
Abstract
Alterations to genes involved in cellular metabolism and epigenetic regulation are implicated in the pathogenesis of myeloid malignancies. Recurring mutations in isocitrate dehydrogenase (IDH) genes are detected in approximately 20% of adult patients with acute myeloid leukemia (AML) and 5% of adults with myelodysplastic syndromes (MDS). IDH proteins are homodimeric enzymes involved in diverse cellular processes, including adaptation to hypoxia, histone demethylation and DNA modification. The IDH2 protein is localized in the mitochondria and is a critical component of the tricarboxylic acid (also called the ‘citric acid’ or Krebs) cycle. Both IDH2 and IDH1 (localized in the cytoplasm) proteins catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG). Mutant IDH enzymes have neomorphic activity and catalyze reduction of α-KG to the (R) enantiomer of 2-hydroxyglutarate, which is associated with DNA and histone hypermethylation, altered gene expression and blocked differentiation of hematopoietic progenitor cells. The prognostic significance of mutant IDH (mIDH) is controversial but appears to be influenced by co-mutational status and the specific location of the mutation (IDH1-R132, IDH2-R140, IDH2-R172). Treatments specifically or indirectly targeted to mIDH are currently under clinical investigation; these therapies have been generally well tolerated and, when used as single agents, have shown promise for inducing responses in some mIDH patients when used as first-line treatment or in relapsed or refractory AML or MDS. Use of mIDH inhibitors in combination with drugs with non-overlapping mechanisms of action is especially promising, as such regimens may address the clonal heterogeneity and the multifactorial pathogenic processes involved in mIDH myeloid malignancies. Advances in mutational analysis have made testing more rapid and convenient, and less expensive; such testing should become part of routine diagnostic workup and repeated at relapse to identify patients who may benefit from treatments that target mIDH.
Similar content being viewed by others
Introduction
Acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) are heterogeneous myeloid disorders with multifactorial pathogenic mechanisms and a broad range of prognoses. AML is characterized by clonal proliferation of poorly differentiated cells of the myeloid lineage.1 MDS reflects the presence of dysplasia and ineffective hematopoiesis commonly leading to bone marrow failure and insufficiency, with a resultant decrease in peripheral blood counts.2 The pathogeneses of both involve recurrent genomic alterations, including somatic gene mutations and/or chromosomal abnormalities, that can define biologically distinct clinical subtypes.3 Comprehensive genomic profiling at the time of diagnosis can inform disease classification, risk stratification and prognosis and ultimately allow for more selective therapeutic interventions.
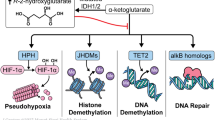
Alterations to cellular metabolism, as well as somatic mutations of genes essential to epigenetic regulation, are implicated in the pathogenesis of several human malignancies.4, 5 Isocitrate dehydrogenases (IDHs) are homodimeric enzymes involved in diverse cellular processes, including adaptation to hypoxia, histone demethylation and DNA modification.6 The IDH2 protein is a critical component of the tricarboxylic acid (also called the ‘citric acid’ or Krebs) cycle, and both IDH2 and IDH1 proteins catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) to produce reduced nicotinamide adenine dinucleotide phosphate (NADPH) from NADP+ (Figure 1). Diverse dioxygenases depend on sufficient levels of α-KG for multiple cellular processes, as well as for epigenetic regulation.7 IDH1 enzymes are localized to the cytoplasm and peroxisomes and IDH2 to the mitochondria.6

IDH mutations in cancer. Mutant IDH1 and IDH2 enzymes result in an increase of the oncometabolite, (R)-2-HG. (R)-2-HG induces a block of cell differentiation by inhibiting the activity of chromatin-modifying histone and DNA demethylases. Inhibition of these epigenetic regulators leads to a ‘hypermethylation signature’ that alters gene expression such that cells lose the ability to progress from immature progenitors to a fully differentiated state.23 (Adapted by permission from Macmillan Publishers Ltd: Prensner and Chinnaiyan,24 copyright 2011).
Somatic mutations in IDH1 (mIDH1) and IDH2 (mIDH2) genes have been described in both solid and hematological malignancies; mIDH1 is more common in solid tumors and mIDH2 is more common in hematological tumors.8 IDH1/2 mutations are heterozygous, retaining one wild-type (wt) allele, suggestive of an oncogenic gain of function. IDH proteins are encoded by the IDH1 gene located at chromosome 2q33 and the IDH2 gene residing at chromosome 15q26.9 An IDH3 isoform is also located in the mitochondria, but no oncogenic mutations in the IDH3 gene have been reported to date.9 Recurrent IDH1/2 mutations are missense variants leading to a single amino-acid substitution of arginine residues at codon 132 in exon 4 of the IDH1 gene and codons 140 or 172 in exon 4 of the IDH2 gene.10 Additionally, a germline-synonymous single-nucleotide polymorphism (rs11554137) located in codon 105 in exon 4 of the IDH1 gene has been reported to have prognostic relevance in AML.9, 11
IDH Mutations in AML and MDS
Mutant IDH enzymes have neomorphic activity, catalyzing NADPH-dependent reduction of α-KG to an oncometabolite, the (R) enantiomer of 2-hydroxyglutarate ((R)-2-HG, also called 2-oxoglutarate) in vitro and in vivo.12, 13, 14 Increased levels of the (S) enantiomer of 2-HG have not been reported in AML or MDS.15 In an AML xenotransplantation model established from a patient with wtIDH1 and mutant nucleophosmin 1 (NPM1), (R)-2-HG but not (S)-2-HG acted as an oncometabolite and daily administration of (R)-2-HG was associated with significantly reduced platelet counts and shorter survival than (S)-2-HG-treated mice.15 High concentrations of (R)-2-HG lead to enhanced proliferation and blocked differentiation of immature hematopoietic cells.16 Cell lines with endogenous IDH mutations (for example, CS-1 chondrosarcoma) or engineered to express mutant IDH proteins (for example, TF-1 human erythroleukemia) show dramatically increased (R)-2-HG levels and impaired cellular differentiation.7, 16, 17 Serum from patients with mIDH AML contains levels of (R)-2-HG that are more than 100-fold higher than expected under normal physiological conditions.14, 18
(R)-2-HG is structurally similar to α-KG and has been shown to competitively inhibit α-KG-dependent enzymes, including members of the ten-eleven-translocation (TET) family of 5-methylcytosine hydroxylases and of the jumonji-domain-containing group of histone lysine demethylases.6, 19, 20 TET2 protein is thought to be involved in both passive and active DNA demethylation by regulating genome-wide and locus-specific hydroxymethylation.21 Similarly, histone demethylases regulate chromatin status, enabling activation or inhibition of gene transcription.22 Inhibition of these epigenetic regulators by (R)-2-HG produces a hypermethylation ‘signature’, altering gene expression and leading to differentiation arrest of hematopoietic progenitors.23, 24 Figueroa et al.25 evaluated the mutational and epigenetic profiles of 385 AML patients aged <60 years; patients with mIDH1/2 AML exhibited a global hypermethylation phenotype associated with significant suppression of gene expression compared with patients with wtIDH1/2 AML.
