
Abstract
Adaptive resistance of myeloma to proteasome inhibition represents a clinical challenge, whose biology is poorly understood. Proteasome mutations were implicated as underlying mechanism, while an alternative hypothesis based on low activation status of the unfolded protein response was recently suggested (IRE1/XBP1-low model). We generated bortezomib- and carfilzomib-adapted, highly resistant multiple myeloma cell clones (AMO-BTZ, AMO-CFZ), which we analyzed in a combined quantitative and functional proteomic approach. We demonstrate that proteasome inhibitor-adapted myeloma cells tolerate subtotal proteasome inhibition, irrespective of a proteasome mutation, and uniformly show an 'IRE1/XBP1-low' signature. Adaptation of myeloma cells to proteasome inhibitors involved quantitative changes in >600 protein species with similar patterns in AMO-BTZ and AMO-CFZ cells: proteins involved in metabolic regulation, redox homeostasis, and protein folding and destruction were upregulated, while apoptosis and transcription/translation were downregulated. The quantitatively most upregulated protein in AMO-CFZ cells was the multidrug resistance protein (MDR1) protein ABCB1, and carfilzomib resistance could be overcome by MDR1 inhibition. We propose a model where proteasome inhibitor-adapted myeloma cells tolerate subtotal proteasome inhibition owing to metabolic adaptations that favor the generation of reducing equivalents, such as NADPH, which is supported by oxidative glycolysis. Proteasome inhibitor resistance may thus be targeted by manipulating the energy and redox metabolism.
Similar content being viewed by others


Introduction
Proteasome inhibition is highly active for the treatment of multiple myeloma (MM).1 Current proteasome-inhibiting drugs comprise the first-in-class, reversible, boronate-type proteasome inhibitor bortezomib and its oral permutation ixazomib and the approved, irreversible, epoxyketone-type inhibitor carfilzomib, as well as next-generation boronate-, epoxyketone- or β-lactone-type of inhibitors.2 Their mechanism of action exploits the highly developed protein biosynthesis machinery of myeloma.3 This extraordinarily active biosynthetic route is controlled by the unfolded protein response (UPR), a complex transcriptional network that balances protein transcription, folding and destruction.4 The IRE1/XBP1 pathway, one of the three key regulatory switches to control UPR activity, also guides plasma cell differentiation.5, 6 MM cells critically rely on timely disposal of misfolded and dysfunctional newly synthesized protein through the endoplasmic reticulum (ER)-associated degradation machinery, of which the proteasome is the rate limiting protease.7 Functional proteasome inhibition disrupts the equilibrium between production and disposal of such protein, which leads to proteotoxic stress and excess activation of the UPR, triggering apoptosis.3
The constitutive proteasome is composed of three pairs of proteolytically active sites (β1c, β2c, β5c) with different substrate specificity.8 Immune cells, including myeloma, may replace these by respective active sites of the immunoproteasome (β1i, β2i, β5i).9, 10 The β5 activity is rate-limiting, and consequently bortezomib and carfilzomib, as well as all synthetic proteasome inhibitors in clinical development, are designed to target β5.2, 11, 12, 13
Proteasome inhibitor resistance of MM is an emerging clinical problem whose biology is poorly understood. Proteasome inhibitor-resistant cell lines generated in vitro by continuous exposure to proteasome-inhibiting drugs serve as models to understand and potentially overcome proteasome inhibitor resistance.14, 15, 16 Mutations in PSMB5 (encoding for β5c) were predicted to lead to impaired inhibitor binding owing to changes in the β5c active site or the inhibitor-binding pocket.14, 17, 18 However, the functional relevance of such mutations on the active site binding of bortezomib or carfilzomib in MM cells has not been demonstrated, and extensive analysis in MM cells derived from patients resistant to proteasome inhibitor therapy failed to identify such PSMB5 mutations.19 Moreover, artificial introduction of mutant PSMB5 in MM cells did not confer bortezomib resistance comparable to bortezomib-selected tumor cells.20 Recently, an alternative biological model for proteasome inhibitor resistance was put forward, supported by respective findings from MM cells of bortezomib-resistant patients. It suggests that bortezomib resistance is the result of changes in the activation status of the UPR, in particular decreased activity of the IRE1/XBP1 axis,21 consistent with high XBP1 being a biomarker for bortezomib sensitivity in the clinic.22
We here dissect the relationship between PSMB5 mutation, proteasome inhibitor target inhibition and resistance to proteasome inhibitor-induced cell death of MM cells. Because our results suggest a complex mechanism of proteasome inhibitor resistance largely independent from either PSMB5 mutations or even significant β5c proteasome activity, we provide a global proteomic comparison of proteasome inhibitor-sensitive vs bortezomib- and carfilzomib-adapted myeloma cells to identify novel potential therapeutic strategies beyond the ubiquitin proteasome pathway.
