
Abstract
Although the therapeutic benefit of proteasome inhibition in multiple myeloma remains unchallenged, drug resistance inevitably emerges through mechanisms that remain elusive. Bortezomib provokes unwanted protein accumulation and the endoplasmic reticulum stress to activate the unfolded protein response (UPR) and autophagy as compensatory mechanisms that restore protein homeostasis. High-throughput screens to detect pharmacologics that modulated autophagy to enhance the anti-myeloma effect of bortezomib revealed metformin, a widely used antidiabetic agent with proven efficacy and limited adverse effects. Metformin co-treatment with bortezomib suppressed induction of the critical UPR effector glucose-regulated protein 78 (GRP78) to impair autophagosome formation and enhance apoptosis. Gene expression profiling of newly diagnosed myeloma patient tumors further correlated the hyperexpression of GRP78-encoding HSPA5 with reduced clinical response to bortezomib. The effect of bortezomib was enhanced with metformin co-treatment using myeloma patient tumor cells and the chemoresistant, stem cell-like side population that may contribute to disease recurrence. The relevance of the findings was confirmed in vivo as shown by metformin co-treatment with bortezomib that delayed the growth of myeloma xenotransplants. Taken together, our results suggest that metformin suppresses GRP78, a key driver of bortezomib-induced autophagy, and support the pharmacologic repositioning of metformin to enhance the anti-myeloma benefit of bortezomib.
Similar content being viewed by others


Introduction
Pharmacologic inhibition of the proteasome leads to disease regression or stabilization in newly diagnosed and treatment-refractory multiple myeloma (MM) patients.1, 2 Bortezomib treatment of myeloma cells promotes the accumulation of misfolded and unfolded proteins that activates apoptotic pathways. However, disruptions that perturb protein degradation also induce endoplasmic reticulum (ER) stress and the unfolded protein response (UPR), a highly conserved signaling network aimed at expanding ER processing capacity and alleviating cellular injury.3, 4, 5, 6 Agents that disrupt the ER stress response represent an attractive approach for selective cancer cell killing and provide the basis to develop targeted drugs that suppress pro-survival adaptations, for example, autophagy.7, 8, 9
Glucose-regulated protein 78 (GRP78) is a central regulator of ER function due to its roles in protein folding and controlling activation of ER stress sensors.10, 11 GRP78 is essential for UPR activation and promotes cytoprotective autophagy through maintenance of ER structural integrity.12 Functional blockade of the proteasome induces GRP78 to promote autophagosome formation and enhance myeloma survival.13 Overexpression of HSPA5, which encodes GRP78, promotes proliferation in many tumor cells and is associated with poor prognosis.14, 15, 16 HSPA5 knockdown also reduces metastatic growth in xenograft models17, 18 whereas GRP78 promotes cancer cell growth and activates the PI3K/Akt pathway.19
Chemical screens to detect UPR modulators revealed that metformin prevented activation of a GRP78-promoter reporter and that metformin inhibited UPR activation to induce cell death under glucose deprivation.7 Although metformin improves glycemic control and is the drug of choice to treat type 2 diabetes (T2D), metformin has also gained attention as it may reduce cancer incidence.20, 21 Preclinical studies have shown that metformin inhibits the growth of cancer cells in vitro and in vivo, however, the mechanism(s) of action remains poorly understood.22, 23, 24 Here, we investigated mechanisms by which metformin exerts its action to show that metformin targets GRP78-dependent autophagy to enhance the effect of bortezomib.
Materials and methods
Myeloma cell lines and patient tumor cells
MM cell lines (MMCLs) were from ATCC (Manassas, Virginia) or the National Cancer Institute (Bethesda, MD, USA). MMCLs were cultured in complete Roswell Park Memorial Institute media (RPMI)-1640 medium and bone marrow stromal HS-5 cells (ATCC) in Dulbecco’s modified Eagle’s medium. Bone marrow aspirates were obtained after approval institutional review board or from ConversantBio (Huntsville, AL, USA) and CD138+ cells then isolated (Miltenyi Biotec, San Diego, CA, USA). Side population (SP) was isolated with Hoechst 33342 dye and flow cytometry as described.25, 26, 27
Chemicals and reagents
Proteasome inhibitors were from ActiveBiochem (Maplewood, NJ, USA), metformin and phenformin from Sigma-Aldrich (St Louis, MO, USA), GRP78 antibodies from Abcam (Cambridge, MA, USA) antibodies to PI3K-p85, PI3Kα/p110, LC3B, Akt and phospho-Akt-Ser-473 from Cell Signaling Technology (Danvers, MA, USA) and poly ADP ribose polymerase from Santa Cruz Technology (Santa Cruz, CA, USA).
