
Abstract
PTEN gene inactivation by mutation or deletion is common in pediatric T-cell acute lymphoblastic leukemia (T-ALL), but the impact on outcome is unclear, particularly in patients with NOTCH1/FBXW7 mutations. We screened samples from 145 patients treated on the MRC UKALL2003 trial for PTEN mutations using heteroduplex analysis and gene deletions using single nucleotide polymorphism arrays, and related genotype to response to therapy and long-term outcome. PTEN loss-of-function mutations/gene deletions were detected in 22% (PTENABN). Quantification of mutant level indicated that 67% of mutated cases harbored more than one mutant, with up to four mutants detected, consistent with the presence of multiple leukemic sub-clones. Overall, 41% of PTENABN cases were considered to have biallelic abnormalities (mutation and/or deletion) with complete loss of PTEN in a proportion of cells. In addition, 9% of cases had N- or K-RAS mutations. Neither PTEN nor RAS genotype significantly impacted on response to therapy or long-term outcome, irrespective of mutant level, and there was no evidence that they changed the highly favorable outcome of patients with double NOTCH1/FBXW7 mutations. These results indicate that, for pediatric patients treated according to current protocols, routine screening for PTEN or RAS abnormalities at diagnosis is not warranted to further refine risk stratification.
Similar content being viewed by others

Introduction
Approximately 25% of pediatric patients with T-cell acute lymphoblastic leukemia (T-ALL) are likely to relapse with recurrent disease, and this presents a clinical challenge as post-relapse prognosis is poor.1 A number of genetic abnormalities, including mutations and copy number changes, have been identified at disease presentation,2 and evidence suggests that most leukemic blast cells at relapse are clonally related to those present at diagnosis.3, 4 This holds out promise, therefore, that molecular profiling at diagnosis and identification of factors that will predict for relapse could improve outcome by facilitating targeted therapy.5 NOTCH1 and FBXW7 gene mutations are frequent in both pediatric and adult T-ALL, but although some studies have demonstrated that their presence is associated with a favorable outcome,6, 7, 8 others found no significant effect.9, 10, 11 However, recent studies have suggested that the impact of NOTCH1/FBXW7 mutations may be modulated by the presence of coincident abnormalities such as in the phosphatase and tensin homolog (PTEN) and RAS genes,12, 13, 14 and this implies that these genes should routinely be characterized at diagnosis.
PTEN is the main negative regulator of the PI3K/AKT signaling pathway and is implicated in regulating downstream effects of NOTCH1 signaling such as proliferation and survival of T-cell progenitors.15 Loss-of-function PTEN mutations leading to constitutive activation of AKT were identified in T-ALL cell lines that were resistant to NOTCH1 inhibition with γ-secretase inhibitors,16 and a number of studies have now described PTEN loss through mutation and/or genomic deletion in up to 35% of pediatric patients with T-ALL,12, 13, 14, 17, 18, 19, 20, 21 although few studies have reported both mutational and copy number status.12, 14, 18, 21 The mutations are predominantly clustered in exon 7 and are usually frameshift indels that are predicted to lead to truncated proteins that retain the 5’ phosphatase domain but are likely to be unstable.22 The genomic deletions generally encompass the whole gene, although microdeletions have recently been reported that result from illegitimate RAG-mediated recombination events.23 In addition, the abnormalities can be either monoallelic or biallelic for mutation and/or deletion. RAS is also an important regulator of proliferation through the RAS/RAF/MEK/ERK pathway, and Kras-induced mouse models of T-cell leukemia/lymphoma show a strong association with the development of NOTCH1 mutations.24, 25 Activating N- and K-RAS mutations have been reported in 4–10% T-ALL patients, particularly in early T-cell precursor ALL.14, 21, 26, 27
These studies suggest that PTEN and RAS abnormalities in T-ALL may be suitable candidates as biomarkers and assist in directing targeted therapy, but their prognostic impact in pediatric patients is unclear, particularly in relation to their NOTCH1/FBXW7 genotype. In general, PTEN abnormalities in pediatric patients are associated with non-significantly worse outcome,12, 13, 23 although in adult patients, the impact was significantly adverse.14 However, whereas one study of pediatric patients found that a trend for adverse impact was not seen in the presence of a NOTCH1 mutation,13 the study of adult patients reported that presence of a PTEN or RAS abnormality ablated the benefit of a NOTCH1 mutation.14 RAS mutations had no impact on clinical outcome in a study of pediatric ALL, but no data were given for the different subgroups.26
In our study of 162 patients treated on the Medical Research Council UKALL 2003 trial, we reported that patients with double NOTCH1 or NOTCH1 and FBXW7 mutations (NOTCH1±FBXW7Double) have a very good outcome and should not be considered for more intensive therapy in first remission, even if slow responders or minimal residual disease (MRD) positive after induction therapy.8 To determine whether PTEN and RAS abnormalities impacted on this good outcome, and whether they could refine stratification of cases with single NOTCH1 mutations (NOTCH1Single) or wild-type NOTCH1 (NOTCH1WT), we investigated PTEN and RAS genotype in samples from 145 patients treated on this trial and evaluated their impact on outcome in the different subgroups.