Although mIDH1 and mIDH2 enzymes both produce (R)-2-HG, they have different enzymatic activities. Cytoplasmic mIDH1 generates less (R)-2-HG than mitochondrial mIDH2 enzymes.13 This may be due to differences in amounts of α-KG substrate, which is found in greater abundance in the mitochondrion than in the cytoplasm. (R)-2-HG production is enhanced in the presence of mIDH1/wtIDH1 heterodimers, suggesting that the retained wtIDH1 enzyme produces some of the α-KG that is reduced to (R)-2-HG.26 In contrast, mIDH2 homodimers can produce abundant (R)-2-HG.13 Mutated IDH2-R172 protein leads to greater accumulation of (R)-2-HG than mIDH2-R140 protein in vitro.17
Epidemiology
Taken together, mIDH1/2 are among the most common mutations in AML (~20% of patients combined, Table 1). IDH mutations increase in frequency with increasing age.27 mIDH are less frequent in MDS (~5%) and myeloproliferative neoplasms, although the frequency increases to ~20% of patients with myeloproliferative neoplasms at leukemic transformation.25, 28, 29 Mutant genes involved in epigenetic regulation, including mIDH, may exist in preleukemic stem cells, which retain the ability to differentiate into multiple lineages, can survive chemotherapy and proliferate during remission, eventually leading to relapse.1, 18, 21, 30, 31
IDH1 mutations are less common than IDH2 mutations in AML and in MDS.1, 8, 10, 32, 33 IDH1 and IDH2 mutations only rarely co-occur in the same patient.34, 35 In myeloid malignancies, IDH1 mutations most often involve a cysteine (R132C) or histidine (R132H) substitution for arginine at R132. IDH2-R140 mutations are more common than IDH2-R172 mutations, representing ~80% of IDH2 mutations in AML.10, 32 IDH2-R172 mutations may be more frequent in older patients.10 With IDH2 mutations, arginine is most often replaced by glutamine at residue 140 (R140Q) and by lysine at residue 172 (R172K).36 Less frequently, other amino-acid substitutions are involved (for example, IDH2-R172W).10, 32
Clinically, several studies suggest that, compared with wtIDH, patients with mIDH are older and tend to have higher platelet and bone marrow blast counts at diagnosis of AML or MDS.10, 35, 37, 38, 39 mIDH are enriched in cytogenetically normal AML (CN-AML; 25–30% of CN-AML cases) and are also associated with cytogenetically intermediate-risk disease, and occur often with trisomy 8.1, 39, 40 mIDH frequently co-occur with NPM1 mutations but are almost always mutually exclusive with TET2 mutations and with mutations in the Wilms’ tumor 1 (WT1) gene.1, 10, 11, 38, 39, 40, 41, 42, 43, 44 mIDH and mutations in TET2 and WT1 result in similar epigenetic alterations and may have overlapping roles in leukemogenesis; all block TET2 enzymatic function, resulting in dysregulated DNA methylation.25, 45 mIDH are frequently accompanied by mutations in the serine/arginine-rich- splicing-factor-2 (SRSF2) gene, which is associated with abnormal splicing of mRNA.1, 46 These mutations have been reported to cluster in primary myelofibrosis, where they are associated with poorer overall survival (OS) and disease-free survival;47 further investigation is needed to elucidate interactions between mIDH and SRSF2 mutations in MDS and AML. Co-occurrence of mIDH and Fms-related tyrosine kinase 3 (FLT3) mutations are less common.44 Marcucci et al.10 reported clinical outcomes for 358 patients with de novo CN-AML; results showed that patients with mIDH1 were less likely to have FLT3-internal tandem duplication (FLT3-ITD). Interestingly, compared with other types of mIDH, mIDH2-R172 is less likely to be accompanied by additional frequently recurring mutations in AML (for example, FLT3-ITD, CCAAT/enhancer-binding-protein-alpha (CEBPA) or NPM1).10, 40, 43
Prognosis
The prognostic impact of mIDH1/2 in AML remains controversial. Several studies have suggested an association with adverse outcomes,10, 37, 38, 48, 49 whereas others have failed to identify any clear influence on clinical response or survival,11, 35, 50, 51 and still others report improved survival (Table 2).40, 44 A meta-analysis that included 8121 patients with AML showed that those with mIDH1 had inferior OS compared with patients without the mutation, and patients with mIDH1 CN-AML had a lower rate of complete remission (CR) with cytotoxic induction chemotherapy.48 Indeed, a preponderance of studies suggest mIDH1 AML confers an adverse prognosis or has no prognostic value (Table 2). One study found no influence of mIDH1-R132 on OS, but the IDH1 single-nucleotide polymorphism rs11554137 variant was associated with an adverse prognosis.11 Analyses of the prognostic impact of IDH2 mutations also show inconsistent results; for example, in one study of patients with CN-AML, mIDH2 had no effect on OS or CR rate compared with wtIDH2,52 but in another study, mIDH2 was associated with lower rates of CR, higher relapse rates and shorter OS.43
Differences in prognostic findings may reflect variations in study methodologies. Some studies evaluate mIDH by point mutation and others combine mIDH1/2 for analysis.49, 51, 53 Specifically, with regard to mIDH2, R172 and R140 mutations are frequently analyzed together, although data suggest that these mutations have different effects on prognosis (Figure 2).1, 10, 44, 54, 55, 56 Recently, Papaemmanuil et al.56 proposed new genomic classifications for AML, including (provisionally) ‘AML with mIDH2-R172’ as a distinct class, and recommended that IDH2 testing be added to prognostic guidelines. In their study of 1540 AML patients, AML with mIDH2-R172 was not accompanied by other class-defining lesions (for example, FLT3, NPM1) and was associated with gene-expression and DNA-methylation profiles seen with more severe abnormalities in metabolic activity, compared with other IDH mutations. Nevertheless, unlike earlier reports,10, 54, 55 investigators found mIDH2-R172 to have a relatively favorable effect on AML prognosis.56 Mutational context may influence AML prognosis, but again, this is unclear. For example, there is conflicting evidence regarding prognosis of patients with mIDH1/2 in the presence of NPM1 mutations and FLT3-ITD−; with some data suggesting a better prognosis and others reporting worsened outcomes or no influence of this