Cell viability
Cells were treated as indicated for 72 h, activated 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) (Sigma-Aldrich) added and absorbance determined.28 Relative cell viability (mean±s.d.) was calculated based on absorbance from untreated cells.
Cell proliferation
The bromodeoxyuridine (BrdU) cell proliferation assay kit (Cell Signaling Technology) was used according to the manufacturer’s instructions.
Apoptosis
Cells were stained with annexin-V (fluorescein isothiocyanate; Thermo-Fischer, Waltham, MA, USA) and analyzed on an LSR-II flow cytometer (BD Biosciences, San Jose, CA, USA).
HSPA5 knockdown
shRNA in pLKO.1-TCR lentiviral cloning vectors were transfected into 293T packaging cells with packaging and envelope vector using lipofectamine 2000 (Thermo-Fisher). Viral supernatants were used to transduce cells then selected in puromycin.
Western blots
Electrophoresed proteins were transferred to nitrocellulose, blocked with buffer and primary antibody added. Membranes were visualized using the LI-COR (Littleton, CO, USA) detection system.
Biostatistics
Data presented are mean±s.d. of independent experiments performed in triplicate. Statistical significance was assessed using the Student t-test and analysis of variance test. Overall survival was determined using the Kaplan–Meier method with 95% confidence intervals. A value of P<0.05 was accepted as statistically significant.
Results
Novel agents that target the proteasome, a proteolytic complex responsible for ubiquitinated protein degradation, achieve remarkable clinical benefit in MM but drug resistance remains a significant obstacle. Current anti-myeloma strategies effectively target the tumor bulk but tumor-initiating subpopulations persist and may be responsible for eventual disease relapse.25, 26 We exploited functional characteristics to isolate the stem cell-like SP by staining with Hoechst 33342 dye. In flow cytometric analysis, SP fall to the side of the bulk of positively Hoechst 33342–stained cells (Figure 1a). A similar approach has been used in a variety of tumor types, including lung, gastric, esophageal, squamous and ovarian carcinoma cell lines.29, 30, 31 The intracellular accumulation of Hoechst dye disappeared when cells were treated with the ABC transporter inhibitors verapamil (100 μM) or reserpine (50 μM). SP and main population (MP) were isolated from the MMCLs and myeloma patient tumor samples and the effect of the proteasome inhibitors bortezomib, carfilzomib and ixazomib were determined on the viability using the XTT assay (Figure 1b). SP was less sensitive to proteasome inhibition than MP (Figure 1c, Supplementary Figure 1). We next determined the effect of bortezomib on cell proliferation by measuring BrdU incorporation.32 Treatment of SP or MP for 8 h with bortezomib generated a dose-dependent increase in absorbance relative to SP or MP that had not been treated with bortezomib (Figure 1d). The results indicated that bortezomib alone slightly increased BrdU incorporation and did not inhibit proliferation during this period of time, consistent with prior studies.33 In previous studies to assess the biologic significance of NF-κB activation during bortezomib-induced growth inhibition, BrdU incorporation was upregulated by bortezomib after 8 h of drug treatment. Importantly, washout experiments after 8-h bortezomib treatment showed that MM cells were destined to die at later times. Although NF-κB was activated by bortezomib in the first 8 h, the results indicated that MM cells were already committed to apoptosis.33 SP and MP were treated with bortezomib at the indicated concentrations for 24 h and the relative percent of cells undergoing apoptosis was determined by annexin-V (fluorescein isothiocyanate) staining and flow cytometry33 (Figure 1e). Bortezomib generated a dose-dependent increase in annexin-positive SP and MP cells to indicate that the bortezomib effect was mediated through apoptosis.

