
Abstract
Serial quantification of BCR–ABL1 mRNA is an important therapeutic indicator in chronic myeloid leukaemia, but there is a substantial variation in results reported by different laboratories. To improve comparability, an internationally accepted plasmid certified reference material (CRM) was developed according to ISO Guide 34:2009. Fragments of BCR–ABL1 (e14a2 mRNA fusion), BCR and GUSB transcripts were amplified and cloned into pUC18 to yield plasmid pIRMM0099. Six different linearised plasmid solutions were produced with the following copy number concentrations, assigned by digital PCR, and expanded uncertainties: 1.08±0.13 × 106, 1.08±0.11 × 105, 1.03±0.10 × 104, 1.02±0.09 × 103, 1.04±0.10 × 102 and 10.0±1.5 copies/μl. The certification of the material for the number of specific DNA fragments per plasmid, copy number concentration of the plasmid solutions and the assessment of inter-unit heterogeneity and stability were performed according to ISO Guide 35:2006. Two suitability studies performed by 63 BCR–ABL1 testing laboratories demonstrated that this set of 6 plasmid CRMs can help to standardise a number of measured transcripts of e14a2 BCR–ABL1 and three control genes (ABL1, BCR and GUSB). The set of six plasmid CRMs is distributed worldwide by the Institute for Reference Materials and Measurements (Belgium) and its authorised distributors (https://ec.europa.eu/jrc/en/reference-materials/catalogue/; CRM code ERM-AD623a-f).
Similar content being viewed by others

Introduction
The BCR–ABL1 fusion gene is the primary pathogenic driver of chronic myeloid leukaemia (CML) and also characterizes a subset of patients with acute lymphoblastic leukaemia. As well as being of diagnostic importance, BCR–ABL1 also serves as a specific marker of the malignant clone, and many laboratories worldwide routinely use serial reverse-transcription quantitative PCR (RT-qPCR) analysis to monitor the response of, individual CML or acute lymphoblastic leukaemia, patients to treatment.1, 2, 3 Indeed, international recommendations for the management of CML include key time-dependent therapeutic milestones based in part on such molecular monitoring.4
For routine testing, two measurements are typically made for each sample under investigation: an estimate of the number of BCR–ABL1 transcripts as a measure of the burden of leukaemia and also the number of transcripts of an internal reference or control gene (CG) as a measure of overall quantity and quality of cDNA. Results for specimens that test positive for BCR–ABL1 are expressed as the ratio of BCR–ABL1/CG transcript numbers in the same volume of cDNA, subject to previously described performance criteria.5,6 For samples that test negative for BCR–ABL1, the number of CG transcripts gives an indication of the sensitivity with which residual disease can be excluded for that particular specimen.1 However, despite the established clinical utility of RT-qPCR for monitoring of CML patients, the comparability of results between testing laboratories may vary widely.7 A major contributor to this variability is the use of different CGs to normalise results.
