
Abstract
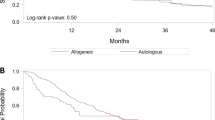
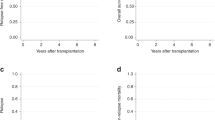
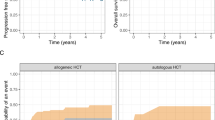
There are limited data on hematopoietic cell transplantation (HCT) in primary plasma cell leukemia (pPCL), an aggressive plasma cell disorder. We report outcomes of 147 patients with pPCL receiving autologous (n=97) or allogeneic (n=50) HCT within 18 months after diagnosis between 1995 and 2006. Median age was 56 years and 48 years for autologous HCT and allogeneic HCT, respectively. Progression-free survival (PFS) at 3 years was 34% (95% confidence interval (CI), 23–46%) in the autologous group and 20% (95% CI, 10–34%) in the allogeneic group. Cumulative incidence of relapse at 3 years was 61% (95% CI, 48–72%) in the autologous group and 38% (95% CI, 25–53%) in the allogeneic group. Overall survival (OS) at 3 years was 64% (95% CI, 52–75%) in the autologous group and 39% (95% CI, 26–54%) in the allogeneic group. Non-relapse mortality (NRM) at 3 years was 5% (95% CI, 1–11%) in the autologous group and 41% (95% CI, 28–56%) in the allogeneic group. The encouraging OS after autologous HCT, establishes the safety and feasibility of this consolidative treatment option after initial induction therapy for pPCL. Allogeneic HCT, although associated with a significantly lower relapse rate, carries a much higher risk of NRM and no OS benefit.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121: 749–757.
Noel P, Kyle RA . Plasma cell leukemia: an evaluation of response to therapy. Am J Med 1987; 83: 1062–1068.
Dimopoulos MA, Palumbo A, Delasalle KB, Alexanian R . Primary plasma cell leukaemia. Br J Haematol 1994; 88: 754–759.
Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008; 22: 1044–1052.
Gertz MA . Managing plasma cell leukemia. Leuk Lymphoma 2007; 48: 5–6.
Fonseca R . Plasma Cell Leukemia. ASH Annu Meet Abstr 2009; 114: SCI–SC4, (22).
Vela-Ojeda J, Garcia-Ruiz Esparza MA, Rosas-Cabral A, Padilla-Gonzalez Y, Garcia-Chavez J, Tripp-Villanueva F et al. Intermediate doses of melphalan and dexamethasone are better than vincristine, adriamycin, and dexamethasone (VAD) and polychemotherapy for the treatment of primary plasma cell leukemia. Ann Hematol 2002; 81: 362–367.
Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant 1995; 15: 825–828.
Lee SJ, Vogelsang G, Gilman A, Weisdorf DJ, Pavletic S, Antin JH et al. A survey of diagnosis, management, and grading of chronic GVHD. Biol Blood Marrow Transplant 2002; 8: 32–39.
Ramsingh G, Mehan P, Luo J, Vij R, Morgensztern D . Primary plasma cell leukemia: a Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer 2009; 115: 5734–5739.
McElwain TJ, Powles RL . High-dose intravenous melphalan for plasma-cell leukaemia and myeloma. Lancet 1983; 2: 822–824.
Saccaro S, Fonseca R, Veillon DM, Cotelingam J, Nordberg ML, Bredeson C et al. Primary plasma cell leukemia: report of 17 new cases treated with autologous or allogeneic stem-cell transplantation and review of the literature. Am J Hematol 2005; 78: 288–294.
Drake MB, Iacobelli S, van Biezen A, Morris C, Apperley JF, Niederwieser D et al. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica 2010; 95: 804–809.
Musto P, Pietrantuono G, Guariglia R, Villani O, Martorelli MC, D'Auria F et al. Salvage therapy with lenalidomide and dexamethasone in relapsed primary plasma cell leukemia. Leuk Res 2008; 32: 1637–1638.
Musto P, Rossini F, Gay F, Pitini V, Guglielmelli T, D'Arena G et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer 2007; 109: 2285–2290.
