
Abstract
Fms-like tyrosine kinase-3 is a commonly mutated gene in acute myeloid leukemia, with about one-third of patients carrying an internal-tandem duplication of the juxtamembrane domain in the receptor (FLT3-ITD). FLT3-ITD exhibits altered signaling quality, including aberrant activation of STAT5. To identify genes affecting FLT3-ITD-mediated STAT5 signaling, we performed an esiRNA-based RNAi screen utilizing a STAT5-driven reporter assay. Knockdowns that caused reduced FLT3-ITD-mediated STAT5 signaling were enriched for genes encoding proteins involved in protein secretion and intracellular protein transport, indicating that modulation of protein transport processes could potentially be used to reduce constitutive STAT5 signaling in FLT3-ITD-positive cells. The relevance of KDELR1, a component involved in the Golgi-ER retrograde transport, was further analyzed. In FLT3-ITD-expressing leukemic MV4-11 cells, downregulation of KDELR1 resulted in reduced STAT5 activation, proliferation and colony-forming capacity. Stable shRNA-mediated depletion of KDELR1 in FLT3-ITD-expressing 32D cells likewise resulted in reduced STAT5 signaling and cell proliferation. Importantly, these cells also showed a reduced capacity to generate a leukemia-like disease in syngeneic C3H/HeJ mice. Together our data suggest intracellular protein transport as a potential target for FLT3-ITD driven leukemias, with KDELR1 emerging as a positive modulator of oncogenic FLT3-ITD activity.
Similar content being viewed by others


Introduction
Fms-like tyrosine kinase-3 (FLT3) belongs to the PDGF-receptor family of receptor tyrosine kinases (RTK), and is involved in proliferation and differentiation of B-cell progenitors, myelomonocytic and dendritic cells, as well as in the maintenance of pluripotent hematopoietic stem cells. FLT3 is expressed in multiple hematopoietic lineages1 and is activated by its specific ligand FL.2 The wild-type receptor signals via the AKT-PI3K and the MAPK/ERK pathways.3, 4 Constitutive activation of FLT3 by mutations occurs in hematological malignancies, most commonly in acute myeloid leukemia (AML), causally contributing to leukemia formation.5, 6 The most prevalent type of mutations are internal tandem duplications of sequence in the juxtamembrane domain (FLT3-ITD) and have been found in ∼30% of acute myeloid leukaemia cases.7, 8 The occurrence of FLT3-ITD mutations is associated with poor prognosis and FLT3 is considered a promising target for therapy of AML.9, 10, 11 FLT3-ITD mutant cells are characterized by constitutive, ligand-independent receptor activity. In addition, FLT3-ITD, but not ligand-activated FLT3 WT, phosphorylates STAT5 and upregulates its targets, such as Pim-1/2 kinases.4, 6, 12 The altered signaling quality of FLT3-ITD is tightly associated with its strong transforming potential, pronounced anti-apoptotic effects, and the suppression of myeloid cell differentiation.4, 12, 13, 14, 15 FLT3-ITD is also characterized by impaired maturation, leading to accumulation of underglycosylated proteins in endoplasmatic reticulum (ER)-associated compartments.11 Insight into the mechanisms of retention, in particular, the identification of factors involved in the biogenesis of FLT3-ITD could possibly be exploited to control its transforming capacity.
In order to identify the components controlling the activity of FLT3-ITD, we performed a genome-wide endoribonuclease-prepared siRNA (esiRNA)16 screen, employing a STAT5-driven luciferase reporter system and a FLT3-ITD-expressing cell line. Among downregulated components reducing STAT5 activity, genes encoding proteins involved in intracellular protein transport were found to be enriched. Selected genes reducing FLT3-ITD activity were further validated in MV4-11 human leukemia cells and in FLT3-ITD-expressing 32D cells. In addition to impaired FLT3-ITD activity upon transient knockdown, stable downregulation of KDELR1, a protein that is involved in the retention of proteins in the luminal endoplasmic reticulum, resulted in reduced proliferation, colony formation and capacity of 32D cells to generate a leukemia-like disease in syngeneic C3H/HeJ mice.
Material and methods
Plasmids and esiRNA
Plasmid pLHRE-firefly-luciferase encoding firefly luciferase under the control of a six times-repeated lactogenic hormone responsive element (LHRE) was from Vincent Goffin, (University Descartes of Paris, France).17 Plasmid pRL-SV40-Renilla-luciferase was purchased from Promega (Mannheim, Germany). EsiRNA preparation was performed as reported previously18 using cDNA derived from HEK293 or MV4-11 cells.
Plasmid pLKO.1 vectors encoding shRNA constructs targeting murine or human KDELR1 and plasmid pLKO.1 encoding a non-targeting control shRNA were obtained from Sigma-Aldrich (MISSION shRNA lentivirus-mediated transduction system; Taufkirchen, Germany). If necessary, the puromycin resistance gene was replaced by a neomycin resistance-conferring gene using standard cloning techniques.
Cell lines
The human embryonal kidney cell line HEK293 (obtained from the DSMZ, Braunschweig, Germany) was cultured in DMEM/F12 supplemented with Glutamax, 10% fetal calf serum (FCS) (Invitrogen, Darmstadt, Germany), 20 mM HEPES, pH 7.3, 50 mM β-mercaptoethanol and 2 mM L-glutamine. Cells were grown at 37 °C, in a 5% CO2-humidified incubator. The human leukemic cell line MV4-11 was cultured in RPMI-1640 supplemented with Glutamax, 10% heat-inactivated FCS. Cells were grown at 37 °C in a 5% CO2-humidified incubator.