Methods
Cell culture
The AMO-1 proteasome inhibitor-resistant cell lines (AMO-BTZ and AMO-CFZ) as well as their single clone-derived derivatives were established and maintained from the AMO-1 myeloma cell line by continuous drug exposure for >12 months.15 Additional information is provided in Supplementary Methods.
Relationship between proteasome inhibition and cytotoxicity
Measurement of proteasome activity was performed as described previously.23 Additional information is provided in Supplementary Methods.
Proteome analysis
Briefly, full-cell lysates were digested with trypsin labeled with light (sensitive cells AMO-1) or intermediate (adapted cells) stable formaldehyde isotopes,24 mixed, fractionated by SCX and analyzed by liquid chromatography-tandem mass spectrometry. Each analysis was performed in triplicate. The MaxQuant (Max Planck Institute of Biochemistry, Martinsried, Germany) was used for identification and quantification of the proteins. A cutoff of log2=0.5 was chosen to categorize differentially expressed proteins. The differentially expressed proteins were subjected to a protein–protein interaction analysis with the Cytoscape (The Cytoscape Consortium, San Diego, CA, USA). Gene ontology (GO) analysis was performed with the BinGo (The Cytoscape Consortium).25 Manual curation was carried out to cluster the GO terms in more general groups (see Supplementary Materials for full protocol).
Statistical analysis
If not stated otherwise, results represent means and s.d. from at least three independent experiments. Additional information is provided in Supplementary Methods.
Results
Subtotal functional proteasome inhibition lacks cytotoxicity in proteasome inhibitor-adapted myeloma cells
Sequencing of the active subunits of the constitutive proteasome revealed a heterozygous A310G mutation leading to a Met45Val change in the S1 pocket of the PSMB5 active site in AMO-BTZ bulk cultures as well as in all single cell-derived clones from these cultures (data not shown). Genetic changes in the PSMB5 (β5c) PSMB6 (β1c) and PSMB7 (β2c) genes could be excluded in AMO-CFZ cells. Therefore, all experiments with AMO-BTZ cells reported here were performed with the Met45Val mutation present, while all experiments in AMO-CFZ were performed in the absence of a mutation. The IC50 for β5c during bortezomib treatment was higher in AMO-BTZ, compared with AMO-1 and AMO-CFZ, while the IC50 for β1c was comparable (Figure 1a; Table 1). Bortezomib increased β2 activity in AMO-1 cells but not in AMO-BTZ or AMO-CFZ. Treatment with 250 nm bortezomib, a concentration in the range of peak bortezomib plasma levels in myeloma patients minutes after intravenous bolus administration, induced approximately 70–80% inhibition of active β5c in AMO-1, AMO-BTZ or AMO-CFZ cells. This resulted in >90% cytotoxicity in AMO-1 cells, while viability in AMO-BTZ or AMO-CFZ remained >90%. Apoptosis was confirmed by propidium iodide/Annexin V staining (Supplementary Figure S1). HL60 acute myeloid leukemia cells similarly adapted to bortezomib (HL60-BTZ) and lacking a PSMB5 mutation showed not only similarly effective proteasome β5c/β5i inhibition by bortezomib, compared with non-adapted control cells, but also a lower sensitivity to bortezomib-induced cytotoxicity (data not shown).

Relationship between cytotoxicity and proteasome β-subunit activity. AMO-1 cells and its derivatives adapted to bortezomib (AMO-BTZ) and carfilzomib (AMO-CFZ) have been cultured for 2 weeks without proteasome inhibitors. Then AMO-1, AMO-BTZ and AMO-CFZ were treated for a 1-h pulse with different concentrations of (a) bortezomib (Btz) or (b) carfilzomib (Cfz). Proteasome activity after drug exposure was measured directly using activity-specific, fluorescent chemical probes.23 Cell viability was determined after cells were grown in fresh medium for 48 h following the 1-h drug exposure. Panels represent (in the order from left to right): cell viability determined by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) test, activity of the proteasome β5c/5i, β1c/1i and β2c/2i subunits, respectively, as assessed by activity-based labeling, for each cell type (AMO-1, AMO-BTZ, AMO-CFZ, horizontal rows) and for each proteasome inhibitor (bortezomib, carfilzomib, panels a and b). Data are presented as a mean±s.d. of three independent replicates.