Materials and methods
Patients and treatment protocol
Diagnostic samples were available from 145 of the 388 (42%) T-ALL patients entered into the UKALL2003 trial, excluding those with bi-phenotypic leukemia or T-cell lymphoma. Ethical approval for the trial was obtained from the Scottish Multi-Centre Research Ethics Committee and informed consent in accordance with the Declaration of Helsinki. The trial opened to patients aged 1–18 years in October 2003. The upper age limit was increased to 20 in September 2006 and 25 in June 2008. Median age of the patients studied was 9 years (range 1–23), 19 were more than 16 years, and only 5 (3%) were more than 18; 111 were male and 34 female. Median follow-up was 6 years 9 months (range, 1–9.8 years).
The trial is registered at http://www.controlled-trials.com under ISRCTN number 07355119. Details of the trial protocol have been published elsewhere,28 and an outline is given in Supplementary Figure 1. Patients with National Cancer Institute (NCI) low-risk score at trial entry (age 1–10 years and white blood cell count <50 × 109/l) were allocated to regimen A, those with high-risk score (age >10 years and/or white blood cell count >50 × 109/l) to regimen B. Patients with a slow early response (defined as >25% bone marrow blasts at day 8 or 15 for high- and low-risk patients, respectively) and <16 years of age were assigned to regimen C. At day 29, patients not in morphological remission but with <25% blasts were also reallocated to regimen C; those with resistant disease (>25% blasts) were taken off protocol and those with a rapid early response in morphological remission were assessed for MRD using quantitative polymerase chain reaction (PCR) analysis of leukemia clone-specific rearranged T-cell receptor γ and δ genes.29 If MRD of more than 1 leukemic cell in 1000 cells (>10−3) was present, patients were classified as MRD-positive and randomized to either remain on their assigned regimen (A or B) or to be reallocated to regimen C. Of the 145 patients included in this study, 13 patients were treated on regimen A, 83 on B and 49 on C.
Mutation screening and mutant quantification
Full experimental details are given in the Supplementary Data. The entire coding sequence of the PTEN gene (exons 1–9) and exons 2 and 3 of the N-RAS and K-RAS genes were screened by PCR of genomic DNA and heteroduplex analysis on the WAVE DNA Fragment Analysis System (Transgenomic, Glasgow, UK). Samples with abnormal chromatograms were sequenced. Where this showed either multiple or low-level mutations, amplicons were cloned using the TOPO TA cloning kit (Invitrogen, Paisley, UK) and sequenced. Exon 7 mutants were sized and quantified by fragment analysis of fluorescently labeled PCR products using both genomic DNA where available and whole genome amplified DNA (see Supplementary Data). Mutant level was expressed as a percentage of total PTEN alleles. Cases with <5% total mutant were scored as wild type (WT).
CytoSNP-850k SNP array analysis
Whole genome amplified DNA was used to determine genome-wide copy number on the Infinium CytoSNP-850k Beadchip array (Illumina, Essex, UK) according to the manufacturer’s protocols. The data were analyzed using the Genotyping Module of the GenomeStudio software to calculate log R intensities and B-allele frequencies for each of the 230 single nucleotide polymorphism (SNP) markers covering the PTEN gene (chromosome 10: 89612850-89721667). Each sample was independently assessed for PTEN copy number changes by two individuals and scored as WT, heterozygous deletion or homozygous deletion.