The coding sequence of the human FLT3 wild type or FLT3-ITD were subcloned into the bicistronic pIREShyg3 vector (Takara Bio Europe/Clontech, St-Germain-en-Laye, France). This ITD allele (36 bp) integrates between codons 598 and 599 in the JM domain of FLT3, and was described previously.19 10 μg plasmid DNA were transfected into HEK293 cells by electroporation. Cells were selected with 50 μg/ml hygromycin B (Calbiochem/Merck Chemicals Ltd., Nottingham, UK) and polyclonal cell lines were used for further experiments. FLT3 expression and phosphorylation were confirmed by immunoblot analysis and flow cytometry as described.19
32D cells stably expressing murine FLT3-ITD were kindly provided by R Grundler and J Duyster (Technical University Munich, Germany).20 The cells were maintained in RPMI-1640 medium supplemented with sodium pyruvate (5 mg/ml), 10% heat-inactivated fetal calf serum (FCS), L-glutamine (2 mM), and 1 ng/ml IL-3 or conditioned medium obtained from murine IL-3 producing BPV cells.21 Cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Antibodies and chemicals
Anti-P-p44/42 MAPK (Thr202/Tyr204, E10, #9106), anti-P-STAT5 (Y694, EPITOMICS, #1208-1), and anti-STAT5 (#9310) antibodies were from Cell Signaling Technology (Frankfurt, Germany). Anti-P-STAT5A/B (Tyr 694/699, 05-886) was purchased from Upstate/Millipore (Schwalbach, Germany), and anti-Erk1 (# M12320) was obtained from BD Transduction Laboratories (Heidelberg, Germany). Anti-GAPDH (Human and Mouse, 1D4) was from Novus Biological (Littleton, CO, USA). For FACS analysis, R-Phycoerythrin (R-PE)-conjugated mouse monoclonal anti-FLT3 (BD, Heidelberg, Germany, Cat # 558996) was used. Antibodies recognizing ß-actin or vinculin were obtained from Sigma-Aldrich. HRP-coupled secondary anti-mouse IgG and anti-rabbit IgG antibodies were from KPL (Gaithersburg, MD, USA). HRP-coupled secondary anti-goat IgG (sc2056) was from Santa Cruz (Heidelberg, Germany). Recombinant human FL and murine IL-3 were purchased from Peprotech, (London, UK).
Genome-scale esiRNA screen
The esiRNA library employed has been described elsewhere.16 For the screen, the pLHRE-firefly-luciferase and the pRL-SV40-Renilla-luciferase plasmids were co-transfected with individual esiRNAs in an arrayed fashion into HEK293 FLT3-ITD cells. For standardization at each microtiter plate 4 replicated samples with esiRNA-targeting human FLT3 (positive control) and esiRNA-targeting GFP (negative control) were applied. Transfection of HEK293 FLT3-ITD for high-throughput screen was performed employing the transfection reagent Effectene (Qiagen, Hilden, Germany). Twenty five nanograms of pLHRE-firefly-luciferase and 25 ng of pRL-SV40-Renilla-luciferase were mixed with 50 ng of esiRNA in 5 μl, 0.6 μl enhancer (Qiagen) and 5.4 μl EC buffer (Qiagen) in 384-well plates (Greiner, Solingen, Germany) and subsequently incubated at room temperature for 5 min. 0.25 μl of Effectene together with 4.75 μl of EC buffer were added, then the plate was centrifuged and incubated for 20 min at room temperature (RT). Thereafter, 4000 cells were seeded in 30 μl of culture medium on top of the formed transfection complex, and after 24 hours, additional 30 μl of medium was added to the wells. Luciferase assays were performed 72 hours after transfection using the Dual Glo luciferase assay (Promega, Madison, WI, USA), according to the procedure recommended by the manufacturer. Z-score values as reference for STAT5 promoter activity were calculated according to z=(x-μ)/δ (x, value of particular sample; μ, mean of all samples; δ, standard deviation of all samples). Z represents the distance between the particular sample score and the population mean in units of the standard deviation. The primers used to generate secondary esiRNAs for the 35 top hits after the primary validation are presented in Supplementary Table 1.
Electroporation of MV4-11 cells was performed as reported by John et al.22 Briefly, 500 000 MV4-11 cells were electroporated with 5 μg of gene-specific esiRNA in 500 μl of culture medium, by a single pulse at 330 V, for 10 ms using the ECM830 electroporator (BTX, Holliston, MA, USA). After the pulse, the cells were incubated at RT for 15 min, before they were seeded in 4.5 ml of pre-warmed culture medium.
RT-qPCR
mRNA was extracted from MV4-11 by Trizol (Invitrogen) according to the protocol of the manufacturer. For reverse transcription of mRNA the Super Script III kit (Invitrogen) was used. Real-time qRT-PCR was performed using the MX300 (Stratagene, Santa Clara, CA, USA) and the ABgene (Hamburg, Germany) SYBR-Green master mix. Gene-specific primers used were: ß-Tubulin-forward, 5′-ATGGCAGTCACCTTCATTGG-3′, ß-Tubulin-reverse, 5′-CCGAAATCCTCCTCCTCTTC-3′; KDELR1-forward 5′-CCTCGCCATCATCTTGCTAC-3′, KDELR1-reverse, 5′-GTGGGAACGACCAGGAACT-3′; FLT3-forward, 5′-GAGAGATGGATTTGGGGCTAC-3′, FLT3-reverse, 5′-GCTCCTCCTGCCTTGTGA-3′. The relative gene expression after different gene knockdown was normalized to ß-Tubulin and calculated with the ddCt method.23
FACS analysis
For quantification of surface-exposed FLT3, the corresponding MV4-11 cells were washed with PBS and stained with PE-labeled anti-CD135 antibody (10 μl/106 cells) by incubating in 250 μl PBS at 4 °C for 30 min, followed by two washes with PBS. Cells were analyzed with a FACS Calibur cytometer (Becton Dickinson, Heidelberg, Germany) and results were analyzed using FlowJo software (Tree Star, Ashland, OR, USA). For determination of surface-localized FLT3 in 32D cells, cells were washed once with PBS and stained with CD135-PE (clone A2F10, catalog # NBP1-43813) from NOVUSBIO (Cambridge, UK). Cells were analyzed with a FACS Canto cytometer (Becton Dickinson). For quantification of surface-exposed FLT3-ITD, the ratio of PE signal to GFP (total FLT3) was calculated.
Production of pseudoviral particles and cell transduction
For generation of lentiviral particles, HEK293T used as packaging cells were maintained in Dulbecco’s modified Eagle medium (DMEM; GIBCO-BRL, Karlsruhe, Germany) supplemented with 10% FCS. The cells were transfected with the pLKO.1 shRNA encoding plasmids and packaging plasmids24 using polyethylenimine (PEI). Media containing lentiviral particles were collected 24, 48 and 72 h after transfection. MV4-11 or 32D cells were infected three times with the pseudotyped particles in presence of 8 μg/ml polybrene (1,5-dimethyl-1,5-diazaundecamethylene polymethobromide, AL-118, #10,768-9, Sigma-Aldrich). The transduced cell pools were selected with 2 μg/ml puromycin or 400 μg/ml G418 48 h post transduction.