Carfilzomib was more selective for β5c/5i activity over β1c/1i activity and, in addition, had some β2c/2i-inhibiting activity, both in contrast to bortezomib (Figure 1b; Table 1, Supplementary Figure S2). The IC50 values of β2c/2i and β1c/1i were in the same order of magnitude for carfilzomib treatment in AMO-1 and AMO-BTZ. By contrast, the IC50 for β5c was approximately five-fold lower in AMO-1 vs AMO-BTZ cells (10 vs 50 nm), suggesting that the PSMB5 A310G mutation also affects β5 binding of carfilzomib, comparable to bortezomib. Inhibition of β5c by carfilzomib was 9 × less effective in AMO-CFZ, compared with AMO-1, while this difference had a factor of only two for bortezomib. Likewise, inhibition of β1/β1i and β2/β2i by carfilzomib was less effective in AMO-CFZ than in AMO-1. When approximately equally effective proteasome inhibition was achieved by carfilzomib in AMO-1, AMO-BTZ and AMO-CFZ (>90% inhibition of β5c/5i, 20% inhibition of β1/1i and β2/2i, achieved by 250 nm carfilzomib in AMO-1 and AMO-BTZ, and 1000 nm carfilzomib in AMO-CFZ), this resulted in >60% cytotoxicity in AMO-1 cells, in contrast to AMO-BTZ or AMO-CFZ.
Proteasome inhibitor-adapted myeloma cells show low IRE1/XBP1 expression
Protein and mRNA expression for IRE1 was uniformly decreased in AMO-BTZ and AMO-CFZ, (Figure 2). Consistent with this, spliced XBP1 was reduced on the mRNA and protein level, while total XBP1 and unspliced XBP1 were increased. Thus AMO-BTZ and AMO-CFZ both match the 'IRE1/sXBP1-low' expression pattern that was observed in myeloma cells from bortezomib-refractory patients. We did not find changes in the expression of protein or mRNA indicative of an activation of autophagy in AMO-BTZ or AMO-CFZ (Supplementary Figure S3).

Protein and RNA expression for UPR signaling pathway. AMO-1 and its derivatives adapted to bortezomib (AMO-BTZ) and carfilzomib (AMO-CFZ) have been cultured for 2 weeks without proteasome inhibitors before RNA and protein was collected. (a) Level of spliced (s), unspliced (u) and total (t) XBP1 in the cell lines. (b) Presence of activated XBP1 depicted as a ratio of spliced XBP1 vs unspliced XBP1 in the cell lines. (c) Representative western blotting bands for protein levels of UPR signaling proteins in the cell lines. For panels a and b, data are presented as mean±s.d. of three independent replicates. Statistical significance <0.05 is marked by asterisks.
We compared the effects of equi-efficient proteasome inhibition on the activation of the UPR-induced apoptotic machinery (Figure 3). AMO-1, AMO-BTZ and AMO-CFZ cells were pulse-treated for 1 h with bortezomib, followed by western blotting analysis of UPR-related proteins. Loss of expression of the active version of the NRF1 transcription factor that is generated by partial proteolysis through the proteasome26 confirmed functional proteasome inhibition for 8 h in all treated cells, independent from the PSMB5 mutation status. Phosphorylation of IRE1 and expression of spliced XBP1 was triggered within 1–2 h upon bortezomib challenge in all cell lines, and increased expression of PKR-like ER kinase was initiated at later time points. Triggering of the UPR-associated apoptotic machinery was observed in all cells, based on the expression of ATF4 and CHOP and activation of caspase 3, albeit with lower signal intensity in AMO-BTZ. By contrast, protein disulfide isomerase (PDI), a key enzyme for the formation of disulfide bonds and protein folding in the ER, showed low expression in AMO-1 at baseline and was strongly induced by bortezomib, while it was constitutively expressed at considerably higher levels in AMO-BTZ or AMO-CFZ cells without significant bortezomib-induced change. This suggested adaptive changes in the reducing and protein-folding machinery of the ER.

Activation of the UPR-induced apoptotic machinery. AMO-1 derivatives adapted to bortezomib (AMO-BTZ) and carfilzomib (AMO-CFZ) have been cultured for 2 weeks without proteasome inhibitors. Then, together with AMO-1 sensitive cell line, they were treated with 500 nm bortezomib for a 1-h pulse. After the pulse, cells were grown in fresh medium for different chase time points (0.5, 1, 2, 4, 8, 16, 24 h). Directly before the addition of the proteasome inhibitor (presample) and immediately after the pulse (postsample), as well as at each chase time point indicated, an aliquot was harvested for protein isolation, and equal amounts of protein from each sample were subsequently processed for western blotting. Data are presented as representative bands of three independent experiments.
Global quantitative protein expression in proteasome inhibitor-adapted myeloma cells
We chose a proteomic approach to identify the full pattern of proteins with significant changes in the expression levels in AMO-1 vs AMO-BTZ and AMO-CFZ cells. In each experiment, >3500 different proteins were identified, of which >2000 proteins showed consistent peptide identification for both labels and in each triplicate, and were thus used for quantification, resulting in >600 protein species identified with statistically significant changes in the expression levels in proteasome inhibitor-adapted cells, compared with the non-adapted controls (three independent replicates, P<0.01, Table 2, Supplementary Figure S4). Interestingly, in the bortezomib-adapted leukemia cell line HL60-BTZ, only 300 proteins showed significantly changed expression levels, although a similar total number of proteins was identified and quantified as in the myeloma cell lines.