SNP allele quantification
Relative allele levels for the A/G SNP rs1903858 in intron 1–2 and the T/G SNP rs555895 in intron 8–9 were quantified using allele-specific restriction enzyme digestion and fragment analysis of fluorescently labeled PCR products (see Supplementary Data). A normal range using samples from 20 SNP-positive hematologically normal controls was first established; the mean allele A% for rs1903858 was 53±2% (range, 50–58%), and the mean allele T% for rs555895 was 53±2% (range, 49–57%). Any deviation from this range was considered to indicate loss of genomic material.
Type 1 microdeletions
To screen for type 1 microdeletions as reported by Mendes et al.,23 PCR was performed with primers in introns 1–2 and 3–4 just outside of the breakpoint junction (see Supplementary Data). Resulting products were sequenced.
Cytogenetic and fluorescence in situ hybridization analysis
Cytogenetic and fluorescence in situ hybridization analyses were performed as previously described.7
Statistical analysis
Kaplan–Meier curves were used to assess overall survival (OS; the time from randomization to death), event-free survival (the time to relapse, secondary tumor or death, whichever came first) and relapse-free survival (the time to relapse or ALL death in those who achieved remission). Patients who did not experience an event were censored at the date last seen. Differences between groups were compared using the log-rank test. All P-values quoted are two-sided. Analyses were performed using Stata version 12.1 (Stata Corp, College Station, TX, USA).
Results
PTEN mutation analysis
Mutation screening
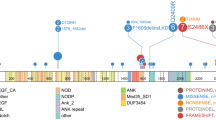
A total of 44 PTEN mutations were identified in 21 of the 145 patients investigated (14%, PTENMUT), 40 in exon 7, 2 in exon 6 and 2 in exon 5 (Supplementary Table 1). Nineteen cases (90%) had exon 7 mutations, two of which also had exon 6 mutations, and two (10%) had just exon 5 mutations (Figure 1a). The mutations included insertions, deletions and indels, with overall size changes ranging between 1 and 20 bps. In 34 (77%), the size change would cause a frameshift, and 10 (23%) were in-frame. Of the 39 with available sequence, 31 (79%) led to truncation of the C-terminus through a nonsense mutation or introduction of a premature stop codon, 8 (21%) were in-frame and non-truncating. Of note, of the 21 mutated cases, only 7 (33%) had a single mutant; 8 (38%) had two mutants, and 3 cases (14%) each had three or four mutants (Figure 1b).

Characteristics of PTEN mutations detected. (a) Schematic representation of the location and type of mutations detected for the 39 mutants with available sequence. (b) Quantification of mutant level by fragment analysis, showing cases with 1–4 mutants. The level is given as a percentage of total alleles. (c) Distribution of total PTEN mutant level detected in mutant-positive patients. Del, deletion; ins, insertion.
Mutant level quantification
Exon 7 mutant levels were quantified using both genomic DNA and whole genome amplified DNA; highly comparable results were obtained (Supplementary Figure 2). The mean mutant level for individual mutants was 20% (range, 2–48%). For the exon 5 and 6 mutants, an estimated mutant level was determined from the sequence peak heights. The median total mutant level for the 21 PTENMUT cases was 48% (range, 10–96%); 5 cases (24%) had ⩽25% total mutant, 6 (29%) had 26–50% and 10 (47%) had >50% (Figure 1c). Where multiple mutations were detected, it was not possible to determine whether these mutants were in the same or different cell populations.
PTEN copy number analysis
SNP arrays
Interpretable results were obtained from Illumina CytoSNP-850k SNP array analysis of 139 samples. Genomic loss of the PTEN gene was detected in 14 patients (10%) and scored as heterozygous loss in 11 cases and homozygous loss in 3 cases (Supplementary Figure 3). A further four cases had evidence of amplification of the PTEN gene, but as they did not demonstrate loss-of-function and the consequence of the amplification was not known, they were scored as WT for the purposes of this study. In all cases, the deletion looked to encompass the whole gene.