mutational profile.35, 38, 44, 49, 51, 55, 57 Patel et al.44 conducted a large study (N=398) to determine the prognostic relevance of frequent somatic mutations in younger patients (<60 years) with AML, which showed a favorable effect of co-occurring mutated NPM1 and mIDH1 or mIDH2. These data were further supported using a proposed integrated prognostic model that combined cytogenetic risk and mutational status using retrospective data from younger patients with newly diagnosed de novo AML.58 In this model (which has not yet been validated clinically), the presence of co-occurring IDH1/2 and NMP1 mutations in patients with intermediate-risk cytogenetics per Medical Research Council criteria was associated with median OS comparable to that of patients with Medical Research Council-defined favorable cytogenetic risk.58 In contrast, Paschka et al.38 reported that the subset of patients with CN-AML with a mIDH/NPM1+/FLT3-ITD− genotype in their study had significantly poorer OS than patients with mIDH CN-AML who did not have that genotype. Generally, mIDH2-R140 is much more likely to be accompanied by mutated NPM1 or other frequently recurring mutation in AML or MDS than mIDH2-R172.10, 55, 56

IDH1/2 mutations appear to have a more consistently negative prognostic impact in MDS and myeloproliferative neoplasms than in AML.59, 60 A study of 193 patients with MDS showed that mIDH1 was associated with shorter OS compared with patients with wtIDH and greater likelihood of leukemic transformation (67% vs 28%, respectively).59 These findings were confirmed by results of a meta-analysis of seven studies that included a total of 1782 MDS patients.60 The relative prognostic impact of type of IDH mutation in MDS is uncertain; Bejar et al.61 reported that, based on survival data for 3200 patients with MDS, mIDH2 was associated with significantly shorter OS, whereas mIDH1 had only a marginal effect on OS. Other studies suggest mIDH1 confers worse prognosis than mIDH2 in MDS.59, 62 The prognostic influence of mIDH type may depend on patients’ overall prognostic risk status.46, 61 Lin et al.46 reported that mIDH2 was a poor prognostic factor in patients with lower-risk MDS, based on International Prognostic Scoring System, revised International Prognostic Scoring System, French–American–British classification or World Health Orgaanization classification, but not in the higher-risk groups.
Serum (R)-2-HG concentration may also serve as a prognostic indicator.63 Of 234 patients with CN-AML, a subgroup of patients who met an (R)-2-HG threshold at diagnosis that the investigators established as ‘high’ (>2.01 μg/ml, log2) were less likely to attain CR and had significantly poorer OS than patients in the ‘normal’ (R)-2-HG group (Figure 3). Posttreatment (R)-2-HG levels may also have prognostic implications. In a study of 223 younger patients with de novo AML treated with standard induction chemotherapy including 62 patients with mIDH, patients in CR who had higher serum levels of (R)-2-HG had poorer OS than patients in CR with lower levels of (R)-2-HG.64 This study showed that (R)-2-HG levels were not significantly different among the different mutation types (mIDH1, mIDH2-R140 or mIDH2-R172); however, there was a trend for poorer OS for the small number of patients (N=9) with mIDH2-R172.64 A different study also found a significant quantitative relationship between (R)-2-HG level and posttreatment clinical outcomes: serum (R)-2-HG concentrations of ⩾2 μmol/l were associated with poorer OS and disease-free survival.65 In the latter study, IDH2-R172 mutations were associated with significantly higher levels of (R)-2-HG compared with the other IDH mutation types.

Further investigation is needed to more clearly elucidate the relationship between (R)-2-HG levels and clinical outcomes.
Detection
Because IDH mutations occur in approximately one in five patients with AML, and even more frequently in patients with CN-AML, mutational testing should be part of routine molecular assessment at diagnosis to identify patients who may in time benefit from targeted treatments currently under clinical study.37 Identification of these mutations at diagnosis may also be pivotal for better risk stratification of MDS patients.60
Testing for mIDH is straightforward, given that nearly all IDH mutations are located on exon 4, and affect IDH1 at a single residue, Arg132, or IDH2 at two residues, Arg140 and Arg172.23 Several methods,66, 67, 68 including PCR, Sanger or next-generation sequencing, are commonly used for mIDH detection.
High-resolution melting (HRM) analysis is a rapid, sensitive and cost-effective method of genotyping and mutational analysis.69 HRM detects sequence differences that change the shape of the melting curve of DNA. A comparison between Sanger sequencing and HRM analysis showed 99–100% concordance of mutation detection but much greater sensitivity with the HRM technique, which detected mutations in samples diluted to only 10% of the mutated DNA.69 As a heterozygous mutation, the highest detectable IDH variant allele fraction (VAF) is 50% and a recent report on 664 adult AML patients by Metzeler et al.70 indicated that, for patients with mIDH2, VAF, on average, approached 50%. At this time, there is no established or standard VAF threshold to identify mIDH as a leukemogenic driver at diagnosis. A too-low VAF positivity threshold (for example, <2%) may have ambiguous clinical relevance and could be a signal of clonal hematopoiesis of indeterminate potential (CHIP). Prevalence of CHIP, a hematological malignancy-associated somatic mutation in the absence of other diagnostic features of MDS or AML, increases with age. Despite relatively high prevalence in older patients, the presence of leukemia-associated mutations is followed by a hematological malignancy in only a minority of cases.71
Many hospitals, particularly those affiliated with academic medical centers, perform mutational analyses with next-generation sequencing using MDS/AML gene panels. Private laboratories also perform these multiplex panels for diagnostic and prognostic purposes. A list of laboratories that conduct mutational analyses is available on the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/gtr/).