The differentially expressed proteins were subjected to a protein–protein interaction search to build a protein–protein interaction network (Supplementary Figure S5). This resulted in a complex network of changes in proteasome inhibitor-adapted cells. Differentially expressed proteins were therefore subjected to a Gene Ontology (GO) analysis, resulting in an average of 30 GO terms for the overexpressed proteins and 10 for the downregulated species in all the three adapted clones (Supplementary Table S1). GO terms involving the ubiquitin proteasome pathway were enriched in all samples, in agreement with proteasome overexpression. GO terms were manually grouped into functionally related clusters resulting in 5–6 groups of differentially expressed proteins (Table 2; see also Supplementary Tables S2–S7 for individual upregulated/downregulated proteins per GO pathway). This supervised clustering showed some general concordance between the adapted cells, suggesting that acquired resistance to proteasome inhibitors is characterized by a complex, but characteristic pattern, of changes in protein expression.
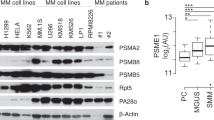
The quantitatively largest group with altered expression levels was a group of proteins involved in metabolic regulation, comprising close to 50% of all polypeptide species with expression changes identified. This suggested that metabolic homeostasis is a major challenge for myeloma cells adapted to proteasome inhibitor treatment. Upregulated proteins included proteins involved in the respiratory chain (for example, CYC1 or UQCR1), the generation of metabolites (for example, BLVRA or APOA1BP), glycolysis (for example, PFKP or PKM) and amino acid (for example, GOT1 or EEFSEC) or nucleic acid metabolism (for example, UMPS or BOP1). The metabolic regulation category also contained proteins (30%) that were significantly downregulated, in particular proteins involved in lipid and cofactor metabolism (for example, ASCT2 or ACLY). Of the top 25 quantitatively downregulated proteins in this cluster, 12 were mitochondrial proteins involved in fatty acid biosynthesis (ACSF2, ACSF3) in AMO-CFZ, and likewise five mitochondrial proteins with similar function were among the top 25 downregulated in AMO-BTZ. Proteins involved in protein catabolism, redox control and protein folding were also overexpressed. This included all proteasome alpha (PSMA 1,2,3,4&7) and beta (PSMB 1,2,3,4,5&7) subunits and corroborated the validity of our analysis. Proteins involved in antioxidant and reactive oxygen species functions (NQO1, PRDX1, SOD1) were also increased in adapted cells, as well as proteins involved in glutathione regulation. Different individual proteins were upregulated in a given functional pathway between AMO-BTZ and AMO-CFZ (for example, GPX1, one of the major enzymes responsible for glutathione peroxidation and thus removal of oxygen peroxide, was only overexpressed in AMO-BTZ, but not in AMO-CFZ, while glutathione transferase 1 (MGST1), which acts by conjugating reduced glutathione to a wide number of exogenous and endogenous hydrophobic electrophiles, was only found upregulated in AMO-CFZ, of which a variant (MGST3) was found upregulated in AMO-BTZ). However, the upregulation of proteins involved in redox pathways overall showed a very consistent pattern between AMO-CFZ and AMO-BTZ cells. Of the 16 proteins significantly upregulated in this cluster in AMO-CFZ cells, 12 were likewise found upregulated in AMO-BTZ cells, suggesting a central role of the redox equilibrium. The protein folding/chaperoning capacity of the adapted cells was also markedly increased: the heat-shock proteins HSP70, HSP90 and HSP105 were consistently among the top 15 most strongly upregulated proteins (2.3–13-fold increase) among proteins involved in folding/chaperoning in both adapted cell clones.
A pattern of downregulation was observed for protein clusters involved in transcription/translation, differentiation, apoptosis and structural/cytoskeletal functions. Transcription and translation regulation proteins comprised about one-fourth of the total downregulated protein species. The apoptosis protein cluster comprised the lowest number of proteins, of which both adapted cells markedly downregulated individual key proteins (decreased BAX in AMO-CFZ, decreased CASP10 and DIABLO in AMO-BTZ). Downmodulation of cytoskeleton and cell differentiation included IKZF3, a transcription factor implicated in lymphocyte differentiation and activity of immunomodulatory myeloma drugs. AMO-BTZ cells were significantly more sensitive to lenalidomide, consistent with a role of IKZF3 levels, while AMO-CFZ were lenalidomide refractory, potentially explained by their high ABCB1 expression (Supplementary Figure S6A).