SNP allele quantification
In 72 of the 145 samples (50%), it was possible to validate the array data and quantify the level of heterozygous loss by measuring the relative allele levels of two common intronic SNPs in introns 1–2 (rs1903858) and 8–9 (rs555895) (Figure 2). Consistent SNP array and SNP allele quantification results were obtained in 70 cases (97%), including 64 WT, 3 with amplification and 3 with heterozygous deletion. However, in two cases that were initially scored as WT on the array, SNP allele quantification indicated loss at the 3’ end of the gene. The deletions were confirmed by re-evaluation of the array data. SNP allele quantification data were also obtained on four cases without array data, three were WT and one had a heterozygous deletion. Of the six quantifiable cases with genomic loss, the SNP allele ratios were consistent with heterozygous deletion in at least half of the cells (range, 46–94%) (Figure 2, Supplementary Table 1).

Representative SNP allele quantification at loci in PTEN intron 1–2 (rs1903858) and intron 8–9 (rs555895) in samples from a normal control and two patients with heterozygous loss.
Type 1 microdeletions
Type 1 microdeletions, removing a 65 kb region encompassing exons 2 and 3,21 were detected in four of the 145 patients (3%) and had not been detected by the SNP array. However, all four patients harbored other PTEN abnormalities. One had a homozygous deletion, one a heterozygous deletion and two had mutations with <20% total mutant level (Supplementary Table 1). Although it was not possible to determine the level of the microdeletions, in each case, the intensity of the PCR product was more likely to indicate their presence in minor sub-clones.
Combined PTEN genotype
Overall, 143 patients had PTEN mutation and deletion status; deletion status was not available for the two remaining patients but both were PTENMUT. In total, therefore, 32 patients (22%) had PTEN abnormalities (PTENABN), comprising 17 PTENMUT (53% of PTENABN cases), 11 (34%) with genomic deletion (PTENDEL) and 4 (13%) with both mutation and deletion (PTENMUT+DEL). Of the 21 PTENMUT cases, 11 were considered to harbor monoallelic mutations (PTENMONO) as the total mutant level was <50% of total alleles and there was either a single heterozygous clone (one mutant, level 37–48%, that is, present in most cells) or multiple sub-clones (more than one mutant, individual level 2–21%) (Supplementary Table 1). The remaining 10 PTENMUT patients were considered to harbor biallelic mutations (PTENBI) as all had >50% total mutant consistent with either a single homozygous or hemizygous mutation or compound heterozygous mutations in at least some of the cells. Therefore, for the combined PTENMUT and PTENDEL genotype, 19 patients (59%) were scored as PTENMONO and 13 patients (41%) as PTENBI.
RAS mutation screening
Missense RAS mutations (RASMUT) were detected in 13 (9%) of the 145 patients analyzed, 8 in N-RAS (4 G12D, 1 each G12V, G13C, G13R and Y64N) and 5 in K-RAS (2 G12C, 1 each G12S, A18D and Y64D). Only 1 of the 32 PTENABN patients (3%) had a RAS mutation. Therefore, 44 patients (30%) were PTEN/RASABN.
Characteristics of T-ALL patients according to PTEN and RAS genotype
There was no significant difference in sex, white blood cell count, age group, NCI risk group or cytogenetic characteristics between PTENABN and PTENWT patients (Table 1). However, PTENABN patients had a significantly higher incidence of CNS disease than PTENWT patients (P=0.04). No significant differences were observed according to RAS genotype.
Prognostic relevance of PTEN abnormalities and RAS mutations
Response to therapy
Response to therapy data was available for all patients, and the incidence of a slow early response did not statistically differ according to the PTEN, RAS or combined PTEN/RAS genotype (Table 2). MRD status at day 29 post diagnosis was available for 134 patients, and no statistically significant differences were seen in the frequency of unfavorable disease according to the different genotype groups (Table 2). When the data were stratified according to the level of PTEN abnormality, there was a suggestion that patients with PTENMONO had the highest levels of MRD, but the subgroups were small and this could have occurred by chance.
