
Abstract
Recessive dystrophic epidermolysis bullosa (RDEB) is a debilitating and ultimately lethal blistering disease caused by mutations to the Col7a1 gene. Development of novel cell therapies for the treatment of RDEB would be fostered by having immunodeficient mouse models able to accept human cell grafts; however, immunodeficient models of many genodermatoses such as RDEB are lacking. To overcome this limitation, we combined the clustered regularly interspaced short palindromic repeats and associated nuclease (CRISPR/Cas9) system with microinjection into NOD/SCID IL2rγcnull (NSG) embryos to rapidly develop an immunodeficient Col7a1−/− mouse model of RDEB. Through dose optimization, we achieve F0 biallelic knockout efficiencies exceeding 80%, allowing us to quickly generate large numbers of RDEB NSG mice for experimental use. Using this strategy, we clearly demonstrate important strain-specific differences in RDEB pathology that could underlie discordant results observed between independent studies and establish the utility of this system in proof-of-concept human cellular transplantation experiments. Importantly, we uncover the ability of a recently identified skin resident immunomodulatory dermal mesenchymal stem cell marked by ABCB5 to reduce RDEB pathology and markedly extend the lifespan of RDEB NSG mice via reduced skin infiltration of inflammatory myeloid derivatives.
Similar content being viewed by others


Main
Recessive dystrophic epidermolysis bullosa (RDEB) is caused by mutations to the Col7a1 gene, resulting in dermal–epidermal separation, extensive blistering, and scar formation. Current treatment for RDEB patients is limited to laborious bandaging regimens and management of pain, itching, and bacterial and fungal infections. In addition, individuals with severe RDEB develop pseudosyndactyly, joint contractures, esophageal strictures, and corneal abrasions, as well as being predisposed to aggressive squamous cell carcinomas in young adulthood.1, 2 This disorder is currently incurable.3, 4 The widespread mucocutaneous wounds in individuals with RDEB trigger systemic inflammatory and immune responses that perpetuate and amplify the symptomatology of RDEB.5, 6
Through experiments originating in RDEB mice,7, 8 our group has shown that allogeneic hematopoietic cell transplantation can ameliorate the symptoms of RDEB.9, 10 However, the therapeutic effects are incomplete, and adjunct cellular therapies may allow for more beneficial outcomes. Although genome modification has been performed in immunocompetent mice11, 12, 13 as part of a platform for novel cellular therapy development, we sought herein to use CRISPR/Cas9 to generate an RDEB model on the NOD/SCID IL2rγcnull (NSG) background to explore the role of the immune system in RDEB. The NSG mouse lacks T, B, and NK immune cells and is therefore permissive to human xenotransplantation.14 Previously, generating knockout NSG mice involved time-consuming and labor-intensive backcrossing or gene editing of recently derived, but not yet widely distributed, NSG embryonic stem cells.15 The CRISPR/Cas9 platform has been used to genetically manipulate NOD-Rag1null IL2rγcnull (NRG) mice using pro-nuclear injection, but in these initial studies the embryos were obtained via labor-intensive in vitro fertilization and yielded low numbers of viable offspring.16 Recently, CRISPR/Cas9 has been used to produce knockout NSG mice by standard embryo injection; however, no dose optimization was reported and average gene disruption rates were only ~40% across 20 F0 animals.17 Therefore, we sought to optimize CRISPR/Cas9-based knockout of Col7a1 in NSG embryos to establish a robust platform for generating an immunodeficient mouse model of RDEB.
Materials and methods
CRISPR Reagents
Guide RNAs (gRNAs) targeting the first coding exon of the murine Col7a1 gene were designed using the MIT CRISPR design tool (http://crispr.mit.edu/). For validation, gRNAs were cloned into a U6 expression vector and co-delivered with a Cas9-expressing plasmid into 3T3 cells followed by determination of nuclease activity by Surveyor assay (Integrated DNA Technologies, Coralville, IA, USA). In vitro transcribed gRNAs for microinjection were produced using the MEGAshortscript T7 Transcription kit (Thermo-Fisher Scientific, Waltham, MA, USA) according to manufacturer’s protocols. Cas9 mRNA for microinjection was obtained from TriLink Biotechnologies (San Diego, CA, USA).
Mice
NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; Stock No. 005557) or C57BL/6 (C57BL/6 J; Stock No. 000664) mice were ordered from The Jackson Laboratory (www.jax.org). CD-1 (Crl:CD1(ICR); Strain No. 022) females were obtained from Charles River (www.criver.com). Animals were housed within the University of Minnesota Mouse Genetics lab facilities in non-SPF conditions. All animal studies were approved by the University of Minnesota Institutional Animal Care and Use Committee.
Embryo Injections
Mouse manipulations were performed as previously described,18 with the modification that the CRISPR/Cas9 RNA was injected into the cytoplasm rather than the pro-nucleus. Briefly, 4- to 5-week-old NSG females were super ovulated using standard methods and housed overnight with >8-week-old NSG males. Plugged females were killed for embryo harvest. Following microinjection, embryos were implanted into pseudo-pregnant CD-1 females.