To help improve the comparability of results, an International Scale (IS) for BCR–ABL1 was proposed,8 which is gradually being implemented by testing laboratories worldwide, most commonly by the derivation of laboratory-specific conversion factors (CFs) or the use of IS-calibrated kits or reagents.9, 10, 11 The IS expresses results as a percentage relative to the standardised baseline established in the International Randomized Study of Interferon and STI571 study; for example, major molecular response (MR), which corresponds to a 3-log reduction from the standardised baseline, is defined as 0.1% BCR–ABLIS.8,12 However, the IS was conceived at a time when most patients had RT-qPCR detectable disease and a major clinical consideration was whether a patient had or had not achieved major MR.13 Second-generation tyrosine kinase inhibitors result in faster and deeper responses compared with imatinib,14,15 and have prompted the need to define levels of deeper MR within the context of the IS. For example MR4, which corresponds to a 4-log reduction from the International Randomized Study of Interferon standardized baseline, has been defined as either (i) detectable disease ⩽0.01% BCR–ABLIS or (ii) undetectable disease in cDNA with ⩾10 000 ABL1 CG transcripts.16 Importantly, many patients treated with second-generation tyrosine kinase inhibitors (as well as an increasing proportion of patients treated long-term with imatinib)17 have undetectable BCR–ABL1 mRNA by RT-qPCR and thus, it has become increasingly important for testing laboratories to estimate comparable and reliable numbers of CG transcripts. Indeed, recent data from the German CML-Study IV have shown that achievement of confirmed MR4,5 at 4 years predicted significantly higher survival probabilities compared with cases who only achieved 0.1–1% BCR–ABLIS.17
Determination of the number of BCR–ABL1 and CG transcripts are typically performed by using a plasmid calibrator, however different calibrators (developed in house or commercially available) are in use worldwide and until now no common reference material exists to which they could be aligned. The aim of this study was to develop an internationally accepted plasmid certified reference material (CRM) that includes BCR–ABL1 and the three most commonly used CGs (ABL1, BCR and GUSB) to help calibrate all measurements of residual disease in CML, and in particular, levels of deep MR.
Materials and methods
Preparation of individual certified plasmid solutions
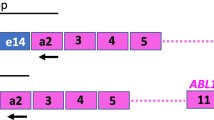
Six plasmid solutions (ERM-AD623a-f), each with a different concentration level, were prepared starting from individual aliquots of the linearised stock pIRMM0099 plasmid (Figure 1 and Supplementary Methods). Dilutions were made in T1E0.01 buffer containing 50 ng/μl Escherichia coli tRNA (Sigma-Aldrich, Gillingham, UK) to yield a range spanning 106–10 copies/μl. The plasmid solutions were sterilised by filtration with a 0.22-μm pore size hydrophilic polyethersulphone membrane (Merck Millipore, Watford, UK) and dispensed into high recovery polypropylene vials. A total of 5000 vials containing approximately 600 μl of plasmid solution were produced for each dilution.

Schematic map of the multitarget plasmid pIRMM0099. The arrows represent the inserts from the transcript fragments of BCR, GUSB and BCR–ABL1. The rectangles show the location of the PCR targets BCR–ABL1 and ABL1 (the ABL1 CG is within the BCR–ABL1 fragment) used quantify the copy number concentration of the plasmid. The single restriction site for EcoRV is also shown.
Digital PCR
Digital PCR was performed by three experienced laboratories: Institute for Reference Materials and Measurements, Geel, Belgium; LGC Limited, Molecular and Cell Biology Team, Teddington, UK and National Measurement Institute, Department of Innovation, Industry, Science and Research, Bioanalysis Group, West Lindfield, NSW, Australia. All three laboratories used the BioMark System (Fluidigm, South San Francisco, CA, USA) and the 12 756 digital array Integrated Fluidic Circuit, which comprises 765 individual partitions of approximately 6 nl volume each with total volume per panel of approximately 4.6 μl. Two PCR targets (one for ABL1 and one for BCR–ABL1 e14a2) were amplified in duplex reaction using the Europe Against Cancer primer/probe sets.5,18 Each sample was analysed on five panels of one digital array and the mean of these five results was considered as one measurement.