IDH mutations detected at diagnosis tend to be stable during disease progression.18, 46 Sequential assessment of 151 patients with MDS demonstrated that all mIDH patients retained the mutation during disease evolution, while none of the wtIDH patients acquired an IDH mutation during follow-up.46 However, variations in detection limits or expansion of the mutant clone over time can account for the presence of seemingly new mutations at relapse not previously detected at diagnosis. In one study, 5.7% of MDS patients were identified as having an IDH mutation at diagnosis, whereas 11.3% of patients had an IDH mutation at the time of leukemic transformation, demonstrating the value of comprehensive molecular profiling at disease progression.39
IDH1/2 mutations may also be suitable molecular markers of minimal residual disease with standard intensive chemotherapy approaches.23 Paired diagnosis and relapse samples demonstrated that mIDH1/2 cells can survive induction chemotherapy and contribute to relapse.18 A study of patients with NPM1-mutant AML with concurrent IDH1/2 (n=17) or DNMT3A (n=15) mutations revealed that IDH1/2 mutations were reliable markers of minimal residual disease for 16 of the 17 patients: 7 of the 8 patients with detectable mIDH1/2 in CR eventually relapsed, whereas all 9 patients with undetectable mIDH1/2 remained in CR for the duration of the study.72 This is distinct from treatment with targeted small-molecule mIDH inhibitors, where emerging data demonstrate CR in the setting of alleviation of maturation arrest–without chemo-ablation or destruction of the mutant clone.73, 74 Mutational persistence during remission and the potential that a low VAF is indicative of CHIP in older patients (a VAF threshold of ⩾2% in peripheral blood has been proposed as a diagnostic criterion of CHIP75) complicate the use of mIDH as a marker of minimal residual disease.
Supranormal levels of (R)-2-HG may serve as a noninvasive biomarker of IDH mutations.64, 65, 76 (R)-2-HG in the serum or plasma can be measured by liquid or gas chromatography coupled with mass spectrometry. Additionally, there is an enzymatic (R)-2-HG assay based on conversion of (R)-2-HG to α-KG in the presence of (R)-2-hydroxyglutarate dehydrogenase and nicotinamide adenine dinucleotide (NAD+) and subsequent detection of generated NADH. This assay was shown to distinguish between (R)-2-HG levels in tumor tissue of patients without mIDH and levels in patients with mIDH-positive AML.77 Currently, no diagnostic or therapeutic (R)-2-HG ‘threshold’ level has been formally established; however, a discriminatory concentration of 700 ng/ml of (R)-2-HG in the serum has been proposed to identify patients with IDH mutations (serum (R)-2-hydroxyglutarate in healthy control subjects was <200 ng/ml in this study).64 At (R)-2-HG levels ⩾700 ng/ml, IDH mutations were detected that were previously missed by Sanger sequencing. Notably, the optimum compartment in which to measure (R)-2-HG has not been determined, although studies are underway to answer this question.27, 78
Treatment
At this writing, there are no approved selective mIDH inhibitor drugs, and consistent with non-mIDH myeloid malignancies, treatment decisions are based on patients’ age, performance status, use of prior treatment and other clinicopathological factors.35 However, the treatment landscape may soon include targeted mIDH enzyme inhibitors and drugs that indirectly target mIDH leukemic cells. Multiple mIDH inhibitors are in preclinical stages of investigation, including HMS-101, which was shown to reduce (R)-2-HG and block colony formation in mIDH1 human AML cells in vitro;79 AGI-026, which reduced (R)-2-HG and was associated with improved survival in a mIDH2-R140 mouse model of R-2-hydroxyglutaric aciduria in vivo;80 and AGI-5198, which reduced (R)-2-HG and induced apoptosis of mIDH1 human chondrosarcoma cells in vitro.81 In addition, several agents are now in various stages of clinical development (Table 3; includes ClinicalTrial.gov study registration information).
Induction chemotherapy
Induction chemotherapy has been the most commonly reported treatment for all AML patients who can tolerate such therapy, including those with mIDH. Compared with patients with wtIDH, rates of response and OS for mIDH patients treated with induction chemotherapy mirror general prognosis, that is, there are reports that outcomes are no different from35, 57 or worse than10 those of patients with wtIDH or are dependent on the presence of NPM1 or other co-mutations.38, 44 In a large retrospective study of patients with AML treated at a single site, patients with mIDH who received front-line induction or salvage chemotherapy had response rates comparable to those of patients with wtIDH regardless of co-mutational status.35 In contrast, in the study by Marcucci et al.10 patients with CN-AML and mIDH1/NPM1+/FLT3-ITD− genotype had significantly shorter postinduction disease-free survival, and mIDH2-R172 patients were significantly less likely to attain CR, than wtIDH patients.
Hypomethylating agents (HMAs)
Because hypermethylation is a pathogenic hallmark of mIDH1/2 in myeloid malignancies, there is a theoretical rationale for treatment with an HMA. Approximately 30–50% of all AML and MDS patients who receive an HMA attain a hematological response of some type.82, 83, 84 However, evidence of increased effectiveness in patients with mIDH has been equivocal.85, 86 A retrospective cohort study that included 11 patients with mIDH MDS revealed that hypomethylating therapy with decitabine was associated with more favorable outcomes than chemotherapy or best supportive care.60 In contrast, a study of 68 older patients (⩾60 years) with de novo, secondary or therapy-related AML found no association between clinical outcomes with front-line HMA treatment, with or without concomitant histone deacetylase inhibitor therapy, in the presence of mIDH1/2.86 Similarly, in a retrospective study of 826 AML patients, 175 patients received front-line therapy with an HMA-based regimen. Of them, 48 mIDH1/2 and 127 wtIDH AML patients showed no significant differences in overall response rate (ORR; 45% vs 58%, P=0.13) or median OS (9.5 vs 10.3 months, P=0.8).35 The equivocal efficacy of HMAs in mIDH AML may be related to leukemogenic effects of excess (R)-2-HG other than hypermethylation of histone and DNA. Cytochrome c oxidase, an enzyme in the mitochondrial electron transport chain, is inhibited by (R)-2-HG and was associated with lowering the apoptotic threshold of THP-1 leukemia cells in vitro, making them dependent on the antiapoptotic effects of B-cell CLL/lymphoma (BCL-2).18 However, Heuser et al.87 predicted that an HMA in combination with an mIDH inhibitor may enhance and accelerate therapeutic response. Clinical trials of combinational therapy with the HMA, azacitidine, and small-molecule mIDH enzyme inhibitors are currently underway (see below).