Long-term outcome
Event-free survival, relapse-free survival and OS were slightly worse for PTENABN compared with PTENWT cases (Figures 3a and b) (Table 2). However, the difference only showed a borderline trend for significance for OS (81 versus 91% at 5 years; hazard ratio (95% confidence interval), 2.27 (0.82–6.24); P=0.10), there were only a small number of events, and the confidence intervals for all analyses were wide. There was no difference in the relative proportion of PTENABN and PTENWT patients treated on the different regimens (Table 1). When patients were stratified according to PTEN mutant level, PTENMONO had the lowest event-free survival and OS, but the number of patients in these groups was small and the differences were not significant across the three groups (Table 2) (Figures 3c and d). Similarly, outcome did not differ in the RAS genotypic groups (Supplementary Figures 4A and B), or in the combined PTEN/RAS groups (Figures 3e and f).

Kaplan–Meier curves for relapse-free survival (a, c, e) and overall survival (b, d, f) stratified according to PTEN and RAS genotype. (a, b) PTENABN and PTENWT. (c, d) PTENWT, PTENMONO and PTENBI. (e, f) PTEN/RASABN and PTEN/RASWT. ABN, abnormal; BI, biallelic; MONO, monoallelic.
NOTCH1/FBXW7/PTEN/RAS genotype of T-ALL patients
We have previously shown that outcome in this cohort differs according to the NOTCH1/FBXW7 genotype, with a very good outcome in the NOTCH1±FBXW7Double group.8 We therefore examined whether PTEN or RAS genotype changed this. The incidence of PTEN abnormalities did not differ according to NOTCH1/FBXW7 genotype (59% PTENABN patients had a NOTCH1 and/or FBXW7 mutation compared with 68% PTENWT patients; P=0.35), nor did RAS genotype (77% for RASMUT versus 65% for RASWT; P=0.54) or the combined PTEN/RAS genotype (59% for PTEN/RASABN versus 65% for PTEN/RASWT; P=0.67). There were also no differences in PTEN and/or RAS genotype when the patients were grouped into the three previously defined NOTCH1/FBXW7 genotype groups, NOTCH1WTFBXW7WT, NOTCH1SingleFBXW7WT and NOTCH1±FBXW7Double, excluding the four patients with an FBXW7 mutation only (Supplementary Table 2).
Of the 37 NOTCH1±FBXW7Double patients, 9 (24%) had PTEN and/or RAS abnormalities (5 PTENABN and 5 RASMUT, 1 with both), but this did not impact on their favorable outcome, as all were alive at 5 years (Table 3). Only two patients relapsed, both in the PTEN/RASWT group. In the NOTCH1SingleFBXW7WT group, 17 of the 55 patients (31%) were PTEN/RASABN (14 PTENABN, 3 RASMUT), and although OS was slightly lower than in the PTEN/RASWT patients, none of the differences were significant (Table 3). Overall, OS and relapse-free survival were lowest for PTEN/RASABN patients in the NOTCH1WTFBXW7WT group (16 of 49 patients (33%); 13 PTENABN, 3 RASMUT), but did not significantly differ from PTEN/RASWT patients in this group (Figure 3f).
In a study of adult cases, a low-risk group, defined as NOTCH1-mutant and/or FBXW7-mutant without a PTEN or RAS mutation, had a significantly better outcome than the high-risk group of all other cases combined together.14 Using this stratification system, there was no significant difference in relapse-free survival in our cohort (87% for both), although there was a slightly improved OS in the low-risk group (94 versus 84%; P=0.06) (Supplementary Figures 5A and B).