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) measurements to assess the homogeneity and stability were performed by the Institute for Reference Materials and Measurements, Geel, Belgium, using the ABI 7900 HT instrument (Applied Biosystems, Lennik, Belgium). The PCR conditions were the same as those used for the digital PCR measurements. For each concentration level, several vials were selected for the homogeneity and stability studies using a random stratified-sampling approach scheme for the whole batch. Each vial was measured three times in separate qPCR runs, and every measurement result was the mean result from three PCR wells (triplicate). A calibration curve with common plasmid solutions of pIRMM0099, produced independently from the stock solutions of ERM-AD623, was included in every qPCR run. This study design avoided 'between-run' effects by using a common calibrant for the calibration curves on each plate. The qPCR results of the BCR–ABL1 PCR target showed the best method repeatability, so these results were used to assess the homogeneity and stability. The set-up of these experiments is described in detail in the certification report of ERM-AD623.19
Statistical analysis
The certified copy number concentration of the six ERM-AD623 plasmid solutions was defined as the mean value of the accepted results from the digital PCR measurements. The combined expanded uncertainties associated with these copy number concentrations consist of uncertainties related to characterisation (uchar), potential between-unit heterogeneity (ubb) and potential degradation during long-term storage(ults). These different contributions were combined to estimate the expanded, relative uncertainty of the certified values (uCRM, rel) with coverage factor k:

Based on the degrees of freedom of the different uncertainty contributions, a coverage factor k of 2 was applied to obtain the expanded uncertainties.20 The calculation of the individual uncertainty contributions (uchar, ubb and ults) are described in the certification report.19
Results
Characterisation of copy number concentration
The certified values for the copy number concentration for the six plasmid solutions (ERM-AD623a-f) were determined by digital PCR measurements, carried out in three experienced laboratories. Eighteen vials of each concentration level were selected using a random stratified-sampling scheme. Each laboratory received six vials of each concentration level and was requested to provide six independent results, one per vial. The content of each vial was diluted gravimetrically and measured on different days and different arrays. For each concentration level, 18 independent results were obtained (Supplementary Table 4). Owing to the technical errors, two independent digital PCR results (one for ERM-AD623a and one for ERM-AD623f) were rejected. For each concentration level, the mean value of the accepted independent digital PCR results was assigned as the certified value for the copy number concentration of the plasmid (Table 1). The uncertainty related to this characterisation exercise, uchar, was also calculated; no statistical difference between the results reported by the three laboratories was found (Supplementary Table 5).
Assessment of homogeneity and stability
Homogeneity and stability studies for each concentration level were performed with qPCR measurements. Inter-unit homogeneity was evaluated to ensure that the certified copy number concentration of each plasmid solution was valid, within a stated uncertainty, for all vials produced for that concentration level. For each concentration level, 23 vials were analysed and statistical analysis of the results showed no outlying results or trends in the filling sequence (Supplementary Figure 3), suggesting that for each concentration level a homogeneous batch was produced. The relative uncertainty related to possible (undetected) heterogeneity, ubb,rel was calculated for each concentration level (Table 1).
Stability testing was performed to establish conditions for long-term and short-term storage using an isochronous design.21 For the short-term stability study, 20 vials of each concentration level were selected and stored at 4 °C for 0, 1, 2 or 4 weeks. All six concentration levels were found to be stable at 4 °C for 4 weeks (Supplementary Figure 4). In the long-term stability study, the stability of each concentration level at −20 °C was tested for nine different time periods with a maximum of 24 months. Based on these results, it can be concluded that the plasmid CRMs can be stored at −20 °C (Supplementary Figure 5). The relative uncertainties related to the stability during long-term storage (24 months) at −20 °C, ults,rel were calculated for each concentration level and are listed in Table 1. In addition, the stability of the plasmid solutions after several freeze-thaw cycles was assessed in a small stability study, with a study design similar to the isochronous study design: 20 vials were exposed to 0, 5, 10 and 20 freeze-thaw cycles. The results obtained for the highest concentration level (ERM-AD623a) showed no significant degradation after 5 or 10 freeze-thaw cycles, however lower copy number concentrations were detected in the vials that were exposed to 20 freeze-thaw cycles. For the other concentration levels that were tested, no degradation was observed after 20 freeze-thaw cycles (Supplementary Figure 6). Although possible degradation was only observed for one concentration level, it was concluded that the plasmid solutions should not be exposed to more than 10 freeze-thaw cycles.