Effect of proteasome inhibitors on myeloma SP and MP viability, proliferation and induction of apoptosis. (a) Representative flow cytometric dot plots of SP analysis. Dot plots show RPMI8226 cells incubated in Hoechst 33342 alone (left), Hoechst 33342 accumulation in the presence of 100 μM verapamil (middle) and Hoechst 33342 accumulation in the presence of 50 μM reserpine (right). Abscissa is Hoechst red fluorescence intensity and ordinate is Hoechst blue fluorescence intensity with the gate representing the SP fraction. (b) Effect of bortezomib, carfilzomib or ixazomib at indicated concentrations on SP and MP viability. SP and MP were isolated from RPMI8226 cells and viability determined using the XTT assay. Error bars represent s.d. values determined from triplicate measurements. (c) Effect of bortezomib, carfilzomib and ixazomib on SP or MP cells isolated from MM patients. Cells were incubated with drugs as indicated and viability determined using the XTT assay. Error bars represent s.d. values determined from triplicate measurements. (d) Effect of bortezomib on SP and MP cell proliferation. Cells were incubated with bortezomib at indicated concentrations for 8 h and BrdU then added for 2 h. BrdU incorporation was determined relative to untreated MP cells. Error bars represent s.d. values determined from triplicate measurements. (e) Effect of bortezomib on the generation of annexin-positive SP and MP cells. Cells were incubated with bortezomib as indicated for 24 h and the percentage of annexin-positive cells determined relative to untreated SP or MP cells. Shown is the percentage increase in annexin-V-positive cells as determined by flow cytometry. Error bars represent s.d. values from triplicate measurements.
Metformin enhances the apoptotic effect of bortezomib
Autophagy represents an alternate protein degradation pathway that may eliminate unwanted proteins upon proteasome inhibition to enhance the anti-myeloma effect of bortezomib.12, 13 U266 cells were transfected with plasmid that expressed the autophagy effector LC3B tagged with the green fluorescent protein. Pharmacologic blockade of autophagy was predicted to promote accumulation of LC3B-green fluorescent protein that was readily detectable by fluorescent imaging. Screening of Food and Drug Administration-approved pharmacologics and bioactive agents (Supplementary Table 1) identified that metformin was one of the most effective drugs that promoted an increase in green fluorescence (Supplementary Table 2). Next, we performed screening to detect drugs that enhanced the effect of bortezomib (5 nM) on myeloma viability. Results indicated that metformin also significantly enhanced the effect of bortezomib on myeloma viability (Supplementary Table 3).
Metformin exhibits anticancer effects in a wide range of in vitro and in vivo models through adenosine monophosphate-activated protein kinase (AMPK)-dependent and -independent pathways.22, 34, 35, 36 When added alone, millimolar concentrations of metformin were required to achieve an appreciable reduction in myeloma viability (Supplementary Figure 2). Importantly, metformin (500μM) co-treatment enhanced the effect of bortezomib to reduce the viability of SP and MP cells isolated from either MMCLs or patient tumors (Figures 2a and b, Supplementary Figure 3). The formation of colonies in solid media by SP cells was reduced with bortezomib treatment (Figure 2c) and the effect was enhanced by metformin co-treatment with bortezomib37 (Figure 2d). SP cells demonstrated a greater capacity to form colonies than MP cells and metformin alone did not significantly reduce colony formation (Supplementary Figure 4). Metformin alone or combined with borteozmib also reduced BrdU incorporation as a measurement of cell proliferation. SP and MP cells treated with 500 μM metformin led to a slight reduction in BrdU incorporation compared with untreated controls (Figure 2e). Metformin co-treatment with bortezomib also reduced BrdU incorporation into either SP or MP cells. The results suggest that whereas bortezomib alone did not inhibit BrdU incorporation, metformin alone or metformin co-treatment with bortezomib reduced proliferation. Cells were then treated with metformin and bortezomib, and those undergoing apoptosis were quantitated using annexin-V staining (Figure 2f). Treatment with metformin (500 μM) did not promote apoptosis but co-treatment with bortezomib increased apoptosis in SP and MP cells.

Effect of metformin co-treatment with bortezomib on myeloma SP and MP cell viability, proliferation and induction of apoptosis. (a) Dose-dependent effect of bortezomib alone or combined with metformin (500 μM) on the viability of SP and MP isolated from RPMI8226 cells. SP and MP cells were incubated with bortezomib alone or combined with metformin (500 μM). Cell viability was determined using the XTT assay. Error bars represent s.d. values determined from triplicate measurements. (b) Dose-dependent effect of bortezomib added at the indicated concentrations alone or combined with metformin (500 μM) on myeloma patient’s SP and MP cell viability. (c) Dose-dependent effect of bortezomib added at indicated concentrations on colony formation. Values were normalized relative to the number of colonies formed from untreated SP. (d) Dose-dependent effect of bortezomib added at indicated concentrations alone or combined with metformin on colony formation. Values were normalized relative to the number of colonies detected with untreated SP cells. (e) Effect of bortezomib on SP and MP cell proliferation. Cells were incubated with bortezomib for 8 h at indicated concentrations and BrdU then added for 2 h. BrdU incorporation was determined relative to untreated MP cells. Error bars represent s.d. values determined from triplicate measurements. (f) Effect of bortezomib on the percentage of annexin-positive SP and MP cells. Cells were incubated with bortezomib at indicated concentrations for 24 h. The relative percentage increase in annexin-positive cells was determined relative to untreated SP or MP cells. Shown is the percentage increase in annexin-positive cells determined by flow cytometry. Error bars represent s.d. values determined from triplicate measurements.