Discussion
PTEN is a tumor-suppressor gene that is frequently inactivated in a wide variety of cancers leading to hyperactivation of the PI3K/AKT signaling pathway.30 It has an important role in the proliferation and survival of T-cell progenitors,15 and its loss may sustain leukemic T-cell viability in T-ALL.31 Furthermore, studies have associated PTEN genetic abnormalities with glucocorticoid resistance, one of the main causes of relapse in T-ALL, and sensitivity can be re-instated by inhibiting PI3K or AKT.32, 33 Knowledge of PTEN genotype may therefore help to refine current risk stratification strategies that are based on response to therapy, and inform clinical decisions. The available information to date suggests that PTEN abnormalities appear to be associated with adverse outcome in T-ALL, but the data are limited; the cohorts in some studies are small, not all studies have examined both mutations and deletions, and most studies have not considered the impact of the mutant level, in particular whether those with complete (biallelic) PTEN loss differ from those with partial (monoallelic) loss. In addition, contradictory results have been reported in pediatric and adult patients for the impact of the abnormalities in the NOTCH1/FBXW7-mutated subgroup,13, 14 which is important for clinical application as at least some studies have shown that patients with NOTCH1/FBXW7 mutations do particularly well.6, 7, 8 There is also no information on the impact of RAS mutations in pediatric T-ALL, although in adult patients, they were associated with worse outcome and were included as poor risk in an oncogenetic classifier strategy.14
In the present study, inactivating PTEN mutations were detected in 14% of the cohort and gene deletions in 10%, with an overall incidence of 22% with PTEN gene abnormalities, which is in keeping with the combined data from other pediatric studies of 16% with mutations,13, 18, 20, 23 6% with deletions,18, 19, 23 and 20% for mutations and/or deletions.18, 23 In common with the other studies, the mutations detected were predominantly located in exon 7 and were truncating mutations predicted to lead to haploinsufficiency, rather than the dominant-negative missense mutations that often occur in other types of cancer and that were associated with a more complete loss of PTEN activity and accelerated tumorigenesis in a mouse model.34 We have also screened for type 1 microdeletions as reported by Mendes et al.,23 but they were only detected in 3% of the patients and did not change the PTEN classification as all four positive cases had other PTEN abnormalities. These results indicate that to evaluate PTEN loss, both mechanisms of inactivation (mutation and deletion) need to be considered, and they confirm that PTEN abnormalities are likely to be a contributory factor in the pathogenesis of a significant proportion of patients with T-ALL.
A striking feature of our study was the high proportion of mutant-positive patients (67%) with more than one PTEN mutation. In some cases, this may reflect compound heterozygosity leading to complete absence of PTEN in a cell, but approximately half of the cases were likely to have multiple minor subclones. Similarly, the deletions in five of the six quantifiable cases (83%) were not present in all cells, and the frequency of gene deletions detected is probably under-estimated as the methodologies used were not sufficiently sensitive to detect low-level deletions. This is in keeping with the intratumoral heterogeneity now being revealed by deep-sequencing studies35 and suggests that acquisition of a PTEN mutation or deletion is more likely to be a later event associated with disease progression. It will therefore be important to determine whether these PTENABN clones are being selected for at relapse. Analysis of paired diagnostic and relapse samples has demonstrated acquisition of PTEN mutations and deletions at relapse,16, 36 but whether these are de novo changes or therapy-induced selection is unclear. Back-tracking has shown that minor PTENABN clones can be clonally expanded to become the dominant clone at relapse.3, 36 Xenotransplantation studies have not consistently demonstrated that loss of PTEN constitutes an adequate selection pressure for driving this expansion,23, 36 although in this setting, primary leukemic cells with PTEN knocked down by short hairpin RNAs did show a competitive engraftment advantage over the unmodified cells.36 The significance of specific abnormalities may also be highly dependent on the presence of other co-incident genetic events, and further work will be required to determine whether this can be predicted from the genetic profile of individual cases.
Despite the data implicating the PI3K/AKT pathway as an important oncogenic driver pathway in T-ALL, in pediatric patients, there is currently little evidence to suggest that the PTEN genotype is a strong prognostic factor. We found no significant association between PTEN abnormalities and either response to therapy or long-term outcome in our cohort, and this absence of an impact could not be attributed to therapy received as the proportion of patients with/without PTEN abnormalities treated on the different regimens did not differ, although it must be acknowledged that the number of events is small and larger studies may reveal more minor effects. There was a suggestion in our study that PTEN abnormalities may be associated with a worse OS and this is in general agreement with other pediatric studies.12, 13, 20 A meta-analysis would be useful, but few studies have reported both PTEN mutational and copy number status. Adding patients with RAS mutations to the abnormal group made no difference. Studies have suggested that the monoallelic loss of PTEN may be more detrimental than biallelic loss, leading to tumor progression rather than Pten-loss-induced cellular senescence.37 However, when our patients were stratified according to the level of mutant or deletion, although PTENMONO patients were associated with unfavorable levels of MRD and a worse OS, the difference was not significant and, in view of the small number of patients in the subgroups, this would require validation in a larger cohort.