Suitability
Two suitability studies were performed to test the performance of the plasmid solutions (as detailed in the Supplementary Methods). In the small scale study, nine laboratories were included which used 14 validated qPCR assays with one of the three CG: ABL1, BCR or GUSB (Supplementary Table 6). The results obtained with each of these 14 assays were considered as separate data sets. Laboratories were asked to generate 16 standard curves; 8 for their CG(s) of choice and 8 for BCR–ABL1 e14a2, using two sets of ERM-AD623. In addition, each lab measured two common cDNA samples using the 16 calibration curves. The data were evaluated for their technical validity and some data were rejected due to one of the following reasons: deviations from the study protocol, degradation of cDNA samples during transport or too much variation6 within triplicate measurements, suggesting pipetting errors. For one data set, all results were rejected due to technical reasons. For the remaining 13 data sets, 174 individual calibration curves were accepted and evaluated for their slope and co-efficient of determination (r2). In total, 171/174 (98.3%) calibration curves had a slope within the range of −3.1 to −3.6; BCR–ABL1 (n= 84), ABL1 (n=36), BCR (n=23) and GUSB (n=28). Three (1.7%) calibration curves had a gradient less than −3.60 (−3.61 (n=2), −3.62 (n=1)). All calibration curves had an r2 above 0.993.
For the cDNA sample with the highest level of BCR–ABL1, 11 data sets provided technically acceptable results. For the other cDNA sample, one data set had to be removed as the cDNA sample was thawed during transportation, and thus 10 data sets were accepted. The estimated copy number concentration of BCR–ABL1 were equivalent among the different data sets obtained by qPCR methods based on the Taqman technology (Supplementary Figure 7). However, for the qPCR method based on the LightCycler technology the measured copy number concentration of BCR–ABL1 had the tendency to be 1.5–2.0 times lower. When comparing the BCR–ABL1/CG copy number ratio measured with Taqman-based or LightCycler-based methods the difference was smaller, as the measured copy number concentrations of CG were also lower with the LightCycler-based methods. (Supplementary Figure 8). However, the number of data sets per CG were too low to obtain conclusive results. Based on this suitability study, it was concluded that the six CRMs performed satisfactorily and can be used to calibrate different qPCR measurements, by determining the copy numbers of BCR–ABL1 e14a2, ABL1, BCR and GUSB.
A larger suitability study was performed where two different levels of BCR–ABL1 e14a2 aRNA diluted in a background of ABL1 aRNA predicted to correspond to approximately 0.1%, and 0.01% BCR–ABL1/ABL1 plus three different HL60/K562 cell line lysates (~5%, 0.05%, 0.005% BCR–ABL1IS) were sent to 57 European laboratories. These laboratories used a variety of different assays (Supplementary Table 7) for analysis. Analysis of the cell line and aRNA samples indicated a good agreement between the BCR–ABL1/ABL1 copy number ratios obtained with the laboratory calibrators and with ERM-AD623. Importantly, the degree of agreement between centres was significantly improved by the use of ERM-AD623 (Figures 2 and 3). As shown in Tables 2 and 3, the percentage of laboratories reporting results within twofold of the median or expected values was better when ERM-AD623 was used for all comparisons. Results within fivefold were equal or better when ERM-AD623 was used. It was not possible to perform a similar comparison for other CGs, as none of the participating laboratories used BCR and only six used GUSB.

Comparison of measured % BCR-ABL1/ABL1 ratio between laboratory calibrators (Lab) and ERM-AD623 for the cell line dilutions. Data from all centres in the second suitability study that used ABL1 as a control gene are included; median values are indicated.

Comparison of measured % BCR-ABL1/ABL1 ratio between laboratory calibrators (Lab) and ERM-AD623 for the aRNA mixtures. Data from all centres in the second suitability study that used ABL1 as a control gene and returned aRNA data are included; median values are indicated.
Estimates of BCR–ABL1 and ABL1 copy number concentrations were very similar with laboratory calibrators and ERM-AD623 for both the cell line and aRNA dilutions (Figures 4 and 5). However, the aRNA samples were each predicted to contain 305 500 ABL1 copies/μl, plus variable amounts of BCR–ABL1. When laboratory data were corrected for the amount of aRNA added to the PCR (which varied between centres as they were asked to use their routine protocols), the median number of ABL1 copies per μl aRNA was 49 347 using the laboratory standard curve and 56 992 using ERM-AD623 as a standard curve.