Metformin suppresses GRP78-dependent autophagy
Accumulation of polyubiquitinated and misfolded proteins within the ER lumen initiates the UPR and autophagosome formation through GRP78 dissociation from ER stress sensors.38 Bortezomib treatment increased GRP78 levels in RPMI8226 lysates (Figure 3a). Metformin has previously been shown to suppress GRP78 induction upon glucose deprivation or proteasome inhibition and, consequently, to disrupt UPR activation.7, 13, 23 Treatment with metformin alone slightly increased GRP78 levels in RPMI8226 lysates whereas bortezomib treatment generated a more significant increase in GRP78 (Figure 3b). Metformin co-treatment suppressed the bortezomib-induced increase in GRP78. Bortezomib also increased GRP78 in SP and MP cell lysates and metformin co-treatment suppressed the induction of GRP78 in these populations (Figure 3c). During autophagosome formation, microtubule-associated protein light chain I (LC3B-I) covalently links to phosphatidyl-ethanolamine and is incorporated into autophagosome membranes where it recruits cargo proteins for degradation. Lipidation converts cytosolic LC3B-I into the active, autophagosome membrane-bound form, LC3B-II. Cells were treated with bortezomib, metformin or both and LC3B-I conversion monitored by western blot (Figure 3d). Bortezomib significantly increased the conversion of LC3B-I to LC3B-II, whereas metformin, alone or in co-treatment with bortezomib, yielded only a slight increase in LC3B-II relative to untreated controls. Bafilomycin A1 was then added for the final 4 h of incubation (lanes 5–8) to prevent autophagosome recycling. The results indicated that treatment with bafilomycin A1 enhanced the accumulation of LC3B-I and LC3B-II to indicate that treatment with bortezomib, metformin or both drugs simultaneously increased autophagic flux. GRP78 levels and autophagosome levels were then monitored by immunohistochemistry and confocal microscopy. GRP78 and autophagosomes were minimal in untreated RPMI8226 cells, but the levels significantly increased after bortezomib treatment (Figure 3e). Metformin alone did not induce the GRP78 or autophagosomes and the effect of bortezomib on GRP78 and autophagosomes was suppressed upon co-treatment with metformin. RPMI8226 cells were treated with the autophagy inhibitors chloroquine, hydroxychloroquine or bafilomycin alone or with bortezomib (Supplementary Figure 5). The autophagy inhibitors slightly reduced viability but co-treatment with bortezomib significantly enhanced the effect to suggest that autophagy inhibition further increases the effect of proteasome inhibition. The effect of metformin co-treatment with bortezomib was also observed after RPMI8826 cells had been cocultured with bone marrow stroma (Supplementary Figure 6).

Effect of metformin on GRP78 and bortezomib-induced autophagy. (a) Western blot to determine the effect of bortezomib treatment on GRP78 levels in RPMI8226 cell lysates. Cells were treated with bortezomib at indicated concentrations for 24 h, lysates prepared and probed using a GRP78-specific antibody. (b) Western blot to determine the effect of metformin co-treatment with bortezomib on GRP78 levels in RPMI8226 cells. Cells were treated with bortezomib and metformin at indicated concentrations for 24 h; lysates prepared and probed using an antibody specific to GRP78. (c) Western blot to determine the effect of metformin co-treatment with bortezomib on GRP78 levels in SP and MP cells isolated from RPMI8226. Cells were treated with bortezomib and metformin at indicated concentrations for 24 h; lysates prepared and probed using an antibody specific to GRP78. (d) Effect of metformin co-treatment with bortezomib on the conversion of LC3B-I to LC3B-II in RPMI8226 cells. Cells were treated with bortezomib (5 nM), metformin (500 μM) for 24 h. Bafilomycin A1 (100 nM), a specific inhibitor of the vacuolar type H+-ATPase was then added for the final 4 h of incubation to inhibit autophagy flux and to prevent autophagosome recycling and lead to LC3B-II accumulation. Lysates were prepared and probed by western blot to detect the relative levels of LC3B-I and LC3B-II. (e) Effect of bortezomib (5 nM) and metformin (500 μM) alone and combined on the levels of GRP78 and autophagosomes in RPMI8226 cells. Cells were treated with drugs for 16 h and GRP78 was then visualized by immunohistochemistry staining and confocal microscopy. Autophagosomes were detected using the cytologic identification (cyto-ID) autophagy detection kit (Enzo Life Sciences, Farmingdale, NY, USA) and visualized using a Zeiss LSM710 confocal microscope (Zeiss Microscopy, Thornwood, NY, USA). Settings for fluorescein isothiocyanate detection were EX488nm and EM550nm. (f) Effect of HSPA5-specific shRNA on autophagosome formation. RPMI8226 cells were transfected with control or HSPA5-specific shRNA and transfectants treated as indicated. Autophagosomes were detected by dye-based staining and confocal microscopy. (g) Autophagosomes were detected by the dye-based method and the relative amount of green fluorescence was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Statistically significant differences in fluorescence are indicated by *. (h) Effect of bortezomib (5 nM) and metformin (500 μM) alone and combined on autophagosome formation in SP cells. SP were treated with drugs as indicated for 16 h and autophagosome formation visualized by dye-based staining and confocal microscopy. Shown are representative images observed in multiple experiments.