Small-molecule mIDH inhibitors
Selective small-molecule mIDH1 and mIDH2 inhibitors are in clinical development;8, 79, 88 these drugs bind within the active catalytic site of mIDH enzymes and prevent the conformational change necessary for mIDH to reduce α-KG to (R)-2-HG.89, 90, 91 In preclinical studies, AGI-6780, a selective inhibitor of mIDH2-R140Q, was shown to rapidly reduce histone hypermethylation and reverse DNA hypermethylation over the course of weeks in TF-1 human erythroleukemia cells engineered to express mIDH2 protein and in IDH2-mutated primary human AML cells in vitro.89, 90 Reduced methylation levels were accompanied by evidence of cellular differentiation.
AG-120, enasidenib (AG-221/CC-90007), AG-881, IDH305 and FT-2102 are small-molecule allosteric inhibitors of mIDH proteins.8, 79, 88 Both AG-120 and enasidenib have shown evidence of promoting differentiation of leukemic cells of AML patients.73, 88, 92 These drugs are not thought to be cytotoxic and may confer lower rates of aplasia, neutropenia and thrombocytopenia than traditional chemotherapeutic agents. Therefore, in theory, these agents may be optimal as salvage therapy, alone or in rational combinations, in mIDH patients with relapsed or refractory (R/R) disease.33
AG-120
AG-120 is an oral inhibitor of mutant IDH1-R132 enzyme that is currently under study in a phase 1 dose-escalation and expansion trial in patients with mIDH1 advanced hematological malignancies.74 Plasma (R)-2-HG levels of patients with mIDH1 AML receiving AG-120 are reduced to levels seen in healthy individuals (~99.7% inhibition).93 AG-120 monotherapy has been generally well tolerated and associated with an ORR of 35% in a study in which the majority of patients (78%) had R/R AML.
Enasidenib
Further along in development, enasidenib is an oral inhibitor of IDH2-R140 and IDH2-R172 enzymes. Enasidenib also reduces (R)-2-HG levels in patients with mIDH2 AML to levels detected in healthy subjects.94 Interim results of a phase 1 dose-escalation and expansion study reported outcomes for 181 patients with advanced hematological malignancies, 128 of whom had R/R AML.73 ORR with enasidenib was 41%, both overall and in the subset of patients with R/R AML.73 There was no meaningful difference in response between R/R AML patients with IDH2-R140Q (36%) or IDH2-R172K (39%) mutations.73 A subgroup of patients without a demonstrable hematological response but with prolonged stable disease showed neutrophil recovery during enasidenib treatment, despite persistence of blasts in peripheral blood and/or bone marrow.73, 95 Of interest, the mIDH2 VAF was not reduced in the majority of patients who attained CR on study, indicating that eradication of the mutant clone was not necessary to attain a response.73 A phase 2 expansion of this study is underway in patients with R/R AML,73 as is the phase 3 IDHentify study, which compares enasidenib with conventional care regimens. At this writing, IDHentify is enrolling older (⩾60 years) patients with mIDH2 AML who are refractory to, or in relapse after, second- or third-line AML therapy. A phase 1/2 study of AG-120 or enasidenib in combination with azacitidine vs azacitidine alone in patients with mIDH1- or mIDH2-positive newly diagnosed AML is underway at this writing.
AG-881
Oral AG-881 inhibits both mIDH1 and mIDH2 proteins and penetrates the blood–brain barrier.23, 96 AG-881 is under evaluation for use in solid tumors and in a phase 1, open-label, dose-escalation and expansion study in patients with advanced hematological malignancies that had progressed prior to mIDH inhibitor therapy.
IDH305 and FT-2102
A small-molecule mIDH1 inhibitor, IDH305 is under evaluation in a phase 1 dose-finding clinical trial for treatment of patients with mIDH1 R/R advanced malignancies, including MDS and AML. Similarly, FT-2102 is an mIDH1 inhibitor under investigation in a phase 1 dose-finding study as a single agent or in combination with azacitidine in patients with AML or higher-risk MDS or who are R/R to prior treatment or ineligible for standard intensive therapy. At this writing, no clinical data associated with IDH305 or FT-2102 treatment have been reported.
Venetoclax (ABT-199)
Venetoclax is an oral, small-molecule BCL-2 inhibitor under investigation for use in AML. Preclinical data demonstrated that expression of mIDH sensitized leukemic cells to venetoclax. This effect was mediated through the intracellular accumulation of (R)-2-HG.18 These preclinical findings are supported by results of recent venetoclax clinical trials in patients with AML. In a phase 2 study of single-agent venetoclax in patients with R/R AML, a CR or CR with incomplete hematological recovery was observed in 3 of the 11 (27%) patients with an IDH mutation, compared with 3 of the 21 (14%) patients without the mutation.97 Additionally, in a trial combining HMAs with venetoclax in elderly patients with untreated AML unfit for intensive chemotherapy, patients with an IDH mutation were more responsive.98 These results suggest that IDH mutations may identify a patient subgroup that is likely to respond to pharmacological BCL-2 inhibition. However, the duration of response was short for most patients, highlighting the need for combination strategies to enhance the efficacy of venetoclax.
CB-839
CB-839 is an oral inhibitor of glutaminase, the enzyme responsible for the production of glutamine. Neoplastic cells depend on glutamine to fuel the tricarboxylic acid cycle and to promote cell growth and proliferation, and glutamine is the primary source of α-KG in mIDH cells.27, 32 In vitro, CB-839 inhibited the growth of mIDH primary human AML cells, and preclinical data have demonstrated that CB-839 preferentially slowed the growth of AML cell lines that ectopically expressed mutant IDH1/2.27, 99 CB-839 is under investigation in a phase 1 trial as a single agent and in combination with azacitidine in patients with R/R AML; while an IDH mutation is not required to enroll, there is a prespecified end point to evaluate response in the subset of patients with mIDH.100
All trans-retinoic acid (ATRA)
(R)-2-HG-related inhibition of lysine-specific demethylases may promote a response to the differentiating agent, ATRA, in non-acute promyelocytic leukemia AML.101 In vitro data show that the combination of ATRA and the tyrosine kinase inhibitor, dasatinib, improved cell differentiation in primary AML samples and in AML cell lines harboring mIDH1-R132H and reduced tumor growth in mutant xenografted mice.102
Conclusion
IDH mutations are frequent in myeloid malignancies, particularly AML; are uniquely associated with elevated levels of the oncometabolite, (R)-2-HG; inhibit epigenetic regulators; and should be included in AML and MDS gene panels for prognostication. Advances in understanding of the genetics underlying myeloid malignancies are igniting an exciting era of development of promising and targeted treatments. Such approaches may be more effective and less toxic than conventional chemotherapy regimens.103 Clinical trials of mIDH inhibitors as monotherapy in the R/R setting have shown much promise, although the emergence of resistant subclones has been observed;73, 74 investigations of use as front-line therapy and in combination regimens are now ongoing.