Reported numbers of BCR–ABL1 and ABL1 transcripts using laboratory-specific methods (which vary with regard to the amount of material analysed), laboratory calibrators (Lab) and ERM-AD623 for cell line dilutions. Data from all centres in the second suitability study that used ABL1 as a control gene are included; median values are indicated. Values for ABL1 are for all three dilutions combined; values for BCR–ABL1 differ between the three levels and are shown separately.

Estimates of copy numbers of BCR–ABL1 and ABL1 transcripts using laboratory calibrators and ERM-AD623 for the aRNA dilutions in the second suitability study. Individual laboratory protocols have been taken into account to derive estimate per μl of aRNA. Data from all centres in the second suitability study that used ABL1 as a control gene and returned aRNA data are included; median values are indicated. Values for ABL1 are for both dilutions combined; values for BCR–ABL1 differ between the two levels and are shown separately. Expected values for ABL1 if the lysis, reverse transcription and qPCR were all perfect is 305 500; expected BCR–ABL1 values for levels 1 and 2 are 300 and 30, respectively.
Discussion
Over the years, several different in-house and commercial plasmid calibrators for BCR–ABL1 and CG measurement by RT-qPCR have been developed. Typically, these calibrators have independently assigned copy number concentrations based on their molecular weight and DNA concentration, but in the absence of an internationally accepted CRM it is inevitable that variation between calibrators has become established. Although the magnitude of this variation is not known, it is likely to adversely affect patient results since our study, as well as a previous study, both found that the use of a common plasmid calibrator substantially improves the comparability of test sample results between centres.22 Although it is likely that this variation may have been at least partly captured by laboratory-specific CFs, for samples with detectable disease, there is an increasing clinical need for laboratories to make comparable estimates of test sensitivity when BCR–ABL1 is undetectable. Thus, there is an increasing need for laboratories to be able to make comparable and reliable estimates of CG transcript numbers.
We have developed a plasmid CRM, ERM-AD623, as a tool to help standardise the measurement of residual disease in CML. ERM-AD623 consists of a set of six plasmid solutions that are certified for the number of specific DNA fragments per plasmid and the copy number concentration of the plasmid. The number of specific DNA fragments per plasmid is defined by the sequence identity of the plasmid, as determined by dideoxy termination sequencing of the entire plasmid. The plasmid contains three inserts, which are all present as a single copy: one DNA fragment specific for the BCR–ABL1 e14a2 transcript, one DNA fragment specific for the GUSB transcript and one DNA fragment specific for the BCR transcript (Supplementary Table 2). The insert from the BCR–ABL1 e14a2 transcript also contains a large fragment from the native ABL1 transcript. As a consequence, the copy number ratios BCR–ABL1/ABL1, BCR–ABL1/GUSB and BCR–ABL1/BCR of the plasmid are 1/1. The uncertainties related to these copy number ratios are considered to be negligible. The copy number concentration of the plasmid in the six plasmid solutions was determined by digital PCR. The expanded uncertainties, uCRM, associated with the certified copy number concentrations include the uncertainties related to characterisation, uchar; potential between-unit heterogeneity, ubb; and potential degradation during long-term storage,ults (Table 1). Homogeneity was demonstrated and the conditions for storage were established by stability testing.