To determine whether the effect of bortezomib on autophagosome formation required GRP78, RPMI8226 cells transfected with scrambled control or HSPA5-specific shRNA. Transfectants were then treated with bortezomib (5 nM), metformin (500 μM) or both drugs. Cells transfected with shRNA to reduce HSPA5 did not form autophagosomes after bortezomib treatment compared with control transfected cells (Figure 3f). Cells transfected with shRNA to inactivate HSPA5 also exhibited reduced viability after bortezomib treatment compared with control transfected cells (Supplementary Figure 7). AMPK is also required for autophagy and genetic knockout of the AMPK catalytic subunits AMPK-α1/α2 in mouse embryonic fibroblasts was shown to similarly enhance the effect of bortezomib on cell viability (Supplementary Figure 8). Quantitation of green fluorescence, indicative of autophagosomes, indicated that bortezomib increased autophagosomes by ~9-fold relative to untreated cells (Figure 3g). Metformin treatment alone increased fluorescence by ~4-fold and autophagosomes formed after metformin treatment were much smaller than those formed with bortezomib. Metformin co-treatment with bortezomib suppressed autophagosome formation in SP cells (Figure 3h).
HSPA5 expression in patient tumors is inversely correlated with clinical response to bortezomib
We reasoned that HSPA5 expression increased during the multi-step process of myelomagenesis and that HSPA5 expression was greater in MM patients that failed bortezomib-based therapy. gene expression profiles analysis indicated that the median expression of HSPA5 was greater in CD138+ plasma cells isolated from newly diagnosed, untreated monoclonal gammopathy of undetermined significance, smoldering multiple myeloma and MM patient populations than the median expression from CD138+ plasma cells isolated from healthy individuals (Supplementary Figure 8). Analysis of HSPA5 in individual patients indicated that HSPA5 expression was increased in a significant subset of monoclonal gammopathy of undetermined significance, smoldering multiple myeloma and MM patients compared with the expression in plasma cells from healthy individuals. As HSPA5 hyperexpression has been linked to chemoresistance, we employed a pharmacogenomic approach to determine whether HSPA5 expression correlated with the lack of clinical response to bortezomib (Supplementary Figure 8). Gene expression profiles were obtained to compare HSPA5 expression in tumors from patients enrolled in a prospective pharmacogenomic study performed to test the efficacy of bortezomib vs dexamethasone. HSPA5 was hyperexpressed in the 86 patients that did not respond to bortezomib relative to the 83 patients that did respond to bortezomib. In addition, HSPA5 was hyperexpressed in patients that did not respond to bortezomib but HSPA5 was not hyperexpressed in patients that failed to respond to dexamethasone to support the notion that HSPA5 hyperexpression specifically correlates with bortezomib resistance.