Assessment of cell proliferation and clonal growth in methylcellulose
To assess proliferation of the different FLT3-ITD-expressing 32D cell pools, cells were washed twice in IL-3-free medium, seeded in RPMI-1640 medium containing 10% heat-inactivated FCS and sodium pyruvate (5 mg/ml) in cytokine-free medium, or supplemented with IL-3 if indicated. For GFP-fluorescence-based proliferation assays, 2 × 105 cells were seeded in 96-well plates (white plates with clear bottom). At time points indicated in the figure legends, proliferation of quadruplicate samples was determined measuring GFP fluorescence intensity with a Tecan reader (Crailsheim, Germany). Alternatively, proliferation was analyzed using CFSE (carboxyfluorescein diacetate, succinimidyl ester) staining and subsequent monitoring of proliferation mediated dilution of CSFE using flow cytometry. Doubling time of the cells was calculated from the reduction of fluorescence intensity. Initial staining was set to 100%. Alternatively, cells were counted manually.
To analyze colony formation of MV4-11 or 32D cell lines, 3 × 104 cells were plated in 1 ml of a culture mixture containing Iscove’s Modified Dulbecco Medium (Life Technologies, Grand Island, NY, USA), 1% methylcellulose, and 20% FCS per well of a 6 well plate. All samples were prepared in triplicate. The plates were incubated at 37 °C, 5% CO2 for the indicated time periods. The total number of colonies in a well was counted using an Olympus CKX41 microscope equipped with a CAMEDIA C-7070 camera (Olympus Deutschland, Hamburg, Germany), and representative sections of the wells were photographed.
Signaling analysis and western blotting
MV4-11 or 32D cells were washed twice with PBS, and starved in serum- and cytokine-free RPMI-1640 medium for 4 h before stimulation at 37 °C. Subsequently, cells were washed once with ice-cold PBS and lysed. Lysis buffers were freshly supplemented with proteinase inhibitors and phosphatase inhibitors (2 μg/ml Leupeptin, 1% Aprotinin; 2 μg/ml Pepstatin A, 1 mM PMSF, 1 mM Pefabloc; 1 mM sodium-orthovanadate; 1 mM glycerol-phosphate), and lysis was allowed on ice for 15 min, before thorough vortexing and centrifugation.
Aliquots of the cell extracts were subjected to SDS-PAGE. Precast 4–12% gradient gel (Invitrogen) and 10% standard PAA gels were used, followed by blotting on nitrocellulose or PVDF membrane (Millipore, Bedford, MA, USA) using a standard protocol,25 and probing with the indicated primary antibodies. After subsequent incubation with horseradish peroxidase-conjugated secondary antibodies, the blots were developed using Western Lightning chemiluminescence detection (Perkin Elmer Life Sciences; Boston, MA, USA) and quantitatively evaluated using a CCD camera-based system (LAS3000, Fuji, Düsseldorf, Germany). For quantification of specific phosphorylation, blots for phosphorylated proteins were stripped and subsequently reprobed with pan-specific antibodies. Specific phosphorylation was calculated as the ratio of the signals for phosphorylated protein to the signal for total protein detected.
Animal experiments
Eight- to ten-week-old male C3H/HeJ mice, which are syngeneic to 32D cells, were used to assess the in vivo development of leukemia-like disease. 32D muFLT3-ITD cells (2 × 106) were injected into the lateral tail vein. The experimental protocols were reviewed and approved by the local Committee on Animal Experimentation. To study expansion of 32D muFLT3-ITD cells, the animals were killed 10 days post injection. Bone marrow cells were flushed from long bones with PBS, and engrafted bone marrow cells were dissolved by incubation of bones in dissociation buffer (DMEM medium containing 10% FCS, 3 mM CaCl2, 10 mM HEPES, Collagenase D, 1 mg/ml) at 37 °C for 45 min. Spleen cells were isolated from minced tissue. The amount of GFP-positive 32D muFLT3-ITD cells was quantified as the ratio to total cell number using flow cytometry. For histology, pieces of liver and spleen were immersion-fixed after necropsy and organ weighing in a neutrally buffered solution containing 4% formalin at 4 °C for at least 10 days, and then embedded in paraffin. Thereafter, they were cut into 7-μm-thick sections and stained with hematoxylin and eosin (H&E) for histological analysis.
Results
Reporter assay to monitor oncogenic FLT3-ITD activity
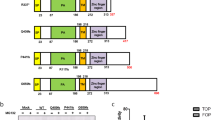
As FLT3-ITD strongly activates STAT5,3, 4, 12, 13, 20, 24, 26 we employed a FLT3-ITD-driven STAT5 activation reporter assay for the screen (Figure 1a). By monitoring the STAT5-driven promoter activity, we aimed at identifying genes modulating the aberrant signaling of FLT3-ITD in response to gene-specific depletion mediated by RNA interference. To allow a streamlined and efficient screening procedure, FLT3-ITD-expressing HEK293 cells were established. Stable expression of FLT3-ITD in HEK293 cells yielded robust activation of STAT5, which could not be observed in cells expressing FLT3 wild-type protein, demonstrating specificity of the receptor-mediated activation (Figure 1b). To validate the specificity of FLT3-ITD-mediated STAT5 activation, we depleted the mutant receptor by RNAi. While a control esiRNA targeting GFP did not alter constitutive STAT5 phosphorylation in HEK293 FLT3-ITD cells, FLT3 esiRNA effectively suppressed the FLT3 receptor level, which was accompanied by abrogation of STAT5 phosphorylation (Figure 1c). To demonstrate the effectiveness of these cells as a STA5 reporter, cells were transfected with the plasmid pLHRE-firefly-luciferase expressing the luciferase gene by the minimal promoter region of the STAT5-responsive lactogenic hormones response element.17 Transient transfection of HEK293 FLT3-ITD cells with pLHRE-firefly-luciferase resulted in strong firefly luciferase activity. In contrast, low luciferase reads were measured in untransfected HEK293 cells or in cells expressing FLT3 WT (Figure 1d). To monitor transfection efficiency, cells were co-transfected with plasmid pRL-SV40, which constitutively expresses Renilla-luciferase from the SV40 enhancer and early promoter elements. Matching Renilla luciferase activity indicated similar plasmid transfection rates in the two cell lines (Figure 1d). Thus, the HEK293 FLT3-ITD reporter system reconstitutes the aberrant FLT3-ITD signaling observed in leukemic cells, and presents a valuable system to conduct the genome-wide RNAi screen.

