
Abstract
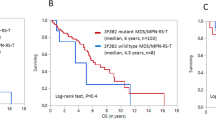
Refractory anaemia with ring sideroblasts (RARS) and marked thrombocytosis (RARS-T) is a provisional entity in the World Health Organisation 2008 classification and has previously been shown to have a high proportion of JAK2V617F (Janus Kinase 2) and SF3B1 (Splicing Factor 3B subunit 1) mutations. The purpose of the present study was to analyse the frequency of SF3B1 mutations in a large cohort of 111 patients with RARS-T and 33 patients with RARS and to explore the prognostic impact of SF3B1 mutational status on RARS-T. The frequency of SF3B1 mutations in RARS-T (96/111, 86.5%) and RARS (28/33, 84.8%) was similar. In RARS-T, median survival was better in SF3B1-mutated patients than in SF3B1-non-mutated patients (6.9 and 3.3 years, respectively, P=0.003). RARS can be differentiated from RARS-T by the frequency of JAK2V617F (0% vs 48.6%). In RARS-T patients, SF3B1 (P=0.021) and JAK2 mutations (P=0.016) were independent factors for a better prognosis. Altogether, our results confirm that RARS-T is an independent entity that should be recognised by the next World Health Organisation classification. The assessment of SF3B1 mutations is of prognostic interest in RARS-T patients. Younger age, JAK2V617F and SF3B1 mutations are the main predicting factors for survival in RARS-T.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Vardiman JW, Bennett JM, Bain BJ, Baumann I, Thiele J, Orazi A . Myelodysplastic/myeloproliferative neoplasms, unclassifiable. In: Swerdlow SH, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H, et al (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue 4th edn. IARC: Lyon, France, 2008; pp 85–86.
Hasserjian RP, Gatterman N, Bennett JM, Brunning RD, Thiele J . Refractory anemia with ringed sideroblasts. In: Swerdlow SH, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H et al (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue 4th edn. IARC: Lyon, France, 2008; pp 96–97.
Thiele J, Kvasnicka HM, Orazi A, Tefferi A, Gisslinger H . Essential thrombocythaemia. In: Swerdlow SH, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H et al (eds) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue 4th edn. IARC: Lyon, France, 2008; pp 48–50.
Ceesay MM, Lea NC, Ingram W, Westwood NB, Gaken J, Mohamedali A et al. The JAK2 V617F mutation is rare in RARS but common in RARS-T. Leukemia 2006; 20: 2060–2061.
Boissinot M, Garand R, Hamidou M, Hermouet S . The JAK2-V617F mutation and essential thrombocythemia features in a subset of patients with refractory anemia with ring sideroblasts (RARS). Blood 2006; 108: 1781–1782.
Flach J, Dicker F, Schnittger S, Kohlmann A, Haferlach T, Haferlach C . Mutations of JAK2 and TET2, but not CBL are detectable in a high portion of patients with refractory anemia with ring sideroblasts and thrombocytosis. Haematologica 2010; 95: 518–519.
Gattermann N, Billiet J, Kronenwett R, Zipperer E, Germing U, Nollet F et al. High frequency of the JAK2 V617F mutation in patients with thrombocytosis (platelet count>600 × 109/L) and ringed sideroblasts more than 15% considered as MDS/MPD, unclassifiable. Blood 2007; 109: 1334–1335.
Hellstrom-Lindberg E, Cazzola M . The role of JAK2 mutations in RARS and other MDS. Hematology Am Soc Hematol Educ Program 2008;, 52–59.
Raya JM, Arenillas L, Domingo A, Bellosillo B, Gutierrez G, Luno E et al. Refractory anemia with ringed sideroblasts associated with thrombocytosis: comparative analysis of marked with non-marked thrombocytosis, and relationship with JAK2 V617F mutational status. Int J Hematol 2008; 88: 387–395.
Remacha AF, Nomdedeu JF, Puget G, Estivill C, Sarda MP, Canals C et al. Occurrence of the JAK2 V617F mutation in the WHO provisional entity: myelodysplastic/myeloproliferative disease, unclassifiable-refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica 2006; 91: 719–720.
Renneville A, Quesnel B, Charpentier A, Terriou L, Crinquette A, Lai JL et al. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia 2006; 20: 2067–2070.
Schmitt-Graeff AH, Teo SS, Olschewski M, Schaub F, Haxelmans S, Kirn A et al. JAK2V617F mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica 2008; 93: 34–40.
Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood 2006; 108: 2173–2181.
Steensma DP, Tefferi A . JAK2 V617F and ringed sideroblasts: not necessarily RARS-T. Blood 2008; 111: 1748.
Wang SA, Hasserjian RP, Loew JM, Sechman EV, Jones D, Hao S et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia 2006; 20: 1641–1644.
Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108: 3472–3476.