Metformin suppresses membrane localization of GRP78 with PI3K
GRP78 exhibits oncogenic functions attributed, in part, to direct binding and activation of PI3K.39 Catalytically active PI3K then phosphorylates protein kinase B/Akt, a Ser/Thr-specific protein kinase that has a key role in cell proliferation and inhibition of apoptosis. Phosphorylation of the PI3K catalytic subunit promotes its plasma membrane localization and increases catalytic activity. Immunohistochemistry demonstrated that PI3K detection was greater in untreated SP than MP cells and that PI3K detection in either SP or MP cells was enhanced with bortezomib treatment. Merge of the fluorescence signals indicated that in the absence of any treatment, GRP78 colocalization with PI3K in SP and MP cells was enhanced by bortezomib and suppressed by metformin (Supplementary Figure 9). Quantification of color-specific pixel counts supported the findings that bortezomib promoted GRP78 association with PI3K and that the association was suppressed by metformin. Furthermore, shRNA-mediated knockdown of HSPA5 followed by bortezomib treatment reduced the colocalization of GRP78 with PI3K. Catalytic activation of PI3K increases levels of the PI3K product PIP3, and Akt subsequently binds to PIP3 at the plasma membrane, allowing PDK1 to phosphorylate Thr308 in the activation loop of Akt leading to partial activation. Akt modification is sufficient to activate mTORC1 and phosphorylation of Akt at Ser473 that then stimulates full Akt activity.40 Bortezomib downregulates Akt phosphorylation in drug sensitive hepatocellular carcinoma cells, whereas Akt phosphorylation was reduced in drug-resistant cells. Bortezomib treatment of MP cells reduced Akt phosphorylation at Ser473, whereas Akt phosphorylation at the same site was maintained in SP cells (Supplementary Figure 9). The level of the PI3K catalytic subunit and the relative fraction of Akt that remained phosphorylated after bortezomib treatment were reduced to a greater extent in MP than SP cells. The level of PI3K 85 kDa adapter was unchanged in SP and MP cells with bortezomib treatment. Co-treatment of SP or MP cells with bortezomib and metformin led to a greater reduction in poly ADP ribose polymerase levels compared with that seen after treatment with either agent alone.
GRP78 suppression reduces PI3K catalytic activity
Transfection of SP and MP cells with HSPA5-specific shRNA enhanced the effect of bortezomib to reduce viability (Supplementary Figure 9). SP cells displayed increased PIP3 production compared with MP cells and HSPA5-specific shRNA also preferentially reduced PIP3 production in SP cells (Supplementary Figure 10). Bortezomib, metformin and co-treatment reduced PIP3 production in SP to a greater extent than in MP. The results suggested that the genetic or pharmacologic suppression of GRP78 reduced PI3K activity.
Metformin enhances the effect of bortezomib in MM patient CD138+ plasma cells
Patient CD138+ plasma cells were treated with bortezomib, metformin or both, and autophagosomes then detected (Supplementary Figure 10). As was seen in MMCLs, bortezomib treatment of patient tumor cells increased staining for autophagosomes and the effect was suppressed with metformin. Treatment of patient CD138+ cells with the same drugs indicated that metformin co-treatment with bortezomib also enhanced the reduction in viability (Supplementary Figure 11). The effect of metformin or phenformin was also observed in AMPK knockout mouse embryonic fibroblasts (Supplementary Figure 12). The effect was also observed in CD138+ cells obtained from bortezomib refractory patients (Supplementary Figure 13).
Metformin enhances the anti-myeloma effect of bortezomib in vivo
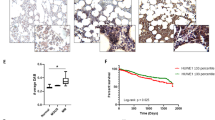
A plasmacytoma model of MM was used to determine the in vivo effect of metformin and bortezomib on tumor burden and overall survival (Figure 4a). MM.1S cells were injected subcutaneously into mice and palpable tumors were detected within 10 days. Bortezomib alone reduced tumor volume relative to vehicle or metformin treated and the effect of bortezomib was enhanced with metformin. The effect was without a significant change in body weight (Supplementary Figure 14). Median survival was 17 days in the vehicle-treated group, 18 days in the metformin-alone-treated group, 22 days in the bortezomib-treated group and 27 days in the bortezomib+metformin-treated group (Figure 4b). The results suggest that metformin augmented the effect of bortezomib on tumor regression with survival benefit.

Effect of bortezomib and metformin on tumor volume (TV) and overall survival. MM.1S cells (3 × 106/injection) were subcutaneously placed in the flanks of 5-week-old nude NCr nu/nu female mice. Mice were then randomly distributed into four groups (10/group) and after the formation of palpable tumors treatment was commenced. Groups received intravenous injection of either vehicle (phosphate-buffered saline containing 10% dimethyl sulfoxide (DMSO)) or bortezomib (0.5 mg/kg in phosphate-buffered saline, 10% DMSO) administered with or without metformin (600 μg/ml diluted in drinking water that contained 5% dextrose). Metformin was administered throughout treatment and vehicle or bortezomib administered on days 1–4. TVmeasurements were made using a vernier caliper and calculated using the formula: TV=0.5(a × b2) where a is the long diameter and b the short diameter. Mice were euthanized when tumors reached 2 cm3, became ulcerated or elicited neurologic or musculoskeletal complications that limited mobility and feeding. TV was evaluated from the first day of treatment. Shown is the average of replicate measurements. (b) Kaplan–Meier survival curves after treated with either vehicle, metformin, bortezomib or both.