Given the genomic complexity of AML and MDS, and the observation that the founding clone can give rise to various subclones during disease evolution, selective agents that target a single mutation are unlikely to be curative in the large majority of patients. However, the use of targeted treatments in combination with drugs with non-overlapping mechanisms of action may address multifactorial pathogenic processes implicated in hematological malignancies and potentially have a revolutionary impact on patient outcomes.
References
Dohner H, Weisdorf DJ, Bloomfield CD . Acute myeloid leukemia. N Engl J Med 2015; 373: 1136–1152.
Albitar M, Manshouri T, Shen Y, Liu D, Beran M, Kantarjian HM et al. Myelodysplastic syndrome is not merely ‘preleukemia’. Blood 2002; 100: 791–798.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114: 937–951.
Cairns RA, Harris IS, Mak TW . Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11: 85–95.
Conway O'Brien E, Prideaux S, Chevassut T . The epigenetic landscape of acute myeloid leukemia. Adv Hematol 2014; 2014: 103175.
Clark O, Yen K, Mellinghoff IK . Molecular pathways: isocitrate dehydrogenase mutations in cancer. Clin Cancer Res 2016; 22: 1–7.
Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJ, Bleeker FE . The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim Biophys Acta 2014; 1846: 326–341.
Stein EM . Molecular pathways: IDH2 mutations-co-opting cellular metabolism for malignant transformation. Clin Cancer Res 2016; 22: 16–19.
Willander K, Falk IJ, Chaireti R, Paul E, Hermansson M, Green H et al. Mutations in the isocitrate dehydrogenase 2 gene and IDH1 SNP 105C>T have a prognostic value in acute myeloid leukemia. Biomark Res 2014; 2: 18.
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010; 28: 2348–2355.
Wagner K, Damm F, Gohring G, Gorlich K, Heuser M, Schafer I et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 2010; 28: 2356–2364.
Reitman ZJ, Parsons DW, Yan H . IDH1 and IDH2: not your typical oncogenes. Cancer Cell 2010; 17: 215–216.
Losman JA, Kaelin WG Jr. . What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 2013; 27: 836–852.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462: 739–744.
Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Goparaju R, Schwarzer A et al. Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia 2016; 30: 1708–1715.
Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013; 339: 1621–1625.
Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem 2013; 288: 3804–3815.
Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 2015; 21: 178–184.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19: 17–30.
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 2011; 12: 463–469.
Chan SM, Majeti R . Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int J Hematol 2013; 98: 648–657.
Accari SL, Fisher PR . Emerging roles of JmjC domain-containing proteins. Int Rev Cell Mol Biol 2015; 319: 165–220.
Yang H, Ye D, Guan KL, Xiong Y . IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 2012; 18: 5562–5571.
Prensner JR, Chinnaiyan AM . Metabolism unhinged: IDH mutations in cancer. Nat Med 2011; 17: 291–293.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18: 553–567.
Jin G, Reitman ZJ, Duncan CG, Spasojevic I, Gooden DM, Rasheed BA et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res 2013; 73: 496–501.
Fathi AT, Wander SA, Faramand R, Emadi A . Biochemical, epigenetic, and metabolic approaches to target IDH mutations in acute myeloid leukemia. Semin Hematol 2015; 52: 165–171.
Abdel-Wahab O, Levine RL . Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013; 121: 3563–3572.
Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A . IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 2010; 24: 1146–1151.
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R . Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA 2014; 111: 2548–2553.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506: 328–333.
Im AP, Sehgal AR, Carroll MP, Smith BD, Tefferi A, Johnson DE et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia 2014; 28: 1774–1783.
Stein EM . IDH2 inhibition in AML: finally progress? Best Pract Res Clin Haematol 2015; 28: 112–115.
Platt MY, Fathi AT, Borger DR, Brunner AM, Hasserjian RP, Balaj L et al. Detection of dual IDH1 and IDH2 mutations by targeted next-generation sequencing in acute myeloid leukemia and myelodysplastic syndromes. J Mol Diagn 2015; 17: 661–668.
DiNardo CD, Ravandi F, Agresta S, Konopleva M, Takahashi K, Kadia T et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol 2015; 90: 732–736.
Molenaar RJ, Thota S, Nagata Y, Patel B, Clemente M, Hirsh C et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia 2015; 29: 2134–2142.
Aref S, Kamel Areida el S, Abdel Aaal MF, Adam OM, El-Ghonemy MS, El-Baiomy MA et al. Prevalence and clinical effect of IDH1 and IDH2 mutations among cytogenetically normal acute myeloid leukemia patients. Clin Lymphoma Myeloma Leuk 2015; 15: 550–555.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 2010; 28: 3636–3643.
DiNardo CD, Jabbour E, Ravandi F, Takahashi K, Daver N, Routbort M et al. IDH1 and IDH2 mutations in myelodysplastic syndromes and role in disease progression. Leukemia 2016; 30: 980–984.
Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 2011; 25: 246–253.
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 2010; 115: 2749–2754.
Patel KP, Ravandi F, Ma D, Paladugu A, Barkoh BA, Medeiros LJ et al. Acute myeloid leukemia with IDH1 or IDH2 mutation: frequency and clinicopathologic features. Am J Clin Pathol 2011; 135: 35–45.
Boissel N, Nibourel O, Renneville A, Gardin C, Reman O, Contentin N et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol 2010; 28: 3717–3723.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–1089.
Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep 2014; 9: 1841–1855.
Lin CC, Hou HA, Chou WC, Kuo YY, Liu CY, Chen CY et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol 2014; 89: 137–144.
Lasho TL, Jimma T, Finke CM, Patnaik M, Hanson CA, Ketterling RP et al. SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012; 120: 4168–4171.