Upregulation of MDR1 and oxidative glycolysis functionally contribute to resistance
ABCB1, the MDR1, was 12-fold upregulated only in AMO-CFZ on the protein and 120-fold by gene expression analysis (data not shown). Likewise, we observed >100 × increased ABCB1 gene expression in carfilzomib-adapted ARH-77 plasma cell leukemia cells (ARH-CFZ) and primary malignant plasma cell samples from a myeloma patient who initially responded to carfilzomib treatment and later showed progressive disease under carfilzomib. The primary patient sample showed more than four-fold upregulated ABCB1 expression upon progression, compared with the baseline (Supplementary Figure S7). The addition of the functional MDR1 inhibitors verapamil and reserpine significantly decreased the LD50 for CFZ-treated AMO-CFZ cells, in contrast to AMO-BTZ cells or AMO-CFZ cells treated with bortezomib, suggesting a high functional significance of ABCB1 overexpression for CFZ resistance (Figure 4 and Supplementary Figure S8). The sensitivity of AMO-CFZ against cyclophosphamide or daunorubicin was unchanged (Supplementary Figures S6C and D), while AMO-BTZ and AMO-CFZ showed decreased sensitivity for panobinostat, consistent with the role of HDAC6 in non-proteasomal protein degradation (Supplementary Figure S6B).

Cytotoxic effect of specific ABCB1 inhibitor verapamil. The AMO-1 cell line and its derivatives adapted to bortezomib (AMO-BTZ) and carfilzomib (AMO-CFZ) have been cultured in the presence of 5 μm verapamil and bortezomib/carfilzomib (Btz/Cfz) for 5 days and viability was assessed by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) test. Absorbance values of treated cells were normalized to the controls without specific inhibitors. Data are presented as a mean±s.d. of three independent replicates.
Oxidative glycolysis is upregulated in proteasome inhibitor-resistant myeloma cells
NADPH dehydrogenase, the quantitatively most important oxidoreductase in eukaryotic cells, was overexpressed 4–6-fold in AMO-BTZ and AMO-CFZ, respectively, and in addition the enzymes that generate NADPH (malate dehydrogenase and enzymes of the oxidative branch of the pentose phosphate pathway (PPP)) were found overexpressed, implicating that increased production of reducing equivalents may serve as a metabolic adaptive response to render cells more proteasome inhibition resistant. Glycolysis can contribute significantly to the synthesis of reducing equivalents via NAPDH (via PPP and mitochondrial pyruvate–malate cycle) and NADH (glycolysis and TCA cycle), which are crucial for maintenance of redox balance and neutralization of reactive oxygen species. Indeed, we observed that the AMO-BTZ and AMO-CFZ cells displayed higher baseline and maximum glycolytic rates than their parental cell line (Figure 5a) as determined by real-time metabolic flux analysis, a finding that is supported by the increased expression of several glycolytic enzymes (Supplementary Tables S2 and S4). Consistent with these findings, high glucose medium decreases the proteasome inhibitor sensitivity of AMO-1 cells, in contrast to AMO-CFZ or AMO-BTZ (Supplementary Figure S9). Malignant cells often display increased glycolytic rates with lactate as end product despite the availability of ample oxygen (Warburg Effect), a phenomenon thought to reflect a metabolic adaptation to maintain cellular ATP levels in the face of often dysfunctional mitochondria. In agreement with the increased expression of several proteins of the electron transport chain in AMO-BTZ and AMO-CFZ cells (Supplementary Tables S2 and S4), we found, however, that these cells showed higher baseline and maximum mitochondrial respiration than the AMO-1 cell line (Figure 5b), indicative of enhanced mitochondrial activity and health. This suggests that the enhanced glycolytic flux in these cells is unlikely merely a bioenergetic adaptation to impaired ox-phos but may have a role in fueling the PPP and TCA to, among other things, aid in the generation of reducing equivalents. To further test this concept, we used FX11, a specific inhibitor of lactate dehydrogenase A that catalyzes the conversion of pyruvate into lactate (Figure 5c), thereby instead promoting the oxidation of pyruvate in the mitochondria. Indeed, this selective block of aerobic glycolysis considerably increased the LD50 for bortezomib in AMO-1, AMO-BTZ and AMO-CFZ cells. This would be in support of the notion that the increased glycolysis in AMO-BTZ and AMO-CFZ cells is not ATP-driven but may rather serve to fuel mitochondrial metabolism and the PPP that could help in the generation of reducing power to mitigate the deleterious effects of proteasome inhibition.