FLT3-ITD induces STAT5 phosphorylation in HEK293 cells. (a) Strategy for the luciferase-based screening assay. FLT3-ITD-mediated STAT5 activation via phosphorylation (P) drives the expression of the firefly luciferase reporter system. The gray box indicates the internal tandem duplication (ITD) in the juxtamembrane domain of FLT3. (b) HEK293 cells stably expressing FLT3-ITD (ITD), FLT3 WT (WT), or no FLT3 (0) were analyzed with FLT3 and STAT5 phospho-specific antibodies (p). Blots were reprobed for total protein (t). The star indicates an unspecific, cross-reacting band. (c) HEK293 cells stably expressing FLT3-ITD were transfected with esiRNA-targeting FLT3 or GFP (negative control). Downregulation of FLT3 was analyzed by immunoblotting with an anti-FLT3 antibody. Activation of STAT5 was analyzed using an anti-P-STAT5 (p) antibody in comparison to an antibody detecting total STAT5 protein (t). (d) Luciferase activity of HEK293 FLT3 wild type or HEK293 FLT3-ITD cells is shown for cells transfected with the STAT5 reporter plasmid pLHRE-firefly-luciferase encoding firefly luciferase (left columns) and the control plasmid pRL-SV40-Renilla-luciferase (right columns), or non-transfected cells. Cells were transfected with identical amount of plasmid DNA. Luciferase assays were performed 72 hours post transfection.
Genome-wide screen for components affecting FLT3-ITD-mediated STAT5 signaling
To identify proteins influencing oncogenic FLT3-ITD activity, HEK293-FLT3-ITD cells were transfected with pLHRE-firefly-luciferase and individual gene-specific esiRNAs. Co-transfection of plasmid pRL-SV40-Renilla-luciferase, which is independent from FLT3-ITD activity (Figure 1d), was carried out for normalization purposes. Specific firefly luciferase activity was then determined as the ratio between firefly and Renilla luciferase.
In total, 17296 esiRNAs were used in our genome-wide approach and transfection was performed in duplicate in 384-well format. After subsequent cultivation of transfected cells for 48 hours, well-specific activity of firefly luciferase was determined and quantified in relation to the activity of Renilla luciferase. Control wells transfected with esiRNA targeting FLT3 were used to demonstrate the functionality of the assay. Knockdown of FLT3 strongly abrogated firefly luciferase activity, demonstrating the efficiency of the esiRNA application. Cell treatment with esiRNA targeting GFP did not affect the specific luciferase activity and served as negative control. The esiRNA-mediated depletion of expression of particular genes resulted in reduction, stimulation, or in no effect on the firefly to renilla luciferase ratios (exemplified in Figure 2a). A graph showing distribution of Z-scores of the overall screen elucidating the range of alteration of specific luciferase is shown in Figure 2b and the primary screening data is presented in Supplementary Table 2.

EsiRNA-mediated screen for modulators of FLT3-ITD-mediated STAT5 activation. (a) Example of a 384-well plate of the array-based screen quantifying the relative firefly luciferase activity. Alteration of luciferase activity is indicated by color changes in particular wells. Elevated relative firefly luciferase activity is indicated in red. Reduced relative firefly luciferase activity is indicated in green. Positions of control transfections are boxed, with the upper wells in the boxes representing esiRNAs targeting GFP (negative control) and the lower wells in the boxes representing esiRNAs targeting FLT3 (positive control). (b) Graph showing the distribution of Z-scores as mean of relative firefly luciferase activity of 17296 tested esiRNAs is presented. (c) Validation of gene targets employing secondary, independent esiRNAs targeting the indicated genes. The graph demonstrates the relative firefly activity of alternative esiRNA pools of 24 selected candidate genes. Error bars indicate the s.d. from three independent experiments. Significance was determined by the Student t-test. *P<0.05. (d) Gene ontology analysis of validated genes resulting in altered FLT3-ITD-mediated STAT5 signaling. Specific ontologies are indicated and the size of the pies represent the frequencies of the genes associated.
The top hits leading to a reduction of STAT5 signaling were assembled from each plate and 380 (Supplementary Table 3) genes were tested in a first validation experiment. The esiRNAs targeting these genes were assembled on two plates and rescreened with six replicates to increase the statistical power to nominate hits. For the best 35 target genes from this first round of validation, we produced a second, independent esiRNA, targeting a different region of the transcript, to validate target specificity in HEK293-FLT3-ITD (Supplementary Table 3). Twenty four of these targets genes showed a consistent reduction of STAT5-dependent promoter activity (Figure 2c; Supplementary Figure 1 and Supplementary Table 3). By using the ‘panther’ database (www.panther.org), the molecular function of their encoded proteins was classified. Certain classes of proteins might be expected to be found in the employed assay. Indeed, ∼20% of these proteins could be classified as transcription factors (Figure 2d), likely because these proteins directly influence the promoter activity of the lactogenic hormones response element. Consistent with this idea, both STAT5a and STAT5b were amongst the knockdowns that produced a pronounced phenotype. Interestingly, another class of proteins that one might not have expected to emerge from the screen was uncovered. Twenty percent of the validated genes that resulted in reduced reporter expression upon knockdown fall into the ontology of intracellular transport and protein secretion (Figure 2d).
Validation of reduction of STAT5 activity in leukemic MV4-11 cells
In order to validate the activity of selected target genes under more physiological conditions, the human AML cell line MV4-11 carrying endogenous homozygote FLT3-ITD was used. First, gene-specific esiRNA for the 24 validated target genes was prepared from MV4-11-derived cDNA samples. Except for Maf23, expression of 23 out of 24 selected genes was confirmed by efficient cDNA amplification (data not shown). Second, we investigated altered STAT5 phosphorylation after knockdown of the 23 candidate genes in these cells as an independent assay, and to reveal genes that interfere with FLT3 ITD signaling upstream of STAT5 activation. EsiRNA-mediated knockdown of six genes showed a reduction of STAT5 phosphorylation of at least 60% as compared with the control cells treated with negative control esiRNA (Figure 3; Supplementary Figure 2), indicating that their effect on FLT3-ITD-mediated STAT5 signaling is cell line-independent. Interestingly, while phosphorylation of STAT5 and AKT was drastically impaired in most samples, effects on activation of ERK were less prominent.