Schnittger S, Bacher U, Haferlach C, Dengler R, Krober A, Kern W et al. Detection of an MPLW515 mutation in a case with features of both essential thrombocythemia and refractory anemia with ringed sideroblasts and thrombocytosis. Leukemia 2008; 22: 453–455.
Wardrop D, Steensma DP . Is refractory anaemia with ring sideroblasts and thrombocytosis (RARS-T) a necessary or useful diagnostic category? Br J Haematol 2009; 144: 809–817.
Malcovati L, Della Porta MG, Pietra D, Boveri E, Pellagatti A, Galli A et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood 2009; 114: 3538–3545.
Broseus J, Florensa L, Zipperer E, Schnittger S, Malcovati L, Richebourg S et al. Clinical features and course of refractory anemia with ring sideroblasts associated with marked thrombocytosis. Haematologica 2012; 97: 1036–1041.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478: 64–69.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011; 365: 1384–1395.
Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 2012; 26: 542–545.
Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 2011; 118: 6239–6246.
Damm F, Thol F, Kosmider O, Kade S, Loffeld P, Dreyfus F et al. SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia 2012; 26: 1137–1140.
Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 2012; 119: 3211–3218.
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G et al. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 2012; 119: 569–572.
Visconte V, Makishima H, Maciejewski JP, Tiu RV . Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia 2012; 26: 2447–2454.
Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F et al. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 2012; 120: 3173–3186.
Jeromin S, Haferlach T, Grossmann V, Alpermann T, Kowarsch A, Haferlach C et al. High frequencies of SF3B1 and JAK2 mutations in refractory anemia with ring sideroblasts associated with marked thrombocytosis strengthen the assignment to the category of myelodysplastic/myeloproliferative neoplasms. Haematologica 2013; 98: 15–17.
Cazzola M, Rossi M, Malcovati L . Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013; 121: 260–269.
Lippert E, Boissinot M, Kralovics R, Girodon F, Dobo I, Praloran V et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood 2006; 108: 1865–1867.
Schnittger S, Bacher U, Kern W, Schroder M, Haferlach T, Schoch C . Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia 2006; 20: 2195–2197.
Schnittger S, Bacher U, Haferlach C, Geer T, Muller P, Mittermuller J et al. Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica 2009; 94: 414–418.
Schnittger S, Bacher U, Haferlach C, Beelen D, Bojko P, Burkle D et al. Characterization of 35 new cases with four different MPLW515 mutations and essential thrombocytosis or primary myelofibrosis. Haematologica 2009; 94: 141–144.
Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012; 119: 3578–3584.
Rymond B . Targeting the spliceosome. Nat Chem Biol. 2007; 3: 533–535.
Webb TR, Joyner AS, Potter PM . The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov Today 2013; 18: 43–49.
Matsuda K, Ishida F, Ito T, Nakazawa H, Miura S, Taira C et al. Spliceosome-related gene mutations in myelodysplastic syndrome can be used as stable markers for monitoring minimal residual disease during follow-up. Leuk Res 2012; 36: 1393–1397.
Acknowledgements
We thank the Medical Doctors from the Haematology Department and Laboratory of the University Hospital of Dijon, the Spanish Group of Hematological Cytology (GECH), as well as Philip Bastable for revising the manuscript. EL and FL are grateful to the Tumor Bank of the CHU of Bordeaux. This work was supported by grants from the association ‘Tulipes contre le cancer’ (Châlon s/Saône, Burgundy, France) and from FEHH (Spain) and 2009 SGR 541 (Generalitat de Catalunya).
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
SS and TH declare part ownership of the MLL Munich Leukemia Laboratory. TA, SJ and VG are employed by the MLL Munich Leukemia Laboratory. All the other authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Broséus, J., Alpermann, T., Wulfert, M. et al. Age, JAK2V617F and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 27, 1826–1831 (2013). https://doi.org/10.1038/leu.2013.120
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2013.120
Keywords
This article is cited by
-
Myelodysplastic/myeloproliferative neoplasms with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T): Mayo-Moffitt collaborative study of 158 patients
Blood Cancer Journal (2022)
-
The ABNL-MARRO 001 study: a phase 1–2 study of randomly allocated active myeloid target compound combinations in MDS/MPN overlap syndromes
BMC Cancer (2022)
-
Molecular pathogenesis of the myeloproliferative neoplasms
Journal of Hematology & Oncology (2021)
-
Chronic myeloid neoplasms harboring concomitant mutations in myeloproliferative neoplasm driver genes (JAK2/MPL/CALR) and SF3B1
Modern Pathology (2021)
-
Improved Variant Detection in Clinical Myeloid NGS Testing by Supplementing a Commercial Myeloid NGS Assay with Custom or Extended Data Filtering and Accessory Fragment Analysis
Molecular Diagnosis & Therapy (2021)