Role of glycolysis in proteasome inhibitor resistance. (a, b) The AMO-1 cell line and its derivatives adapted to bortezomib (AMO-BTZ) and carfilzomib (AMO-CFZ) were seeded in a 96-well XF Extracellular Flux Analyzer to simultaneously quantify (a) extracellular acidification rates (ECAR) and (b) oxygen consumption rates (OCR) as measures of glycolysis and mitochondrial respiration, respectively. (a) Real-time baseline glycolytic rate of the different cell lines was determined by subtracting the difference in levels in ECAR before and after injection of 10 mm glucose. Reserve glycolytic capacity was calculated by subtracting baseline ECAR readings from ECAR values following treatment with ETC blockers antimycin A/rotenone. (b) Real-time OCR was determined during sequential treatments with oligomycin (ATP-synthase inhibitor), FCCP (uncoupler) and antimycin-A/rotenone (ETC inhibitors). Maximum mitochondrial respiration is reached following treatment with FCCP that leads to uncoupling of the mitochondria. Data represent means±s.e.m. of eight replicates. One representative experiment of three is shown. (c) The AMO-1 cell line and its derivatives adapted to bortezomib (AMO-BTZ) and carfilzomib (AMO-CFZ) have been cultured in the presence of 40 μm FX11, and bortezomib (Btz) for 5 days, and viability was assessed by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) test. Absorbance values of treated cells were normalized to the controls without specific inhibitors. Data are presented as a mean±s.d. of three independent replicates.
Discussion
Proteasome PSMB5 gene mutations are believed to explain clinical proteasome inhibitor resistance, although attempts have failed to identify them in patient-derived material. This 'mutation model' was challenged when downmodulation of IRE1/sXBP1 was shown to result in proteasome inhibitor resistance while 'IRE1/XBP1-low' myeloma cells accumulate in bortezomib-resistant patients. We demonstrate that proteasome inhibitor-adapted myeloma cells tolerate near-complete inhibition of proteasome activity, irrespective of the presence of a PSMB5 mutation. Furthermore, AMO-BTZ and AMO-CFZ showed an 'IRE1/XBP1-low' pattern of UPR activation, independently from proteasome mutations. These data argue against individual PSMB5 mutations as key factor for proteasome inhibitor resistance and support the 'IRE1/XBP1-low' model. They also suggest that proteasomal proteolysis is no longer the 'Achilles’ heel' of proteasome inhibitor-resistant myeloma but that its functionality is almost dispensable.
Because our findings suggested a complex biology of proteasome inhibitor resistance, we undertook the first quantitative proteome analysis of proteasome inhibitor-resistant myeloma. Our results show that adaptation of myeloma to proteasome inhibitors involves complex changes in hundreds of proteins. The gross pattern observed was similar between AMO-BTZ and AMO-CFZ cells and may be understood as characteristic adaptive response to proteasome inhibition, balancing the equilibrium between proteasomal load and capacity at a new functional level of very low proteasome activity. Although different proteins were involved in the adaptation process between both cell lines, the pattern of changes in particular included proteins involved in energy homeostasis or the production, folding or destruction of protein. Our data are consistent with the observation that proteasome overexpression mediated by nuclear factor 2 (NRF2) can contribute to adaptive resistance to proteasome inhibitors,27 although in our model NRF2 was not upregulated (data not shown).
To our knowledge, while all bortezomib-adapted myeloma cells with a biologically meaningful resistance factor >5 × that tolerate high (100 nm) bortezomib concentrations contain a PSMB5 mutation,28 carfilzomib-resistant myeloma cells have not yet been reported. AMO-CFZ cells described here lack a mutation in the PSMB5, PSMB6 or PSMB7 genes, have a resistance factor of approximately >20 × and >7 × for carfilzomib and bortezomib, respectively, and tolerate very high concentrations of each of the proteasome-inhibiting drugs, so that they represent the first comprehensive cell line model for proteasome inhibitor resistance and at the same time mirror the mutation status as well as the 'IRE1/XBP1-low' biology found in resistant patients. The A310G mutation at Met45Val directly affects the S1-binding pocket in the β5 substrate channel and significantly impairs chymotryptic proteasome activity and binding to bortezomib or carfilzomib.17 We demonstrate that the heterozygous Met45Val mutation results in 3–5-fold lower β5-inhibiting activity of bortezomib and carfilzomib in human myeloma cells.
The MDR ABCB1 was the most significantly upregulated protein in AMO-CFZ cells. MDR-mediated resistance to carfilzomib has been observed in solid tumor cells,29 in contrast to bortezomib, and we did not observe ABCB1 upregulation in AMO-BTZ cells. The increased IC50 for inhibition of all individual proteasome subunits in AMO-CFZ cells after carfilzomib treatment is consistent with drug export. Indeed, co-treatment of AMO-CFZ with carfilzomib and MDR inhibitors decreased carfilzomib resistance.