Knockdown of validated hits changes signaling in MV4-11 cells. The phosphorylation status in relation to the total protein of STAT5, AKT and ERK in esiRNA-transfected MV4-11 cells is shown. Cells were transfected with esiRNAs targeting indicated genes. Forty eight hours post transfection, cells were harvested, lysed and subjected to immunoblotting.) Whole-cell lysates were blotted and phosphorylation (p) of STAT5, AKT and ERK was analyzed using phospho-specific antibodies (left panel). The same blots were reprobed for total proteins (t) and FLT3. GAPDH served as loading control. Representative blots from three replicated experiments are shown. The relative rates of phosphorylation were quantified by division of phosphospecific signals by total protein (right panel). Data obtained from GFP esiRNA treated cells (negative control) were set to 100%. Significance was determined by the Student t-test. *P<0.05.
Despite similar effects on FLT3-ITD signal transduction, the validated six genes encode proteins of different functional categories. EP300 acts as a molecular integrator of transcription in hematopoietic cells. It regulates transcription via chromatin remodeling27 and by interacting with PU.1 and PML IV, thereby enhancing C/EBP epsilon transcription, which is essential in granulocyte differentiation.28 Connexin-26 (Cx-26) is a component of cellular junctions, VDAC3 is a mitochondrial ion channel and RBM15 acts as a RNA-binding protein. Finally, KDELR1 and SLC30A8 have specific roles in protein transport and secretion. Thus, FLT3-ITD-mediated activation of STAT5 is influenced by the perturbation of various pathways. In the further analysis, we have focused on KDLER1 because it has a role in the retention of ER-resident proteins. A possible link to the previously observed ER retention of FLT3-ITD,29 which is associated with STAT5 activation, was intriguing.
Knockdown of KDELR1 stimulates surface localization of FLT3-ITD
As constitutively active FLT3 is retained in cells and its intercellular localization promotes STAT5 activation,11, 30 we addressed the question, if KDELR1 depletion alters the subcellular localization of FLT3-ITD. To investigate this possibility, MV4-11 cells transfected with esiRNA targeting KDELR1 were stained with PE-labeled CD135-PE antibody, and surface loacalization of FLT3 was analyzed in comparison to esiRNA-treated cells transfected with a negative control esiRNA. Interestingly, while the overall FLT3 levels did not change after KDELR1 depletion (Figure 3), we observed an increased accumulation of the receptor at the cell surface (Figure 4). Hence, reduced levels of KDELR1 change the ratio of intracellular to cell surface expression of FLT3-ITD. While esiRNA targeting FLT3 RNA efficiently impaired receptor level, depletion of KDELR1 only enhanced level of surface-exposed FLT3-ITD. Interestingly, depletion of KDELR1 also mildly raised the expression of PU.1 (Supplementary Figure 3), consistent with the attenuation of FLT3-ITD signaling.31

Depletion of KDELR1 enhances surface localization of FLT3-ITD. MV4-11 cells were transfected by esiRNA targeting FLT3, GFP (negative control), or KDELR1. Forty eight hours post transfection, cells were labeled with PE-labelled anti-CD135 antibody and analyzed by flow cytometry (left panel). The intensity of the labeling, reflecting the amount of FLT3-ITD on the cell surface is shown. The result represents a representative example out of three different experiments. A quantification of the flow cytometry data is shown in the right panel. Error bars indicate the s.d. from three independent experiments. Significance was determined by the Student’s t-test. *P<0.05.
Downregulation of KDELR1 impairs proliferation and transformation of haematopoietic FLT3-ITD cells
To elucidate the function of KDELR1 in FLT3-ITD signaling further, we downregulated KDELR1 in human and murine cell systems using stable shRNA expression. A set of non-validated human KDELR1-targeting MISSION shRNA expression cassettes (designated hK01-hK03) was packaged into lentiviral pseudotypes, and used to transduce MV4-11 cells. As demonstrated by qRT-PCR, shRNAs hK02 and hK03 mediated reasonable downregulation of gene expression as compared with cells transduced with particles encoding a nontargeting control shRNA (Figure 5a), with hKO2 showing the most potent knockdown measured at mRNA level.

Stable shRNA-mediated depletion of KDELR1 in MV4-11 cells impairs FLT3-ITD activity. MV4-11 cells were lentivirally transduced with shRNA cassettes targeting KDELR1 (hK01, 02, 03) or a nontargeting shRNA cassette (con) as indicated. (a) qRT-PCR for KDELR1 mRNA in the above cell lines. Mean±s.d. of three independent experiments. One-way ANOVA was performed and post hoc comparisons were made with the Holm–Sidak test. *P<0.05, significant differences to control (con). (b) Signaling activity of MV4-11 shKDELR1 cells. Total cell lysates were subjected to SDS-PAGE, blotted to PVDF membrane and analyzed with antibodies recognizing phosphorylated STAT5. Blots were reprobed for total STAT5. Chemiluminescence signals were detected using a CCD camera-based chemiluminescence detection system. The blots shown are representative for at least three experiments with consistent results. Numbers under the blots represent rates of specific phosphorylation (amount of phosphorylated divided by total signal protein). (c) Proliferation of MV4-11 shKDELR1 lines (hK01, rhombi; hK02, triangles; hK03, squares) and MV4-11 sh control cells (filled circles). CFSE stained cells were grown in RPMI medium supplemented with 10% FCS without cytokines. At the indicated time points, the amount of cell bound CFSE was analyzed using flow cytometry. The graph demonstrates CFSE levels and mean doubling time of cells (in hours). Cells were counted at indicated time points and evaluated by calculating the area under the curve (AUC), respectively. One-way ANOVA was performed and post hoc comparisons were made with the Holm–Sidak test. *P<0.05, significant differences to control (con). Data are given in mean±s.d. The graph shows a representative experiment.