Survival Experiments
Within 24 h of birth, neonatal mice were visually inspected for characteristic blistering on paws. Non-blistered animals were separated from blistered littermates and retained for genotyping and use as potential founders for establishing a breeding colony. Blistered animals were observed daily over the course of the experiment, and deaths were recorded as they occurred. Animal health was monitored closely and with consultation with veterinary staff. Moribund animals that did not respond to treatment with gentamicin were killed and counted as a death. Survival experiments were conducted independently at least twice.
ABCB5+ Cell Isolation
Human ABCB5+ dermal cells were obtained as described previously.19 Briefly, full thickness human skin was obtained from skin biopsies of healthy donors with informed consent. Single-cell suspensions were generated as described previously, and ABCB5+ cells were isolated by positive selection using anti-ABCB5 mAb (clone 3C2-1D12),20 labeling, and magnetic bead cell sorting according to GMP standards (Ticeba, Heidelberg, Germany) as described previously.19 These cells are plastic adherent and reveal the characteristic surface marker expression profile of MSCs.19 ABCB5+ cells fulfill the requirements of the International Society for Cellular Therapy regarding the definition of minimal criteria of MSCs,19 and represent a European Medicines Agency-classified advanced therapy medicinal product currently already undergoing clinical evaluation in a phase I/II human clinical trial in an unrelated disease indication, chronic venous stasis skin ulcer (ClinicalTrials.gov Identifier: NCT02742844).
ABCB5+ Cell Transplantation
Blistered mice were randomly separated into experimental and control groups. ABCB5+ cells were re-suspended in phosphate-buffered saline (PBS) and 5 × 105 cells were injected in 10 μl volume directly into the facial vein.
Immunohistochemistry and Microscopy
Skin from a control mouse and Col7a1−/− NSG mouse was frozen at optimal cutting temperature (OCT, Sakura Finetek USA, Torrance, CA, USA) and cut at 6 μm on a cryostat. Sections were fixed for 5 min in room temperature acetone followed by blocking with 10% normal donkey serum for 1 h (Jackson ImmunoResearch Labs Cat# 017-000-121 Lot# RRID:AB_2337258). Primary antibody collagen VII (1:2000, a kind gift from Drs. David Woodley and Mei Chen) was applied for 1 h, washed, and then secondary antibody donkey anti-rabbit cy3 1:500, (Jackson ImmunoResearch Labs Cat# 711-165-152 Lot# RRID:AB_2307443) was added for 1 h. Slides were washed with 1 × PBS and coverslipped with hard-set DAPI (4,6-diamidino-2-phenylindole, Vector Laboratories Cat# H-1500 Lot# RRID:AB_2336788). Slides were examined by confocal fluorescence microscopy (Fluoview 1000, Olympus BX61, Olympus Optical, Tokyo, Japan). For quantification of CD68+ macrophages, blinded counts were performed on equivalent areas spanning to 200 μm below the epidermis on duplicate tissue sections on at least two animals.
Statistics
Statistical differences in survival were determined by log-rank test using GraphPad Prism software (Graphpad Prism, RRID:SCR_002798). All other statistical analyses were performed using the Student’s t-test with P-values <0.5 being considered significant.
Results
We pursued standard superovulation followed by mating and subsequent embryo collection, embryo injection, and implantation into pseudo-pregnant female surrogates (Figure 1a). To generate RDEB NSG mice, we employed a gene knockout strategy using two gRNA targeting exon 1 of Col7a1. Both gRNAs were delivered simultaneously to maximize the probability of generating a null allele (Figure 1b).21 We designed two high-scoring gRNAs that we subsequently validated in vitro prior to the injection experiments. Both gRNAs had high on-target activity as determined by Surveyor nuclease assay (Figure 1c). In vitro transcribed gRNAs were co-delivered with Cas9 mRNA into single-cell NSG embryos, which were subsequently transferred into pseudo-pregnant surrogates. Our initial dose of CRISPR/Cas9 (50 ng/μl Cas9 and 25 ng/μl each gRNA), resulted in a high level (69%) of biallelic null animals as evidenced by severe blistering and death shortly after birth (Figure 2a; Table 1). Blistered pups showed a complete loss of C7 protein at the dermal-epidermal junction in skin and the mucosal epithelium of the esophagus (Figure 2b). Targeted insertions and deletions (indels) within the first exon of Col7a1 were confirmed by sequencing (Figure 2c). Furthermore, we observed several mice containing biallelic mutations that did not result in frameshift, so the actual frequency of mutation is probably slightly higher than is represented by blistered pups. As our initial goal was to generate animals harboring mono-allelic frameshift mutations that would survive for subsequent breeding, we decided to lower the dose of CRISPR/Cas9 (25 ng/μl Cas9 and 12.5 ng/μl of each gRNA) in subsequent injections. This resulted in a decreased frequency of biallelic knockout animals (34%) and thus a higher number of surviving animals suitable for genotyping and subsequent breeding (Table 1). In our previous experiences using high-quality gRNA such as employed here, off-target activity is extremely low.22 However, in situations where high fidelity gRNAs are not available, the lower dose strategy described here, or a strategy employing a dual nickase system could be employed to minimize off-target mutations.23 Flow cytometric analysis of peripheral blood showed the lack of B, T, and NK lymphoid cells and confirmed that CRISPR/Cas9-modified animals retained the NSG phenotype (Supplementary Figure S1).