Feng JH, Guo XP, Chen YY, Wang ZJ, Cheng YP, Tang YM . Prognostic significance of IDH1 mutations in acute myeloid leukemia: a meta-analysis. Am J Blood Res 2012; 2: 254–264.
Yamaguchi S, Iwanaga E, Tokunaga K, Nanri T, Shimomura T, Suzushima H et al. IDH1 and IDH2 mutations confer an adverse effect in patients with acute myeloid leukemia lacking the NPM1 mutation. Eur J Haematol 2014; 92: 471–477.
Chotirat S, Thongnoppakhun W, Promsuwicha O, Boonthimat C, Auewarakul CU . Molecular alterations of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) metabolic genes and additional genetic mutations in newly diagnosed acute myeloid leukemia patients. J Hematol Oncol 2012; 5: 5.
Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 2010; 116: 2122–2126.
Thol F, Damm F, Wagner K, Gohring G, Schlegelberger B, Hoelzer D et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 2010; 116: 614–616.
Abdel-Karim I, Plunkett WK Jr, O'Brien S, Giles F, Thomas D, Faderl S et al. A phase I study of pemetrexed in patients with relapsed or refractory acute leukemia. Invest New Drugs 2011; 29: 323–331.
Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H, Preudhomme C . Differential prognosis impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood 2011; 117: 3696–3697.
Green CL, Evans CM, Zhao L, Hills RK, Burnett AK, Linch DC et al. The prognostic significance of IDH2 mutations in AML depends on the location of the mutation. Blood 2011; 118: 409–412.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374: 2209–2221.
Ravandi F, Patel K, Luthra R, Faderl S, Konopleva M, Kadia T et al. Prognostic significance of alterations in IDH enzyme isoforms in patients with AML treated with high-dose cytarabine and idarubicin. Cancer 2012; 118: 2665–2673.
Sloan CE, Luskin MR, Boccuti AM, Sehgal AR, Zhao J, Daber RD et al. A modified integrated genetic model for risk prediction in younger patients with acute myeloid leukemia. PLoS One 2016; 11: e0153016.
Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M et al. IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica 2010; 95: 1668–1674.
Jin J, Hu C, Yu M, Chen F, Ye L, Yin X et al. Prognostic value of isocitrate dehydrogenase mutations in myelodysplastic syndromes: a retrospective cohort study and meta-analysis. PLoS One 2014; 9: e100206.
Bejar R, Papaemmanuil E, Haferlach T, Garcia-Manero G, Maciejewski J, Sekeres M et al. Somatic mutations in MDS patients are associated with clinical features and predict prognosis independent of the IPSS-R: Analysis of combined datasets from the International Working Group for Prognosis in MDS-Molecular Committee. Blood 2015; 126: 907.
Patnaik MM, Hanson CA, Hodnefield JM, Lasho TL, Finke CM, Knudson RA et al. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia 2012; 26: 101–105.
Wang JH, Chen WL, Li JM, Wu SF, Chen TL, Zhu YM et al. Prognostic significance of 2-hydroxyglutarate levels in acute myeloid leukemia in China. Proc Natl Acad Sci USA 2013; 110: 17017–17022.
DiNardo CD, Propert KJ, Loren AW, Paietta E, Sun Z, Levine RL et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 2013; 121: 4917–4924.
Janin M, Mylonas E, Saada V, Micol JB, Renneville A, Quivoron C et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol 2014; 32: 297–305.
Mahdieh N, Rabbani B . An overview of mutation detection methods in genetic disorders. Iran J Pediatr 2013; 23: 375–388.
Patel KP, Barkoh BA, Chen Z, Ma D, Reddy N, Medeiros LJ et al. Diagnostic testing for IDH1 and IDH2 variants in acute myeloid leukemia an algorithmic approach using high-resolution melting curve analysis. J Mol Diagn 2011; 13: 678–686.
Horbinski C, Kelly L, Nikiforov YE, Durso MB, Nikiforova MN . Detection of IDH1 and IDH2 mutations by fluorescence melting curve analysis as a diagnostic tool for brain biopsies. J Mol Diagn 2010; 12: 487–492.
Gorniak P, Ejduk A, Borg K, Makuch-Lasica H, Nowak G, Lech-Maranda E et al. Comparison of high-resolution melting analysis with direct sequencing for the detection of recurrent mutations in DNA methyltransferase 3A and isocitrate dehydrogenase 1 and 2 genes in acute myeloid leukemia patients. Eur J Haematol 2016; 96: 181–187.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016; 128: 686–698.
McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, Stephens J et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep 2015; 10: 1239–1245.
Debarri H, Lebon D, Roumier C, Cheok M, Marceau-Renaut A, Nibourel O et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: a study by the Acute Leukemia French Association. Oncotarget 2015; 6: 42345–42353.
Stein EM, DiNardo C, Altman JK, Collins R, DeAngelo DJ, Kantarjian HM et al. Safety and efficacy of AG-221, a potent inhibitor of mutant IDH2 that promotes differentiation of myeloid cells in patients with advanced hematologic malignancies: Results of a phase 1/2 trial. Blood 2015; 126: 323.
DiNardo C, de Botton S, Pollyea DA, Stein EM, Fathi AT, Roboz GJ et al. Molecular profiling and relationship with clinical response in patients with IDH1 mutation-positive hematologic malignancies receiving AG-120, a fi rst-in-class potent inhibitor of mutant IDH1, in addition to data from the completed dose escalation portion of the phase 1 study. Blood 2015; 126: Abstract 1306.
Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015; 126: 9–16.
Fathi AT, Sadrzadeh H, Borger DR, Ballen KK, Amrein PC, Attar EC et al. Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood 2012; 120: 4649–4652.
Balss J, Pusch S, Beck AC, Herold-Mende C, Kramer A, Thiede C et al. Enzymatic assay for quantitative analysis of (D)-2-hydroxyglutarate. Acta Neuropathol 2012; 124: 883–891.
Brunner AM, Neuberg D, Wander SA, Sadrzadeh H, Ballen KK, Amrein PC et al. Use of 2HG levels in the serum, urine, or bone marrow to predict IDH mutations in adults with acute myeloid leukemia. Blood 2015; 126: 2597–2597.
Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013; 122: 2877–2887.
Wang F, Travins J, Lin Z, Si Y, Chen Y, Powe J et al. A small molecule inhibitor of mutant IDH2 rescues cardiomyopathy in a D-2-hydroxyglutaric aciduria type II mouse model. J Inherit Metab Dis 2016; 39: 807–820.
