Desire S, Balasubramanian P et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: long-term follow-up data. J Clin Oncol 2010; 28: 3866–3871.
Ghavamzadeh A, Alimoghaddam K, Rostami S, Ghaffari SH, Jahani M, Iravani M et al. Phase II study of single-agent arsenic trioxide for the front-line therapy of acute promyelocytic leukemia. J Clin Oncol 2011; 29: 2753–2757.
Zhou J, Zhang Y, Li J, Li X, Hou J, Zhao Y et al. Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood 2010; 115: 1697–1702.
Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013; 369: 111–121.
Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA 2004; 101: 5328–5335.
Dos Santos GA, Kats L, Pandolfi PP . Synergy against PML-RARa: targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J Exp Med 2013; 210: 2793–2802.
Sanz MA, Lo-Coco F . Modern approaches to treating acute promyelocytic leukemia. J Clin Oncol 2011; 29: 495–503.
Tomita A, Kiyoi H, Naoe T . Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O 3) in acute promyelocytic leukemia. Int J Hematol 2013; 97: 717–725.
Gallagher RE . Retinoic acid resistance in acute promyelocytic leukemia. Leukemia 2002; 16: 1940–1958.
Schachter-Tokarz E, Kelaidi C, Cassinat B, Chomienne C, Gardin C, Raffoux E et al. PML-RARalpha ligand-binding domain deletion mutations associated with reduced disease control and outcome after first relapse of APL. Leukemia 2010; 24: 473–476.
Gallagher RE, Moser BK, Racevskis J, Poire X, Bloomfield CD, Carroll AJ et al. Treatment-influenced associations of PML-RARalpha mutations, FLT3 mutations, and additional chromosome abnormalities in relapsed acute promyelocytic leukemia. Blood 2012; 120: 2098–2108.
Gurrieri C, Nafa K, Merghoub T, Bernardi R, Capodieci P, Biondi A et al. Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood 2004; 103: 2358–2362.
Goto E, Tomita A, Hayakawa F, Atsumi A, Kiyoi H, Naoe T . Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood 2011; 118: 1600–1609.
Lehmann-Che J, Bally C, de The H . Resistance to therapy in acute promyelocytic leukemia. N Engl J Med 2014; 371: 1170–1172.
Zhu HH, Qin YZ, Huang XJ . Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med 2014; 370: 1864–1866.
Faust GG, Hall IM . SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 2014; 30: 2503–2505.
Shiraishi Y, Sato Y, Chiba K, Okuno Y, Nagata Y, Yoshida K et al. An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res 2013; 41: e89.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576.
Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol 2014; 10: e1003665.
Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res 2012; 22: 1589–1598.
Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 2015; 6: 6042.
Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012; 481: 506–510.
Parkin B, Ouillette P, Li Y, Keller J, Lam C, Roulston D et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood 2013; 121: 369–377.
Kronke J, Bullinger L, Teleanu V, Tschurtz F, Gaidzik VI, Kuhn MW et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013; 122: 100–108.
Shyr C, Tarailo-Graovac M, Gottlieb M, Lee JJ, van Karnebeek C, Wasserman WW . FLAGS, frequently mutated genes in public exomes. BMC Med Genomics 2014; 7: 64.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499: 214–218.
Chiba K, Shiraishi Y, Nagata Y, Yoshida K, Imoto S, Ogawa S et al. Genomon ITDetector: a tool for somatic internal tandem duplication detection from cancer genome sequencing data. Bioinformatics 2015; 31: 116–118.
Ding W, Li YP, Nobile LM, Grills G, Carrera I, Paietta E et al. Leukemic cellular retinoic acid resistance and missense mutations in the PML-RARalpha fusion gene after relapse of acute promyelocytic leukemia from treatment with all-trans retinoic acid and intensive chemotherapy. Blood 1998; 92: 1172–1183.
Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 2010; 328: 240–243.
Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, Duffort S et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell 2010; 18: 88–98.
Wu JN, Roberts CW . ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 2013; 3: 35–43.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486: 400–404.
Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D et al. Genomic analyses of gynaecologic carcinosarcomas reveal frequent mutations in chromatin remodelling genes. Nat Commun 2014; 5: 5006.
Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368: 2059–2074.
Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet 2015; 47: 1061–1066.
Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015; 526: 519–524.
Dietrich S, Hullein J, Lee SC, Hutter B, Gonzalez D, Jayne S et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 2015; 126: 1005–1008.
Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet 2012; 90: 565–572.
Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet 2012; 44: 379–380.
Lekstrom-Himes JA, Dorman SE, Kopar P, Holland SM, Gallin JI . Neutrophil-specific granule deficiency results from a novel mutation with loss of function of the transcription factor CCAAT/enhancer binding protein epsilon. J Exp Med 1999; 189: 1847–1852.
Federzoni EA, Valk PJ, Torbett BE, Haferlach T, Lowenberg B, Fey MF et al. PU.1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood 2012; 119: 4963–4970.
Wells RA, Catzavelos C, Kamel-Reid S . Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 1997; 17: 109–113.
Homan CC, Kumar R, Nguyen LS, Haan E, Raymond FL, Abidi F et al. Mutations in USP9X are associated with X-linked intellectual disability and disrupt neuronal cell migration and growth. Am J Hum Genet 2014; 94: 470–478.
Acknowledgements
We thank Dr Elizabeth Moran (Rutgers New Jersey Medical School, The State University of New Jersey) for useful discussions and Dr Patrick Tan (Singapore) for sharing laboratory facilities. We also thank Dr Zang Zhi Jiang, Lim Su Lin and Mehnaz Elias (Singapore) for technical help. This work was funded by the Singapore Ministry of Health’s National Medical Research Council (NMRC) under its Singapore Translational Research (STaR) Investigator Award to HPK, NMRC Centre Grant awarded to National University Cancer Institute, Singapore (NCIS), NCIS Centre Grant Seed Funding, the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centers of Excellence initiative. The study was also funded by NIH/NCI R01CA026038-35 awarded to HPK. We also acknowledge the funding support from NCI Grant CA31946. The study was supported by Grant HE 5240/5-1 from Deutsche Forschungsgemeinschaft awarded to MH. The study was also supported by Grants OMRPG3C0021 and MOHW104-TDU-B-212-124-006 awarded to L-YS. V Mathews was supported by Senior Fellowship program of Wellcome DBT India Alliance (IA/S/11/2500267), New Delhi, India and SG received Senior Research Fellowship from Council for Scientific and Industrial Research (CSIR), New Delhi, India. This research was supported by the Samuel Oschin Comprehensive Cancer Institute (SOCCI) at Cedars-Sinai Medical Center through the Eleanor and Glenn Padnick Discovery Fund in Cellular Therapy Project. We also acknowledge a generous donation from Melamed family.
Author contributions
V Madan conceived the study, performed experiments, analyzed data and wrote the manuscript; PS, LH, JS, DK and NF performed experiments; AM, YN, KY, K-TT, MS, L-ZL, YS, SM and HY performed bioinformatics and statistical analyses; SG, NH, TA, SR, JM, EC, HH, GM, TM, TI, AB, RAL, BLP, M Lübbert, WJC, H-FT, MH, A Ganser, MK-M, SMK, HMK, DN, W-KH, WS, A Ghavamzadeh, KA and TH collected and processed patient samples and provided clinical data; M-CK and L-YS collected, processed and analyzed patient samples and provided clinical data; MG, L-WD, Q-YS, WC and M Lill contributed reagents and helped in data analysis; SO contributed clinical samples and supervised bioinformatics analysis; V Mathews conceived the study and provided clinical samples and data; HPK conceived and supervised the study, interpreted the data and wrote the manuscript. All authors reviewed and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Haferlach declares part ownership of MLL Munich Leukemia Laboratory. Dr Alpermann is an employee of MLL Munich Leukemia Laboratory. The other authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Madan, V., Shyamsunder, P., Han, L. et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia 30, 1672–1681 (2016). https://doi.org/10.1038/leu.2016.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.69
This article is cited by
-
SWI/SNF complexes in hematological malignancies: biological implications and therapeutic opportunities
Molecular Cancer (2023)
-
Advances in the Treatment of Hairy Cell Leukemia Variant
Current Treatment Options in Oncology (2022)
-
A novel RARA–SNX15 fusion in PML–RARA-positive acute promyelocytic leukemia with t(11;17;15)(q13;q21.2;q24.1)
International Journal of Hematology (2022)
-
Current views on the genetic landscape and management of variant acute promyelocytic leukemia
Biomarker Research (2021)
-
Therapy-related leukaemias with balanced translocations can arise from pre-existing clonal haematopoiesis
Leukemia (2021)