The suitability of ERM-AD623 as a calibrator for qPCR-methods quantifying the level of BCR–ABL1 e14a2 transcript in cDNA samples was also investigated in two multicentre studies. The plasmid CRM is intended to calibrate the qPCR measurement and not the whole RT-qPCR process, including RNA extraction and reverse transcription. Therefore, no formal commutability studies could be performed. Instead, the suitability studies showed that the analytical behaviour (defined by the r2 and the slope of the calibration curve) of the plasmid solutions in different qPCR assays is within previously defined recommendations.6,23 In the small-scale suitability study, ERM-AD623 was used to calibrate qPCR measurements of two common cDNA samples. The BCR–ABL1/CG copy number ratios thus obtained were equivalent between the different qPCR assays used. However, when comparing the copy number concentration of BCR–ABL1, these results seem to confirm the previously reported variability between methods using TaqMan platforms versus methods using LightCycler platforms.22 This variation may be partly due to the lower input of cDNA in many LightCycler protocols and is likely captured by CFs for detectable disease for which a BCR–ABL1/CG ratio can be calculated. This difference might, however, may need to be taken into consideration in attempts to standardise measurement of undetectable disease. In the large-scale suitability study, we found that the use of ERM-AD623 improved the degree of agreement of results when BCR–ABL1 is detectable for both cell lines and aRNAs. However, there were still large differences in estimates of CG copy numbers for the aRNA samples, for which the initial number of RNA molecules was known. Overall, the median estimates of ABL1 and BCR–ABL1 aRNA numbers were five- to sixfold lower than expected (Figure 5), suggesting that there is substantial room for most laboratories to improve the efficiency of reverse transcription and consequently the sensitivity of their assays.
ERM-AD623 is provided as a set of six plasmid solutions that should be used to construct calibration curves for both BCR–ABL1 e14a2 and the CG of choice. As the sequences of BCR–ABL1 and the CG are located on the same plasmid, the contribution of the calibrator to the measurement uncertainty associated with the measured value for the copy number ratio BCR–ABL1/CG is negligible. Furthermore, we anticipate that having BCR–ABL1 and the CG on the same construct will help to reduce variability compared to assays that use different plasmid calibrators for different targets. Nevertheless, the uncertainty associated with the certified copy number concentrations of the ERM-AD623 solutions must be taken into account when reporting results.
Roughly 98% of CML patients express a p210 BCR–ABL protein, which is encoded in about half of cases by an e14a2 mRNA fusion (BCR exon 14 spliced to ABL1 exon 2; also known as b3a2). A similar proportion expresses the smaller e13a2 (also known as b2a2) variant. About 10% of cases express both e14a2 and e13a2. The remaining 2% of cases are accounted for by multiple-atypical variants, the most common of which are e14a3, e13a3, e6a2, e19a2 and e1a2. Most (70%) cases of BCR–ABL1-positive acute lymphoblastic leukaemia cases express e1a2, with the remaining 30% split, expressing e14a2 and/or e13a2. As e14a2 and e13a2 only differ by 75bp, many laboratories use a common probe/primer set to amplify cDNA derived from both transcripts. However, it should be noted that ERM-AD623 is only certified for the measurement of e14a2 BCR–ABL1; for e13a2 the suitability of ERM-AD623 has to be verified at each laboratory by determining that e14a2 and e13a2 are amplified with equal efficiencies, which can be determined from the standard curve equations by looking specifically at the gradient and intercept components. Importantly, the certified copy number concentrations of ERM-AD623 refer to numbers of double-stranded plasmid molecules and thus should be doubled for use as a qPCR calibrator for single-stranded cDNA.
It is important to note that the use of ERM-AD623 does not by itself produce results on the IS; instead it helps to improve the accuracy of results prior to conversion as well as the accuracy of CG copy-number estimates for samples where BCR–ABL1 is not detected. Conversion to the IS requires established CFs processes9 or the use of secondary reference materials that are traceable to the World Health Organisation International Genetic Reference Panel for the Quantification of BCR–ABL translocation.10 The combination of CFs and a common plasmid calibrator should help testing laboratories to generate standardized results. However, some lack of agreement between results from different laboratories using diverse methodologies and CGs will remain. Whether this remaining disagreement is acceptable, depends on the effect it has on clinical interpretation. When evaluating the performance characteristics of a method, two factors should be considered: trueness (that is the degree of closeness of mean measured-quantity value and the true-quantity value) and the precision (that is the degree to which repeated measurements under unchanged conditions show the same results). The trueness of a method can be estimated by comparing the average value obtained from several replicate measurements on a reference material with an established IS value. The precision of a method can be estimated from the 95% limit of agreement of all the individual measurement results obtained for the reference material.24 Existing experience with the set-up and validation of laboratory-specific CFs has shown that an average difference within ±1.2-fold of the established value and 95% limits of agreement within ±5-fold of the established value were achieved by the best performing methods.9 This led to an major MR concordance rate of 91%, a level of agreement which probably represents the maximum that can be achieved using the current RT-qPCR technology.