As for the esiRNA-mediated knockdowns, the effect of KDELR1 shRNA knockdown on FLT3-ITD signaling activity was first assessed by analyzing activation of STAT5. MV4-11 cells expressing KDELR1-directed shRNA showed a moderate but reproducible reduction of STAT5 phosphorylation (Figure 5b). Note that, consistent with hKO2 providing the best knockdown, it also showed the most pronounced effect on STAT5 phosphorylation (Figure 5b). Furthermore, reduced KDELR1 levels in MV4-11 were associated with a marked reduction of proliferation in the absence of cytokines as compared with the control cells (Figure 5c).
To further analyze the role of KDELR1 for FLT3-ITD activity, we employed the murine myeloid cell line 32D, stably expressing FLT3-ITD.20 Expression of FLT3-ITD confers growth factor-independent cell growth and transformation of these cells, providing a valuable assay system. KDELR1 levels in these cells were again downregulated using the lentivirus-mediated shRNAs. qRT-PCR revealed downregulation by two shRNAs targeting the murine KDELR1, named mK3 and mK5 (Figure 6a). As knockdown of KDELR1 by the single shRNAs was only relatively modest, shRNA gene cassettes mK3 and mK5 were also simultaneously expressed, using a strategy described previously.24 In the stable cell line 32D, FLT3-ITD mK3+5 double shRNA-targeting of KDELR1 indeed yielded a slightly more pronounced depletion of KDELR1 (Figure 6a).

Growth, signaling and proliferation of 32D FLT3-ITD cell lines depleted for KDELR1. (a) Investigation of knockdown capacity of shRNA constructs. 32D FLT3-ITD cells were lentivirally transduced with shRNA constructs mK3, mK5, mK3 and mK5 (mK3+5), or a nontargeting control shRNA (con). qRT-PCR analyses for KDELR1 mRNA is shown. Mean of at least three independent experiments are presented. One-way ANOVA was performed and post hoc comparisons were made with the Holm–Sidak test. *P<0.05, significant differences to control (con). (b) Signaling activity of 32D FLT3-ITD of KDELR1-depleted cells. Total cell lysates were subjected to SDS-PAGE, blotted to PVDF membrane and analyzed with antibodies recognizing phosphorylated STAT5. Blots were reprobed for total STAT5 and ß-actin. The blots shown are representative for at least three experiments, with consistent results. Numbers under the blots represent rates of specific phosphorylation (amount of phosphorylated divided by total signal protein). (c) Growth capacity of 32D FLT3-ITD cells expressing indicated shKDELR1 cell lines. The cells were seeded in 96-well plates in RPMI medium supplemented with 10% FCS without cytokines. Cell growth was measured by monitoring GFP activity of particular wells at the indicated time points. Values are means of triplicates. The data set shown is a representative of three experiments with consistent results. Differences between cell lines evaluated were compared by unpaired t-tests using Bonferroni correction for multiple uses if appropriate. Significant differences are indicated with * (P<0.05). Values are represented by means and their s.e.
As in the systems tested before, in KDELR1-depleted FLT3-ITD-expressing 32D cells the phosphorylation of STAT5 was reduced as compared with control shRNA-expressing cells. Likewise, we noted again a modest, but significant increase of surface-exposed FLT3-ITD in cells after KDELR1 depletion (Supplementary Figure 4). The extent of reduction of STAT5 activation largely correlated with the extent of downregulation of KDELR1: more efficient downregulation yielded in more pronounced reduction of STAT5 phosphorylation (Figure 6b). Proliferation of the 32D cells was monitored by GFP-mediated fluorescence. GFP is co-expressed with FLT3-ITD in a bicistronic expression system. In absence of cytokines, knockdown of KDELR1 reduced proliferation as compared with the control cells. When FLT3-ITD-dependent proliferation was bypassed by addition of IL-3 (Figure 6c), or when experiments in the parental 32D cells lacking FLT3-ITD were carried out (Supplementary Figure 5), knockdown of KDLER1 did not significantly affect proliferation of these cells, demonstrating that the reduced proliferation is not due to general proliferation impairment upon KDELR1 knockdown.
As FLT3-ITD-induced cell transformation correlates with the capacity of the cells to form colonies in semisolid media, we next asked if reduced activation of STAT5 would translate into an impaired colony formation of KDELR1-depleted cell lines. 32D cells expressing FLT3-ITD and KDELR1-targeting shRNA showed no significant alteration of colony numbers in methyl cellulose plates, although a slight reduction in the size of colonies was observed in cell line expressing shRNA mK3+5 (data not shown). The capacity of colony formation in MV4-11 hK02 cells was, however, reproducibly reduced as compared with MV4-11 cells expressing the control shRNA (Figure 7a).

Depletion of KDELR1 reduces transforming capacity. (a) Clonal growth of MV4-11 cells expressing shKDELR1 (hK02) and shRNA control cells were subjected to colony formation assays in methylcellulose in RPMI medium with 10% FCS. Number of colonies per well were counted after 8 days. Means of normalized values for three separate experiments ±s.d., significance tested by t-test. (b) Knockdown of KDELR1 enhances survival in mice transplanted with 32D FLT3-ITD cells. Kaplan–Meier plot of survival of mice injected with 32D FLT3-ITD cells expressing indicated shRNAs. Development of a leukemia-like disease was analyzed (n=11). For comparison, 32D FLT3-ITD cells expressing a nontargeting shRNA (n=10) were injected. The percentage of surviving mice (y-axis) is plotted with respect to time in days (x-axis). Statistic significance of different survival of mice was P<0.002 (Gehan–Breslow Test). (c) Liver from naive C3H mice (left panel) shows a regular architecture of lobuli, portal field and terminal hepatic venule (black arrow). Mice injected with 32D muFLT3-ITD control shRNA cells (middle panel) exhibit pronounced destruction in liver and spleen with moderate-to-severe hepatomegaly with massive diffuse and nodular leukemic infiltrates (black arrow), focal hepatocellular damage, dilated sinusoids, focal fatty degeneration of hepatocytes (black arrowhead) and massive intravascular leukemic infiltrates in portal and terminal hepatic venule with focal vascular occlusions (black/white arrow). (d) Severe splenomegaly with massive diffuse leukemic infiltrates in red pulp and moderate-to-severe atrophy in white pulp (black arrowhead). Mice injected with 32D muFLT3-ITD control shRNA cells with KDELR1 knockdown (right panel) exhibit a less pronounced pathology, that is, moderate hepato- and splenomegaly, leukemic infiltrates located inside dilated sinusoids in liver (black arrow) as well as diffuse leukemic infiltrates in red pulp (black arrow) and only a moderate atrophy of white pulp (black/white arrow) within the spleen.