Li L, Paz AC, Wilky BA, Johnson B, Galoian K, Rosenberg A et al. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS One 2015; 10: e0133813.
Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015; 126: 291–299.
Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012; 30: 2670–2677.
Fenaux P, Ades L . Review of azacitidine trials in Intermediate-2-and High-risk myelodysplastic syndromes. Leuk Res 2009; 33 (Suppl 2): S7–S11.
Emadi A, Faramand R, Carter-Cooper B, Tolu S, Ford LA, Lapidus RG et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol 2015; 90: E77–E79.
DiNardo CD, Patel KP, Garcia-Manero G, Luthra R, Pierce S, Borthakur G et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk Lymphoma 2014; 55: 1925–1929.
Heuser M, Araujo Cruz MM, Goparaju R, Chaturvedi A . Enigmas of IDH mutations in hematology/oncology. Exp Hematol 2015; 43: 685–697.
Yaqub F . Inhibition of mutant IDH1 in acute myeloid leukaemia. Lancet Oncol 2015; 16: e9.
Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013; 340: 622–626.
Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 2015; 125: 296–303.
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340: 626–630.
de Botton S, Pollyea DA, Stein EM, Dinardo C, Fathi AT, Roboz GJ et al. Clinical safety and activity of AG-120, a first-in-class potent inhibitor of the IDH1 mutant protein, in a phase 1 study of patients with advanced, IDH1-mutant hematological malignancies. Haematologica 2015; 100: Abstract P563.
Fan B, Le K, Manyak E, Liu H, Prahl M, Bowden C et al. Longitudinal pharmacokinetic/pharmacodynamic profile of AG-120, a potent inhibitor of the IDH1 mutant protein, in a phase 1 study of IDH1-mutant advanced hematologic malignancies. Blood 2015; 126: Abstract 1310.
Fan B, Chen Y, Wang F, Yen K, Utley L, Almon C et al. Pharmacokinetic/pharmacodynamic (PK/PD) evaluation of AG-221, a potent mutant IDH2 inhibitor, from a phase 1 trial of patients with IDH2 mutation-positive hematologic malignancies. Haematologica 2015; 100: Abstract 379.
Stein EM, Tallman MS . Emerging therapeutic drugs for AML. Blood 2016; 127: 71–78.
Agios Pharmaceuticals. Agios Pharmaceuticals selects third novel IDH mutant inhibitor, AG-881, for clinical development. (News release). 2015 (cited 28 March 2016); available from http://investor.agios.com/phoenix.zhtml?c=251862&p=irol-newsArticle&ID=2041339.
Konopleva M, Pollyea DA, Potluri J, Chyla BJ, Busman T, McKeegan E et al. A phase 2 study of ABT-199 (GDC-0199) in patients with acute myelogenous leukemia (AML). Blood 2014; 124: Abstract 118.
Pollyea DA, DiNardo CD, thirman MJ, Letai A, Wei AH, Jonas BA et al. Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥65 years ineligible for standard induction therapy. J Clin Oncol 2016; 34 (Suppl): Abstract 7009.
Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV . Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol 2014; 42: 247–251.
Wang ES, Frankfurt O, Orford KW, Bennett M, Flinn IW, Maris M et al. Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with relapsed/refractory leukemia. Blood 2015; 126: Abstract 2566.
Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 2012; 18: 605–611.
Boutzen H, Saland E, Cathebras M, Larrue C, Farge T, Serhan N et al. The combination of ATRA and dasatinib for differentiation therapy in acute myeloid leukemias with IDH mutations. Blood 2015; 126: Abstract 2542.
Estey E, Levine RL, Lowenberg B . Current challenges in clinical development of ‘targeted therapies’: the case of acute myeloid leukemia. Blood 2015; 125: 2461–2466.
Graubert T, Walter MJ . Genetics of myelodysplastic syndromes: new insights. Hematology: The Education Program of the American Society of Hematology 2011; 2011: 543–549.
Chotirat S, Thongnoppakhun W, Wanachiwanawin W, Auewarakul CU . Acquired somatic mutations of isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) in preleukemic disorders. Blood Cells Mol Dis 2015; 54: 286–291.
Schnittger S, Haferlach C, Ulke M, Alpermann T, Kern W, Haferlach T . IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 2010; 116: 5486–5496.
Acknowledgements
We received editorial support during manuscript development from Sheila Truten and Kelly Dittmore of MC2 Inc., Wynnewood, PA, USA who were funded by Celgene Corporation. Celgene did not contribute to content or participate in development of this paper. The authors are fully responsible for all content and editorial decisions.
Author Contributions
All authors contributed to, revised and approved the manuscript content and gave approval for submission to the journal.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
BCM has received research funding from Celgene and Agios and has received remuneration for Advisory Board participation from Celgene and Agios. ATF is a consultant for and receives clinical research funding from Celgene and declares Advisory Board participation for Agios. CDD has received research funding from Novartis, Celgene, Agios and Abbvie/Genentech and participates in Advisory Boards for Celgene and Agios. DAP has received research funding from Celgene and is a consultant for Celgene, Pfizer, Alexion, Ariad and Karyopharm. SMC has received research funding from Celgene, Agios and Abbvie/Genentech. RS declares Advisory Board participation for Novartis.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Medeiros, B., Fathi, A., DiNardo, C. et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 31, 272–281 (2017). https://doi.org/10.1038/leu.2016.275
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.275
This article is cited by
-
IDH2/R140Q mutation confers cytokine-independent proliferation of TF-1 cells by activating constitutive STAT3/5 phosphorylation
Cell Communication and Signaling (2024)
-
Hypomethylating agents (HMAs) show benefit in AML rather than in intermediate/high-risk MDS based on genetic mutations in epigenetic modification (EMMs): from a retrospective study
Annals of Hematology (2024)
-
Acute myeloid leukemia: from NGS, through scRNA-seq, to CAR-T. dissect cancer heterogeneity and tailor the treatment
Journal of Experimental & Clinical Cancer Research (2023)
-
Isocitrate dehydrogenase 1 mutation drives leukemogenesis by PDGFRA activation due to insulator disruption in acute myeloid leukemia (AML)
Leukemia (2023)
-
Metabolic determinants of tumour initiation
Nature Reviews Endocrinology (2023)