Importantly, we found no evidence that the presence of a PTEN abnormality impacted on the highly favorable outcome that we previously reported in NOTCH1±FBXW7Double patients.8 Of note, these results are in keeping with those from the pediatric ALL-BFM 2000 trial,13 but in distinct contrast to adult patients where a significantly adverse impact of PTEN/RAS abnormalities was found, irrespective of NOTCH1/FBXW7 genotype.14 Applying the NOTCH1/FBXW7/RAS/PTEN-based oncogenetic classification criteria proposed for adults to our pediatric cohort, which does not discriminate between NOTCH1/FBXW7 double- and single-mutated patients,14 there was no difference in relapse rate between the high-risk and low-risk groups and only a borderline difference in OS, which could be attributed to better salvage of relapsed patients in the low-risk group. This difference (94 versus 84%) was small compared with the major difference seen in adult patients (82 versus 44%) and with the small number of events involved, it could have occurred by chance. Larger studies will be needed to determine whether this classification system is informative in pediatric patients.
These results would therefore suggest that, at present, despite PTEN loss being common in pediatric T-ALL, screening for PTEN abnormalities at diagnosis would not add further information to refine the currently available risk-adapted therapy strategies. However, with the current development and clinical evaluation of many PI3K inhibitors,38 the PTEN genotype may serve as a potential biomarker for response to these agents.
References
Herold R, von Stackelberg A, Hartmann R, Eisenreich B, Henze G . Acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Munster Group. (ALL-REZ BFM) experience: early treatment intensity makes the difference. J Clin Oncol 2004; 22: 569–570.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446: 758–764.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008; 322: 1377–1380.
Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood 2008; 112: 4178–4183.
Pui CH, Carroll WL, Meshinchi S, Arceci RJ . Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol 2011; 29: 551–565.
Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood 2009; 113: 3918–3924.
Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig WD et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia 2010; 24: 2005–2013.
Jenkinson S, Koo K, Mansour MR, Goulden N, Vora A, Mitchell C et al. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia 2013; 27: 41–47.
Mansour MR, Sulis ML, Duke V, Foroni L, Jenkinson S, Koo K et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J Clin Oncol 2009; 27: 4352–4356.
Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia 2010; 24: 2014–2022.
Clappier E, Collette S, Grardel N, Girard S, Suarez L, Brunie G et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia 2010; 24: 2023–2031.
Zuurbier L, Petricoin EF 3rd, Vuerhard MJ, Calvert V, Kooi C, Buijs-Gladdines JG et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 2012; 97: 1405–1413.
Bandapalli OR, Zimmermann M, Kox C, Stanulla M, Schrappe M, Ludwig WD et al. NOTCH1 activation clinically antagonizes the unfavorable effect of PTEN inactivation in BFM-treated children with precursor T-cell acute lymphoblastic leukemia. Haematologica 2013; 98: 928–936.
Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, Lambert J, Beldjord K, Lengline E et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol 2013; 31: 4333–4342.
Ciofani M, Zuniga-Pflucker JC . Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol 2005; 6: 881–888.
Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 2007; 13: 1203–1210.
Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007; 447: 966–971.
Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009; 114: 647–650.
Remke M, Pfister S, Kox C, Toedt G, Becker N, Benner A et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-beta and PI3K-AKT pathways and deletions at 6q15-16.1 as a genomic marker for unfavorable early treatment response. Blood 2009; 114: 1053–1062.
Jotta PY, Ganazza MA, Silva A, Viana MB, da Silva MJ, Zambaldi LJ et al. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia 2010; 24: 239–242.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012; 481: 157–163.
Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H . The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci USA 1999; 96: 10182–10187.