Although ERM-AD623 can be used by testing laboratories on a day-to-day basis, it may be used to align local plasmid calibrators, if that is preferred. It can also be used for assay optimisation, for example the cycle threshold or crossing point values for BCR–ABL1 e14a2, and each CG should be identical for each dilution as they are present in a 1/1 ratio. It should be noted that the certified values refer to the use of 5 μl of each dilution; users may wish to use smaller volumes but the uncertainty of the certified value in that case would need to be determined and would be expected to be greater than that stated. In particular, for the lowest level dilution (CRM code ERM-AD623f) Poisson distribution effects would have to be taken into consideration. The plasmid dilutions can be obtained from the Institute for Reference Materials and Measurements or its authorised distributors (https://ec.europa.eu/jrc/en/reference-materials/catalogue/; CRM code ERM-AD623a-f)
References
Cross NC . Standardisation of molecular monitoring for chronic myeloid leukaemia. Best Pract Res Clin Haematol 2009; 22: 355–365.
Press RD, Kamel-Reid S, Ang D . BCR–ABL1 RT-qPCR for monitoring the molecular response to tyrosine kinase inhibitors in chronic myeloid leukemia. J Mol Diagn 2013; 15: 565–576.
Branford S, Prime J . Chronic myelogenous leukemia: monitoring response to therapy. Curr Hematol Malig Rep 2011; 6: 75–81.
Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia. Blood 2013; 122: 872–884.
Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N et al. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe Against Cancer program. Leukemia 2003; 17: 2318–2357.
Foroni L, Wilson G, Gerrard G, Mason J, Grimwade D, White HE et al. Guidelines for the measurement of BCR–ABL1 transcripts in chronic myeloid leukaemia. Br J Haematol 2011; 153: 179–190.
Zhang T, Grenier S, Nwachukwu B, Wei C, Lipton JH, Kamel-Reid S et al. Inter-laboratory comparison of chronic myeloid leukemia minimal residual disease monitoring: summary and recommendations. J Mol Diagn 2007; 9: 421–430.
Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR–ABL transcripts and kinase domain mutations and for expressing results. Blood 2006; 108: 28–37.
Branford S, Fletcher L, Cross NC, Muller MC, Hochhaus A, Kim DW et al. Desirable performance characteristics for BCR–ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood 2008; 112: 3330–3338.
White HE, Matejtschuk P, Rigsby P, Gabert J, Lin F, Lynn Wang Y et al. Establishment of the first World Health Organization International Genetic Reference Panel for quantitation of BCR–ABL mRNA. Blood 2010; 116: e111–e117.
White HE, Hedges J, Bendit I, Branford S, Colomer D, Hochhaus A et al. Establishment and validation of analytical reference panels for the standardization of quantitative BCR–ABL1 measurements on the international scale. Clin Chem 2013; 59: 938–948.
Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 2003; 349: 1423–1432.
Muller MC, Cross NC, Erben P, Schenk T, Hanfstein B, Ernst T et al. Harmonization of molecular monitoring of CML therapy in Europe. Leukemia 2009; 23: 1957–1963.
Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010; 362: 2251–2259.
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010; 362: 2260–2270.
Cross NC, White HE, Muller MC, Saglio G, Hochhaus A . Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012; 26: 2172–2175.