Given the redundancy of signaling and the genetic architecture of advanced myeloma, targeting individual signaling proteins may lack significant activity in proteasome inhibitor-refractory myeloma. However, most of these patients still have measurable paraprotein levels, indicating that bortezomib-adapted myeloma cells in vivo usually continue to produce high amounts of secretory protein. Our results suggest that such myeloma cells may secure continued paraprotein production by entertaining a highly optimized machinery of protein folding via increased chaperone expression and stringent redox conditions, thus minimizing defective folding that would otherwise increase proteasomal load. Indeed, AMO-CFZ maintained fully functional IgA secretion levels, whereas paraprotein secretion was absent from AMO-BTZ (Supplementary Figure S10). Adequate supply of energy and oxidizing equivalents are key requisites toward proper redox conditions. Tumor cells often use aerobe glycolysis instead of oxidative generation of ATP, called 'Warburg Effect'. However, the oxidative glucose turnover is also one of the major providers of NADPH, the most important oxidizing agent, regenerated from NADP when malate from the Krebs cycle is converted into pyruvate. Indeed, several of our results suggest that the metabolism of proteasome inhibitor-resistant myeloma cells has been geared to provide NADPH for reducing activity with high efficiency: NADPH dehydrogenase, the most important oxidoreductase in eukaryotes was overexpressed 4–6-fold in AMO-BTZ and AMO-CFZ cells, similarly to malate dehydrogenase and enzymes of the PPP, the most important providers of NADPH. At the same time, several enzymes involved in synthesis of fatty acids and lipids from pyruvate/citrate backbones in the mitochondria via Acetyl-CoA were among the top downregulated proteins in both AMO-CFZ and AMO-BTZ cells, so that mitochondrial pyruvate is, to a smaller fraction, used as building blocks for lipid biosynthesis and can, to a larger fraction, undergo oxidative glycolysis. These characteristics suggest a quantitative adaptation in the flow of metabolic intermediates in proteasome inhibitor-resistant myeloma cells, optimizing for the provision of energy and oxidizing equivalents via oxidative glycolysis (see proposed metabolic model of proteasome inhibitor-adapted myeloma cells, Figure 6). Proteasome inhibitor-resistant myeloma may thus be sensitive to the manipulation key metabolic processes, that is, oxidation of glucose, or the generation of NADPH and/or ATP. A comparison of the metabolomes between AMO-1, AMO-BTZ and AMO-CFZ is currently underway.

Model of redox homeostasis and metabolic changes during the adaptation to proteasome inhibitors. Excessive proteotoxic stress is the major mechanism of cytotoxicity of proteasome inhibitors in myeloma. The level of proteotoxic stress results from the degree of imbalance between proteasomal load and proteasomal proteolytic capacity. The proteasomal load of misfolded protein is tightly connected to the protein-folding capacity of the ER, which in turn relies on proper redox conditions. NAD(P)H is critical for providing oxidizing equivalents and thus for minimizing the proteasomal load in proteasome inhibitor-adapted myeloma cells that are viable with extremely low proteasome activity while producing high paraprotein amounts. NAD(P)H is mostly provided from intermediates of the TCA (Krebs Cycle), for example, malate–pyruvate and mitochondrial isocitrate dehydrogenase reactions, which we found consistently upregulated in proteasome inhibitor-adapted myeloma cells. In line with this, multiple other enzymes that are involved in NAD(P)H generation were upregulated, while enzymes mediating lipid biosynthesis from Acetyl-CoA, which removes tricarboxylic acid intermediates from the Krebs Cycle and thus decreases NAD(P)H generation, are downregulated. This way, we hypothesize that maximum NAD(P)H-producing intermediates may be provided, limiting proteotoxic stress in adapted/resistant cells. Cancer cells often use the faster, but less efficient, pathway for ATP production to generate ATP via pyruvate–lactate conversion (Warburg effect). When this is inhibited using the specific inhibitor of LDHA, FX-11, the sensitivity of myeloma cells to proteasome inhibitors decreases, presumably because glucose metabolism entirely switches to the TCA cycle, increasing NAD(P)H production and reducing proteotoxic stress. Upregulation of ABCB1 decreases proteotoxic stress via reduced intracellular concentrations of proteasome inhibitors, which can be reversed by specific ABCB1 inhibitors Verapamil or Reserpine. ABCB1, ATP-binding cassette, sub-family B (MDR/TAP) member 1; ACLY, ATP Citrate Lyase; ACSF2, Acyl-CoA synthetase family member 2; ACSF3, Acyl-CoA synthetase family member 3; ARE, antioxidant response elements; BVLRA, biliverdin reductase; CoQ, coenzyme Q; CoQH2, coenzyme Q-reduced; CytC, cytochrome C; GSSG, oxidized glutathione; GSH, reduced glutathione; GOT1, glutamic-oxaloacetic transaminase 1; GPX, glutathione peroxidase; LDHA, lactate dehydrogenase; NQO1, NAD[P]H dehydrogenase, quinone1; NRF2, nuclear factor (erythroid-derived 2)-like 2; PDI, protein disulfide isomerase; PEP, phosphoenolpyruvate; PI, proteasome inhibitors; PKM, pyruvate kinase, muscle; PRDX, peroxiredoxin; ROS, reactive oxygen species; SOD, superoxide dismutase; UQ, ubiquinone.