Discussion
The discovery of new therapeutics to treat cancers that are resistant to existing therapies remains a challenge. Pharmacologic screens identified metformin as an agent that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Our results are consistent with prior studies, that have established that autophagy is activated upon ER stress as a defensive mechanism for survival41, 42 and that the UPR regulator GRP78 is an obligatory component of stress-induced autophagy.12 Although several cellular conditions have identified autophagy as a pro-survival mechanism that attenuates ER stress and antagonizes apoptosis, autophagy and apoptosis may also cooperate, in parallel or in sequence, to drive cell death.43, 44 Components of the extrinsic apoptosis pathway TNF-related apoptosis-inducing ligand, tumor necrosis factor and fas-associated protein with death domain have been shown to simultaneously induce autophagy.45 The autophagy and apoptosis pathways also share a number of molecules, for example, beclin, bcl2, caspases, ATG5 and ATG12, that facilitate crosstalk and coordinated regulation of these pathways.46 Based upon the spectrum of results observed in diverse model systems, the cellular response to external cues that dictates the outcome as either autophagy or apoptosis may be context-dependent and reflect the cell type, growth conditions as well as the intensity and duration of insult.
The orally-administered, well-tolerated and inexpensive agent metformin enhanced the anti-myeloma effect of bortezomib in vitro and in vivo to suggest that the drug combination offers a therapeutic advantage for myeloma patients. Epidemiological studies indicate that a subset of patients that received metformin to treat T2D exhibited a lower cancer risk and reduced cancer-related mortality.47 Meta-analyses suggest that patients with T2D have a greater incidence of malignancies, including MM.20 Analysis is ongoing to compare myeloma patient outcomes for those concurrently diagnosed with T2D and for those that received bortezomib vs bortezomib and metformin. The risk of bortezomib-induced peripheral neurotoxicity is greater in patients that present with peripheral neuropathy secondary to a history of T2D.48 Bortezomib-induced peripheral neuropathy is a dose-dependent phenomenon and dose reduction may prevent worsening of symptoms. Metformin may not only provide better glucose control but also may allow for reduced bortezomib dosing to prevent the development or worsening of neuropathy.
The effects of metformin observed here required doses much higher than those needed for the same effects with phenformin.49 However, the risk/benefit ratio clearly favors metformin over phenformin. Phenformin was associated with an elevated incidence of lactic acidosis linked to fatal outcomes that prompted removal from the US market. Currently there are >60 clinical studies evaluating the therapeutic effects of metformin in cancer.50
References
Kyle RA, Rajkumar SV . Multiple myeloma. Blood 2008; 111: 2962–2972.
Palumbo A, Anderson K . Multiple myeloma. N Engl J Med 2011; 364: 1046–1060.
Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107: 4907–4916.
Chen Y, Brandizzi F . IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 2013; 23: 547–555.
Hetz C . The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13: 89–102.
Cao SS, Kaufman RJ . Unfolded protein response. Curr Biol 2012; 22: R622–R626.
Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-Ya K et al. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res 2009; 69: 4225–4234.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14: 230–239.
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26: 9220–9231.
Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res 2011; 71: 2848–2857.
Fu Y, Lee AS . Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol Ther 2006; 5: 741–744.
Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS . The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 2008; 15: 1460–1471.
Abdel Malek M, Jagannathan S, Malek E, Sayed DM, Elgammal SA, Driscoll JJ . Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 2015; 6: 3098–3110, advanced online publication.
Lee AS . GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res 2007; 67: 3496–3499.
Wang Q, He Z, Zhang J, Wang Y, Wang T, Tong S et al. Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detect Prev 2005; 29: 544–551.
Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N et al. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol 2008; 10: 236–243.
Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastas 2006; 23: 401–410.
Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res 2008; 68: 498–505.
Liu P, Cheng H, Roberts TM, Zhao JJ . Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009; 8: 627–644.
Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010; 3: 1451–1461.
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD . Metformin and reduced risk of cancer in diabetic patients. BMJ 2005; 330: 1304–1305.
Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N . Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 2007; 67: 10804–10812.
Leclerc GM, Leclerc GJ, Kuznetsov JM, DeSalvo J, Barredo JC . Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS One 2013; 8: e74420.