Taken together, stable shRNA-mediated downregulation of KDELR1 resulted in impaired phosphorylation of STAT5 and in reduced proliferation in both cellular FLT3-ITD systems tested. These results indicate that KDELR1 depletion indeed results in reduction of the transforming capacity of FLT3-ITD. Therefore, we finally addressed the question, whether the altered KDELR1 levels would affect cell transformation assessed in vivo. 32D cells expressing FLT3-ITD and KDELR1-targeting shRNA mK3+5 cells were analyzed for their capacity to generate a leukemia-like disease in syngeneic C3H/HeJ mice. FLT3-ITD expressing 32D cells caused a rapid disease development as shown previously.32 In contrast, animals injected with KDLER1-depleted cells showed a significant retardation of the development of the myeloproliferative disease when compared with mice injected with control cells (Figure 7b). All mice developed signs of myeloproliferative disease, as depicted by enlarged liver and spleen (Table 1). However, organ enlargement was significantly less pronounced in mice injected with 32D muFLT3-ITD cells with KDELR1 knockdown. The delayed progress in development of myeloproliferative disease was also reflected by the histological features of disease-related organ pathologies, with only moderate manifestation of leukemic infiltrates within dilated sinusoids, central and portal veins in liver as well as moderate leukemic infiltrates in red pulpa and moderate atrophy in white pulpa of spleen from mice injected with 32D muFLT3-ITD KDELR1 knockdown cells (Figures 7c and d). In contrast, mice injected with 32D muFLT3-ITD cells exhibited more pronounced destruction in liver and spleen with massive diffuse and nodular leukemic infiltrates and additional focal hepatocellular damage and focal fatty degeneration of hepatocytes as well as focal splenic infarction owing to vascular occlusion by leukemic infiltrates and perisplenitis (Figures 7c and d). Furthermore, depletion of KDELR1 significantly blocked expansion of 32D FLT3-ITD cells in the spleen and the soluble bone marrow as demonstrated by the amount of GFP-positive 32D muFLT3-ITD cells at day 10 post infection (Table 1). Insignificant differences in the solid bone marrow (Table 1), possibly due to local high IL-3 concentrations, indicated lesser consequences of KDLER1 knockdown in this compartment.
Discussion
The oncogenic transformation of cells expressing FLT3-ITD proteins is mediated by constitutive activity of this RTK.3, 4, 13, 14, 26, 30 The cytokine-independent kinase activity of the RTK results in accumulation in cytosolic compartments on the ER-trans-Golgi-biogenesis route.11 Associated with intracellular retention, FLT3-ITD induces aberrant STAT5 activation29, 30 leading to the induction of a panel of downstream targets.4, 12, 13, 14, 15 Interestingly, our genome-wide RNAi screen identified several factors implicated in protein transport and secretion processes that influence FLT3-ITD-driven STAT5 signaling. While only 2% of the proteome of the mammalian cell can be classified as proteins involved in protein transport, about 20% of the hits of our RNAi-based screen presented components, which are known to be involved in cellular protein transport. This finding suggests that influencing protein transport might be a useful strategy to disrupt this fatal signaling pathway.
A role of KDELR1 for FLT3-ITD-mediated transformation could be validated in further investigations; notably, its knockdown delayed the development of MPD in mice. KDELR1 is a luminal ER protein that acts as a receptor for proteins undergoing retrograde transport from the Golgi to the ER. It recognizes the motif KDEL (Lys-Asp-Glu-Leu) in its client proteins and effectuates the maintenance of protein composition of the ER lumen. Overexpression of KDELR1, while producing an imbalance in the ER–Golgi traffic, resulted in the disruption of the Golgi-mediated protein transport (reviewed in Capitani et al.33). Owing to the abrogation of protein transport from the ER to the Golgi, enhanced KDELR1 results in a phenotype similar to Brefeldin A (BFA) treatment.34 Intriguingly, also treatment of FLT3-ITD-expressing cells with BFA resulted in the accumulation of immature FLT3-ITD and increased selectively STAT5 activation.30
As FLT3 does not contain a KDEL sequence motif, it is unlikely to be a direct client protein of KDELR1. However, effects of KDLER1-depletion on FLT3-ITD trafficking and function may be mediated indirectly. In yeast, the homolog KDEL receptor protein (yeast KDELR shows 50% homology to human KDELR1) seems to have a rather general role for ER function. As Yamamoto et al.35, 36 showed, yeast KDLER mediates a retrieval mechanism that contributes to the quality control at the ER and mediates the ER stress response. In human T-cells, BIP mediates the retrograde transport of the T-cell antigen receptor alpha chain (TCRα) from the Golgi to the ER. BiP, a KDELR1 client, is a KDEL protein that is involved in the retrograde transport to the ER and induction of ER stress response. The disruption of the ligand-binding function of KDELR released TCRα from the early secretory system to the cell surface, leading secondarily to the missorting of BiP. It may be speculated that similar mechanisms have a role in the case of the KDLER1-dependent regulation of FLT3-ITD. The BiP-KDELR1-mediated retrieval from post-ER compartments to the ER may contribute to the observed entrapment of FLT3-ITD. As oncogenic activation of STAT5 has been related to ER-entrapped FLT3-ITD molecules,11, 26, 29, 30 cell treatments restoring transport efficiency of the mutant receptor to the cellular surface might open novel ways to abrogate its transforming capacity.
Taken together, our esiRNA-based screen identified novel mediators of FLT3-ITD signaling. It will be interesting to unravel the molecular mechanisms on how these genes influence the signaling quality of FLT3-ITD in future studies. Furthermore, the identified pathways may offer new avenues to change the pathogenic signaling of this oncogene. As exemplified for KDELR1, factors affecting the aberrant biogenesis are possibly novel molecular targets to impair the transforming capacity of FLT3-ITD-expressing cells.
References
Matthews W, Jordan CT, Wiegand GW, Pardoll D, Lemischka IR . A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 1991; 65: 1143–1152.