We document that proteasome activity is largely dispensable in bortezomib- or carfilzomib-adapted myeloma cells, suggesting that nonproteasomal targets are required to overcome proteasome inhibitor resistance. PSMB5 mutations are likewise not required for high-level proteasome inhibitor resistance of myeloma, while an important role of the 'IRE1/XBP1-low' activation status of the UPR is supported. Our full proteomic analysis of proteasome inhibitor-resistant myeloma cells reveals a complex adaptation pattern that in particular involves changes in glycolysis, protein folding and redox function. Our data suggest exploring these metabolic pathways in proteasome inhibitor-resistant myeloma.
References
Adams J . The proteasome: a suitable antineoplastic target. Nat Rev Cancer 2004; 4: 349–360.
Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012; 120: 947–959.
Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH . Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107: 4907–4916.
Walter P, Ron D . The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334: 1081–1086.
Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001; 412: 300–307.
Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL . High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol 2012; 189: 3328–3338.
Mayor T . Navigating the ERAD interaction network. Nat Cell Biol 2011; 14: 46–47.
Ciechanover A . Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 2005; 6: 79–87.
Singh AV, Bandi M, Aujay MA, Kirk CJ, Hark DE, Raje N et al. PR-924, a selective inhibitor of the immunoproteasome subunit LMP-7, blocks multiple myeloma cell growth both in vitro and in vivo. Br J Haematol 2011; 152: 155–163.
Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M et al. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012; 148: 727–738.
Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC et al. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat Methods 2005; 2: 357–362.
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007; 67: 6383–6391.
Huber EM, Groll M . Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew Chem Int Ed Engl 2012; 51: 8708–8720.
Franke NE, Niewerth D, Assaraf YG, van MJ, Vojtekova K, van Zantwijk CH et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012; 26: 757–768.
Ruckrich T, Kraus M, Gogel J, Beck A, Ovaa H, Verdoes M et al. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009; 23: 1098–1105.
Tornatore L, Sandomenico A, Raimondo D, Low C, Rocci A, Tralau-Stewart C et al. Cancer-selective targeting of the NF-kappaB survival pathway with GADD45beta/MKK7 inhibitors. Cancer Cell 2014; 26: 495–508.
Huber EM, Heinemeyer W, Groll M . Bortezomib-resistant mutant proteasomes: structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015; 23: 407–417.
Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Z I, Berkers CR et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008; 112: 2489–2499.
Lichter DI, Danaee H, Pickard MD, Tayber O, Sintchak M, Shi H et al. Sequence analysis of beta-subunit genes of the 20 S proteasome in patients with relapsed multiple myeloma treated with bortezomib or dexamethasone. Blood 2012; 120: 4513–4516.
Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A et al. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010; 24: 1506–1512.
Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE et al. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013; 24: 289–304.
Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, Iland H et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 2012; 97: 64–72.
Li N, Kuo CL, Paniagua G, van den Elst H, Verdoes M, Willems LI et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nat Protoc 2013; 8: 1155–1168.
Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ . Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 2009; 4: 484–494.
Maere S, Heymans K, Kuiper M . BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005; 21: 3448–3449.
Sha Z, Goldberg AL . Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr Biol 2014; 24: 1573–1583.
Li B, Fu J, Chen P, Ge X, Li Y, Kuiatse I et al. The nuclear factor (erythroid-derived 2)-like 2 and proteasome maturation protein axis mediates bortezomib resistance in multiple myeloma. J Biol Chem 2015; 290: 29854–29868.
Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J . Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat 2015; 18: 18–35.
Ao L, Wu Y, Kim D, Jang ER, Kim K, Lee DM et al. Development of peptide-based reversing agents for p-glycoprotein-mediated resistance to carfilzomib. Mol Pharm 2012; 9: 2197–2205.
Acknowledgements
We thank Alexander Kisselev, Dartmouth College and Arun P Wiita, UCSF for critical reading of the manuscript. This work was supported by the Swiss National Research Foundation SNF (grant 31003A_143924/1 to CD).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Soriano, G., Besse, L., Li, N. et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 30, 2198–2207 (2016). https://doi.org/10.1038/leu.2016.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.102
This article is cited by
-
The cytotoxic activity of carfilzomib together with nelfinavir is superior to the bortezomib/nelfinavir combination in non-small cell lung carcinoma
Scientific Reports (2023)
-
Lipid metabolic vulnerabilities of multiple myeloma
Clinical and Experimental Medicine (2023)
-
Mitochondrial dysfunction and drug targets in multiple myeloma
Journal of Cancer Research and Clinical Oncology (2023)
-
A clinically relevant pulse treatment generates a bortezomib-resistant myeloma cell line that lacks proteasome mutations and is sensitive to Bcl-2 inhibitor venetoclax
Scientific Reports (2022)
-
Clonal evolution after treatment pressure in multiple myeloma: heterogenous genomic aberrations and transcriptomic convergence
Leukemia (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}