Esparis-Ogando A, Alegre A, Aguado B, Mateo G, Gutierrez N, Blade J et al. Bortezomib is an efficient agent in plasma cell leukemias. Int J Cancer 2005; 114: 665–667.
Al-Nawakil C, Tamburini J, Bardet V, Chapuis N, Bourry E, Roux C et al. Bortezomib, doxorubicin and dexamethasone association is an effective option for plasma cell leukemia induction therapy. Leuk Lymphoma 2008; 49: 2012–2014.
Palumbo A, Gay F, Falco P, Crippa C, Montefusco V, Patriarca F et al. Bortezomib as induction before autologous transplantation, followed by lenalidomide as consolidation-maintenance in untreated multiple myeloma patients. J Clin Oncol 2010; 28: 800–807.
Acknowledgements
The CIBMTR is supported by Public Health Service Grant/Cooperative Agreement U24-CA76518 from the National Cancer Institute (NCI), the National Heart, Lung and Blood Institute (NHLBI), and the National Institute of Allergy and Infectious Diseases (NIAID); a Grant/Cooperative Agreement 5U01HL069294 from NHLBI and NCI; a contract HHSH234200637015C with Health Resources and Services Administration (HRSA/DHHS); two Grants N00014-06-1-0704 and N00014-08-1-0058 from the Office of Naval Research; and grants from AABB; Allos, Inc.; Amgen, Inc.; Anonymous donation to the Medical College of Wisconsin; Astellas Pharma US, Inc.; Be the Match Foundation; Biogen IDEC; BioMarin Pharmaceutical, Inc.; Biovitrum AB; BloodCenter of Wisconsin; Blue Cross and Blue Shield Association; Bone Marrow Foundation; Buchanan Family Foundation; CaridianBCT; Celgene Corporation; CellGenix, GmbH; Children's Leukemia Research Association; ClinImmune Labs; CTI Clinical Trial and Consulting Services; Eisai, Inc.; Genentech, Inc.; Genzyme Corporation; Histogenetics, Inc.; HKS Medical Information Systems; Hospira, Inc.; Kirin Brewery Co., Ltd.; The Leukemia & Lymphoma Society; Merck & Company; The Medical College of Wisconsin; Millennium Pharmaceuticals, Inc.; Miller Pharmacal Group; Milliman USA, Inc.; Miltenyi Biotec, Inc.; National Marrow Donor Program; Nature Publishing Group; Novartis Oncology; Oncology Nursing Society; Osiris Therapeutics, Inc.; Otsuka America Pharmaceutical, Inc.; Pall Life Sciences; Pfizer Inc; Schering Corporation; Sigma-Tau Pharmaceuticals; Soligenix, Inc.; StemCyte, Inc.; StemSoft Software, Inc.; Sysmex America, Inc.; THERAKOS, Inc.; Vidacare Corporation; ViraCor Laboratories; ViroPharma, Inc.; and Wellpoint, Inc.
DISCLAIMER
The views expressed in this article do not reflect the official policy or position of the National Institute of Health, the Department of the Navy, the Department of Defense or any other agency of the US Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Mahindra, A., Kalaycio, M., Vela-Ojeda, J. et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia 26, 1091–1097 (2012). https://doi.org/10.1038/leu.2011.312
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2011.312
Keywords
This article is cited by
-
Validation of the revised diagnostic criteria for primary plasma cell leukemia by the Korean Multiple Myeloma Working Party
Blood Cancer Journal (2022)
-
A clinical perspective on plasma cell leukemia; current status and future directions
Blood Cancer Journal (2021)
-
First-line treatment and survival of newly diagnosed primary plasma cell leukemia patients in the Netherlands: a population-based study, 1989-2018
Blood Cancer Journal (2021)
-
Allogeneic Stem Cell Transplantation in Patients with High-Risk Multiple Myeloma: Utopia or Continuous Challenge in Aiming for Cure?
Current Treatment Options in Oncology (2021)
-
Successful upfront cord blood transplantation for plasma cell leukemia in the first complete response after daratumumab therapy
International Journal of Hematology (2021)