Lyman SD, James L, Vanden Bos T, de Vries P, Brasel K, Gliniak B et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell 1993; 75: 1157–1167.
Choudhary C, Schwable J, Brandts C, Tickenbrock L, Sargin B, Kindler T et al. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with FLT3-ITD mutations. Blood 2005; 106: 265–273.
Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000; 19: 624–631.
Gilliland DG, Griffin JD . The roles of FLT3 in hematopoiesis and leukemia. Blood 2002; 100: 1532–1542.
Stirewalt DL, Radich JP . The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 2003; 3: 650–665.
Nakao M, Yokota S, Iwai T, Okamoto T, Taniwaki M, Onodera N et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996; 10: 1911–1918.
Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002; 99: 4326–4335.
Knapper S . The clinical development of FLT3 inhibitors in acute myeloid leukemia. Expert Opin Investig Drugs 2011; 20: 1377–1395.
Schmidt-Arras D, Schwable J, Böhmer FD, Serve H . Flt3 receptor tyrosine kinase as a drug target in leukemia. Curr Pharm Des 2004; 10: 1867–1883.
Schmidt-Arras DE, Böhmer A, Markova B, Choudhary C, Serve H, Böhmer FD . Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol 2005; 25: 3690–3703.
Mizuki M, Fenski R, Halfter H, Matsumura I, Schmidt R, Müller C et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000; 96: 3907–3914.
Mizuki M, Schwable J, Steur C, Choudhary C, Agrawal S, Sargin B et al. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 2003; 101: 3164–3173.
Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T . Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002; 21: 2555–2563.
Rocnik JL, Okabe R, Yu JC, Lee BH, Giese N, Schenkein DP et al. Roles of tyrosine 589 and 591 in STAT5 activation and transformation mediated by FLT3-ITD. Blood 2006; 108: 1339–1345.
Kittler R, Surendranath V, Heninger AK, Slabicki M, Theis M, Putz G et al. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat Methods 2007; 4: 337–344.
Goffin V, Shiverick KT, Kelly PA, Martial JA . Sequence-function relationships within the expanding family of prolactin, growth hormone, placental lactogen, and related proteins in mammals. Endocr Rev 1996; 17: 385–410.
Kittler R, Heninger AK, Franke K, Habermann B, Buchholz F . Production of endoribonuclease-prepared short interfering RNAs for gene silencing in mammalian cells. Nat Methods 2005; 2: 779–784.
Kindler T, Breitenbuecher F, Kasper S, Estey E, Giles F, Feldman E et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood 2005; 105: 335–340.
Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J . Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood 2003; 102: 646–651.
Molineux G, Testa NG . Haemopoiesis—A Practical Approach. Oxford University Press: USA, 1992.
John M, Geick A, Hadwiger P, Vornlocher HP, Heidenreich O . Gene silencing by RNAi in mammalian cells. Curr Protoc Mol Biol. 2003; Chapter 26: Unit 26.2.
Winer J, Jung CK, Shackel I, Williams PM . Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem 1999; 270: 41–49.
Arora D, Stopp S, Böhmer SA, Schons J, Godfrey R, Masson K et al. Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J Biol Chem 2011; 286: 10918–10929.
Towbin H, Staehelin T, Gordon J . Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 1979; 76: 4350–4354.
Choudhary C, Muller-Tidow C, Berdel WE, Serve H . Signal transduction of oncogenic Flt3. Int J Hematol 2005; 82: 93–99.
Blobel GA . CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood 2000; 95: 745–755.
Yoshida H, Ichikawa H, Tagata Y, Katsumoto T, Ohnishi K, Akao Y et al. PML-retinoic acid receptor alpha inhibits PML IV enhancement of PU.1-induced C/EBPepsilon expression in myeloid differentiation. Mol Cell Biol 2007; 27: 5819–5834.
Schmidt-Arras D, Böhmer SA, Koch S, Müller JP, Blei L, Cornils H et al. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood 2009; 113: 3568–3576.
Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Böhmer FD et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell 2009; 36: 326–339.
Choudhary C, Brandts C, Schwalbe J, Tickenbrock L, Sargin B, Ueker A et al. Activation mechanism of STAT5 by oncogenic Flt3-ITD. Blood 2007; 110: 370–374.
Müller JP, Schonherr C, Markova B, Bauer R, Stocking C, Böhmer FD . Role of SHP2 for FLT3-dependent proliferation and transformation in 32D cells. Leukemia 2008; 22: 1945–1948.
Capitani M, Sallese M . The KDEL receptor: new functions for an old protein. FEBS Lett 2009; 583: 3863–3871.
Fullekrug J, Sonnichsen B, Schafer U, Nguyen Van P, Soling HD, Mieskes G . Characterization of brefeldin A induced vesicular structures containing cycling proteins of the intermediate compartment/cis-Golgi network. FEBS Lett 1997; 404: 75–81.
Yamamoto K, Fujii R, Toyofuku Y, Saito T, Koseki H, Hsu VW et al. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J 2001; 20: 3082–3091.
Yamamoto K, Hamada H, Shinkai H, Kohno Y, Koseki H, Aoe T . The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J Biol Chem 2003; 278: 34525–34532.
Acknowledgements
We thank Vincent Goffin (University Descartes of Paris, France) for providing pLHRE-firefly-luciferase plasmid and Drs R Grundler and J Duyster (Technical University Munich, Germany) for providing 32D cells stably expressing FLT WT or FLT3-ITD proteins. This study was supported by grants from the Deutsche Forschungsgemeinschaft Sonderforschungsbereich 655 (BU1400/3-1) and the Deutsche Krebshilfe (106685).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Caldarelli, A., Müller, J., Paskowski-Rogacz, M. et al. A genome-wide RNAi screen identifies proteins modulating aberrant FLT3-ITD signaling. Leukemia 27, 2301–2310 (2013). https://doi.org/10.1038/leu.2013.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2013.83
Keywords
This article is cited by
-
Towards integrated oncogenic marker recognition through mutual information‐based statistically significant feature extraction: an association rule mining based study on cancer expression and methylation profiles
Quantitative Biology (2017)
-
FLT3–ITD and its current role in acute myeloid leukaemia
Medical Oncology (2017)
-
NOX4-driven ROS formation mediates PTP inactivation and cell transformation in FLT3ITD-positive AML cells
Leukemia (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}