Hehlmann R, Muller MC, Lauseker M, Hanfstein B, Fabarius A, Schreiber A et al. Deep molecular response is reached by the majority of patients treated with imatinib, predicts survival, and is achieved more quickly by optimized high-dose imatinib: results from the randomized CML-study IV. J Clin Oncol 2014; 32: 415–423.
Beillard E, Pallisgaard N, van der Velden VH, Bi W, Dee R, van der Schoot E et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using 'real-time' quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)—a Europe against cancer program. Leukemia 2003; 17: 2474–2486.
Deprez L, Mazoua S, Corbisier P, Trapmann S, Schimmel H, White H et al. The certification of the copy number concentration of solutions of plasmid DNA containing a BCR–ABL b3a2 transcript fragment. Certified reference material: ERM-AD623a, ERM-AD623b, ERM-AD623c, ERM-AD623d, ERM-AD623e ERM-AD623f. Luxembourg: Publications Office of the European Union, 2012; Report number EUR 25248; ISBN 978-92-79-23343-2. 2012.
Joint Committee for Guides in Metrology. Evaluation of measurement data: Guide to expression of uncertainty in measurement (GUM) http://www.bipm.org/utils/common/documents/jcgm/JCGM_100_2008_E.pdf. 2008.
Linsinger TP, Pauwels J, Lamberty A, Schimmel HG, van der Veen AM, Siekmann L et al. Estimating the uncertainty of stability for matrix CRMs. Fresenius J Anal Chem 2001; 370: 183–188.
Muller MC, Erben P, Saglio G, Gottardi E, Nyvold CG, Schenk T et al. Harmonization of BCR–ABL mRNA quantification using a uniform multifunctional control plasmid in 37 international laboratories. Leukemia 2008; 22: 96–102.
van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, van Dongen JJ et al. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia 2003; 17: 1013–1034.
Jennings LJ, Smith FA, Halling KC, Persons DL, Kamel-Reid S . Molecular Oncology Resource Committee of the College of American P. Design and analytic validation of BCR–ABL1 quantitative reverse transcription polymerase chain reaction assay for monitoring minimal residual disease. Arch Pathol Lab Med 2012; 136: 33–40.
Acknowledgements
Initial development of the plasmid was supported by the UK Department of Health. The second suitability study was funded by the European LeukemiaNet (Heidelberg, Germany) and Novartis Pharmaceuticals, Basel, Switzerland, via the ‘European Treatment and Outcome Study’, EUTOS. We thank LGC Limited, Molecular and Cell Biology Team, Teddington, UK, the National Measurement Institute (NMI), Department of Innovation, Industry, Science and Research, Bioanalysis Group, West Lindfield, Australia and Asuragen Inc., 2150 Woodward St, Austin, TX 78744, USA, for their contributions to this work and Paul Strike (NHS Research Design Service South West) for statistical advice.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukaemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
White, H., Deprez, L., Corbisier, P. et al. A certified plasmid reference material for the standardisation of BCR–ABL1 mRNA quantification by real-time quantitative PCR. Leukemia 29, 369–376 (2015). https://doi.org/10.1038/leu.2014.217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2014.217
This article is cited by
-
Estimating prognostic relevant cutoff values for a multiplex PCR detecting BCR::ABL1 in chronic myeloid leukemia patients on tyrosine kinase inhibitor therapy in resource-limited settings
Annals of Hematology (2023)
-
Designing highly multiplex PCR primer sets with Simulated Annealing Design using Dimer Likelihood Estimation (SADDLE)
Nature Communications (2022)
-
Impact of BCR::ABL1 transcript type on RT-qPCR amplification performance and molecular response to therapy
Leukemia (2022)
-
Standardization of molecular monitoring of CML: results and recommendations from the European treatment and outcome study
Leukemia (2022)
-
Development of certified reference material NMIJ CRM 6205-a for the validation of DNA quantification methods: accurate mass concentrations of 600-bp DNA solutions having artificial sequences
Analytical and Bioanalytical Chemistry (2019)