DeSalvo J, Kuznetsov JN, Du J, Leclerc GM, Leclerc GJ, Lampidis TJ et al. Inhibition of Akt potentiates 2-DG-induced apoptosis via downregulation of UPR in acute lymphoblastic leukemia. Mol Cancer Res 2012; 10: 969–978.
Jakubikova J, Adamia S, Kost-Alimova M, Klippel S, Cervi D, Daley JF et al. Lenalidomide targets clonogenic side population in multiple myeloma: pathophysiologic and clinical implications. Blood 2011; 117: 4409–4419.
Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC . Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 1996; 183: 1797–1806.
Kim KH, Cheong HJ, Kim SJ, Kim SH, Yoon J, Kim HJ et al. Side population of multiple myeloma and multiple myeloma stem cell. Blood, American Society of Hematology Annual Meeting 2014; 124: 5786.
Scudiero DA . Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988; 48: 4827–4833.
Fukuda K, Saikawa Y, Ohashi M, Kumagai K, Kitajima M, Okano H et al. Tumor initiating potential of side population cells in human gastric cancer. Int J Oncol 2009; 34: 1201–1207.
Huang D, Gao Q, Guo L, Zhang C, Jiang W, Li H et al. Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev 2009; 18: 465–473.
Loebinger MR, Giangreco A, Groot KR, Prichard L, Allen K, Simpson C et al. Squamous cell cancers contain a side population of stem-like cells that are made chemosensitive by ABC transporter blockade. Br J Cancer 2008; 98: 380–387.
Kee N, Sivalingam S, Boonstra R, Wojtowicz JM . Utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods 2002; 115: 97–105.
Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K et al. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009; 114: 1046–1052.
Kahn BB, Alquier T, Carling D, Hardie DG . AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005; 1: 15–25.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167–1174.
Sahra IB, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008; 27: 3576–3586.
Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C . Clonogenic assay of cells in vitro. Nat Protoc 2006; 1: 2315–2319.
Richardson PG, Mitsiades C, Hideshima T, Anderson KC . Bortezomib: Proteasome inhibition as an effective anticancer therapy. Future Oncol 2005; 1: 161–171.
Manning BD, Cantley LC . AKT/PKB signaling: navigating downstream. Cell 2007; 29: 1261–1274.
Chen K-F, Yeh P-Y, Yeh K-H, Lu Y-S, Huang S-Y, Cheng A-L . Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res 2008; 68: 6698–6707.
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26: 9220–9231.
Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282: 4702–4710.
Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280: 20722–20729.
Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis 2011; 2: e199.
Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K et al. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J Cell Mol Med 2013; 17: 12–29.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM . New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 2009; 32: 1620–1625.
Badros A, Goloubeva O, Dalal JS, Can I, Thompson J, Rapoport AP et al. Neurotoxicity of bortezomib therapy in multiple myeloma: a single-center experience and review of the literature. Cancer 2007; 110: 1042–1049.
Pentikainen PJ, Neuvonen PJ, Penttila A . Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol 1979; 16: 195–202.
Quinn BJ, Kitagawa H, Memmott RM, Gillis JJ, Dennis PA . Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab 2013; 24: 469–480.
Acknowledgements
JJD is a recipient of the UC Hematology and Oncology Translational Studies Award and a mentor in the Egyptian–US Joint Supervision Program. MAYAM is a recipient of the Egyptian Cultural and Educational Bureau Award in the Egyptian–US Joint Program.
Author contributions
JJD, SJ, MAYAM, NV, KCA and EM designed the experiments. JJD, SJ, MAYAM, EM, NV, TL and KCA analyzed the results and JJD, EM and MAYAM wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Jagannathan, S., Abdel-Malek, M., Malek, E. et al. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 29, 2184–2191 (2015). https://doi.org/10.1038/leu.2015.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.157
This article is cited by
-
Multiple myeloma, a quintessential malignant disease of aging: a geroscience perspective on pathogenesis and treatment
GeroScience (2023)
-
NEDD4L binds the proteasome and promotes autophagy and bortezomib sensitivity in multiple myeloma
Cell Death & Disease (2022)
-
A functional screen with metformin identifies microRNAs that regulate metabolism in colorectal cancer cells
Scientific Reports (2022)
-
Metformin exerts anti-AR-negative prostate cancer activity via AMPK/autophagy signaling pathway
Cancer Cell International (2021)
-
Crosstalk between endoplasmic reticulum stress and oxidative stress: a dynamic duo in multiple myeloma
Cellular and Molecular Life Sciences (2021)