Mendes RD, Sarmento LM, Cante-Barrett K, Zuurbier L, Buijs-Gladdines JG, Povoa V et al. PTEN microdeletions in T-cell acute lymphoblastic leukemia are caused by illegitimate RAG-mediated recombination events. Blood 2014; 124: 567–578.
Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE et al. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood 2008; 112: 3373–3382.
Kong G, Du J, Liu Y, Meline B, Chang YI, Ranheim EA et al. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J Biol Chem 2013; 288: 18219–18227.
Perentesis JP, Bhatia S, Boyle E, Shao Y, Ou Shu X, Steinbuch M et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia 2004; 18: 685–692.
Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med 2011; 208: 2571–2579.
Qureshi A, Mitchell C, Richards S, Vora A, Goulden N . Asparaginase-related venous thrombosis in UKALL 2003- re-exposure to asparaginase is feasible and safe. Br J Haematol 2010; 149: 410–413.
Goulden NJ, Knechtli CJ, Garland RJ, Langlands K, Hancock JP, Potter MN et al. Minimal residual disease analysis for the prediction of relapse in children with standard-risk acute lymphoblastic leukaemia. Br J Haematol 1998; 100: 235–244.
Hollander MC, Blumenthal GM, Dennis PA . PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer 2011; 11: 289–301.
Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest 2008; 118: 3762–3774.
Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell 2013; 24: 766–776.
Blackburn JS, Liu S, Wilder JL, Dobrinski KP, Lobbardi R, Moore FE et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 2014; 25: 366–378.
Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014; 157: 595–610.
Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, Swanton C . Cancer: evolution within a lifetime. Annu Rev Genet 2014; 48: 215–236.
Clappier E, Gerby B, Sigaux F, Delord M, Touzri F, Hernandez L et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med 2011; 208: 653–661.
Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436: 725–730.
Thorpe LM, Yuzugullu H, Zhao JJ . PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015; 15: 7–24.
Acknowledgements
We are grateful to the clinical investigators who entered and managed patients in the trial. Primary childhood leukemia samples used in the study were provided by the Leukaemia & Lymphoma Research Childhood Leukaemia Cell Bank working with the laboratory teams in the Bristol Genetics Laboratory, Southmead Hospital, Bristol, UK, Molecular Biology Laboratory, Royal Hospital for Sick Children, Glasgow, UK, Molecular Haematology Laboratory, Royal London Hospital, London, UK and Molecular Genetics Service and Sheffield Children’s Hospital, Sheffield, UK. We would also like to thank Dr Paul Smith, Department of Haematology, UCL Cancer Institute, and Kerra Pearce, UCL Genomics, for technical assistance. This work was supported by Cancer Research UK, UK Leukaemia & Lymphoma Research, and the UK Children’s Cancer and Leukaemia Group, and was undertaken at UCL, which receives a proportion of funding from the Department of Health’s NIHR Biomedical Research Centre’s funding scheme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jenkinson, S., Kirkwood, A., Goulden, N. et al. Impact of PTEN abnormalities on outcome in pediatric patients with T-cell acute lymphoblastic leukemia treated on the MRC UKALL2003 trial. Leukemia 30, 39–47 (2016). https://doi.org/10.1038/leu.2015.206
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.206
This article is cited by
-
PTEN/PI3K/Akt pathway alters sensitivity of T-cell acute lymphoblastic leukemia to l-asparaginase
Scientific Reports (2022)
-
Prognostic value of Oncogenetic mutations in pediatric T Acute Lymphoblastic Leukemia: a comparison of UKALL2003 and FRALLE2000T protocols
Leukemia (2022)
-
Prognostic impact of chromosomal abnormalities and copy number alterations in adult B-cell precursor acute lymphoblastic leukaemia: a UKALL14 study
Leukemia (2022)
-
Targeted sequencing to identify genetic alterations and prognostic markers in pediatric T-cell acute lymphoblastic leukemia
Scientific Reports (2021)
-
ROS play an important role in ATPR inducing differentiation and inhibiting proliferation of leukemia cells by regulating the PTEN/PI3K/AKT signaling pathway
Biological Research (2019)