CRISPR/Cas9-based disruption of type VII collagen by embryo injection. (a) Strategy using the CRISPR/Cas9 nuclease system to produce Col7a1−/− NSG mice. CRISPR guide RNA and Cas9 mRNA are injected into cytoplasm of single-cell NSG embryos, which are then transferred to CD-1 pseudo-pregnant female surrogates. Upon birth, visibly blistered animals were used for transplantation and/or survival experiments, whereas the non-blistered animals were kept for genotyping and subsequent breeding colony establishment. (b) First coding exon of murine Col7a1. To maximize the frequency of frameshift mutations, two guide RNAs (gRNAs) (green text) were designed to cut near each other within first coding exon. Start codon in red, PAM sequences in orange. Cut site indicated by red triangle. (c) Surveyor assay from transient transfection used to validate the nuclease assay of each gRNA. Red triangles indicate the Surveyor cleavage products.

Phenotypic manifestations of recessive dystrophic epidermolysis bullosa (RDEB) in CRISPR/Cas9 knockout NSG mice. (a) Phenotypic manifestations of RDEB in Col7a1−/− NSG mice. Neonatal mice exhibit blistered paws shortly after birth, followed by formation of the more severe blisters and open wounds characteristic of skin fragility. (b–e) Immunofluorescence staining of type VII collagen expression in Col7a1−/− NSG mice. Cross-sections of skin and esophagus in wild-type (b, d) and knockout (c, e) neonates showing the absence of type VII (red) in the esophageal membrane and at the dermal–epidermal junction in skin. (f) Representative patterns of Col7a1 mutations produced by CRISPR/Cas9 nuclease activity after embryo injection. Indels are observed at both guide RNA (gRNA) target sites independently and simultaneously. gRNA-spanning deletions are also observed.
Interestingly, we observed that Col7a1-null NSG mice appeared to live longer than the previously reported Col7a1 knockout models in our hands, suggesting that the CRISPR/Cas9-mediated gene disruption may result in reduced disease severity via an unknown mechanism. To rigorously test this, we performed side-by-side CRISPR/Cas9-mediated knockout of Col7a1 in both NSG and C57Bl/6 mice using a high dose (100 ng/μl Cas9 and 50 ng/μl of each gRNA) strategy aimed at maximizing the production of biallelic knockout animals. In this setting, we achieved biallelic knockout frequencies of 83% (N=79) and 75% (N=36) for NSG and C57Bl/6 mice, respectively, with no substantial differences in embryo viability (number of viable pups born/number injected embryos implanted) (Table 1). However, in the resulting knockout animals we observed a statistically significant (P<0.001) increase in survival in NSG compared with the C57Bl/6 mice (Figure 3a), suggesting that strain-specific or immune system-related differences in pathology were the cause of the discrepancy in survival rather than the method for gene disruption.

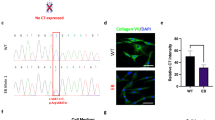
Immunomodulation alleviates recessive dystrophic epidermolysis bullosa (RDEB) pathology. (a) Survival of Col7a1−/− NSG (n=15) and immune-competent C57BL/6 (n=10) mice, created by CRISPR/Cas9 embryo injection (P<0.0001). (b) Survival of Col7a1−/− NSG mice infused with ABCB5+ cells (n=17) versus control (n=8, P=0.01). (c) Immunofluorescence staining for type VII collagen (red) in the mice living past 60 days (long-term survivor, LTS). (d) Immunofluorescence staining of murine CD68+ macrophages within the dermis of Col7a1−/− control (top row), wild-type (middle row), and Col7a1−/− treated with ABCB5+ cells (bottom row). Left column are sections stained for mouse CD45 (green), middle for mouse CD68 (red), and right column are images merged with DAPI nuclear stain. (e) Quantification of intradermal CD68+ macrophages by immunofluorescence staining in neonatal NSG mice (*P<0.05).
Importantly, we found that the high efficiency and robustness of the CRISPR/Cas9 system coupled with the scalability of embryo injection allowed for us to routinely obtain large numbers (>30) of Col7a1−/− animals. Coupled with the relatively precise timing of birth afforded by direct embryo implantation, we found this system to be highly conducive to cellular transplantation experiments. This capability allowed us to conduct proof-of-concept studies in neonatal Col7a1-null NSG mice transplanted with human-dermis-derived cells expressing the ATP-binding cassette sub-family B member 5 (ABCB5) glycoprotein.19, 24 We chose ABCB5+ cells because they share many similarities with mesenchymal stromal/stem cells (MSCs), including the expression of C7 (western blots available online, Supplementary Figure S2), previously demonstrated potent immunosuppressive and anti-inflammatory capabilities,19 and the ability to preferentially home to the skin.25, 26 In blistered mice receiving ABCB5+ cells, we observed a dramatic improvement in survival compared to non-transplanted controls, with 7/17 mice surviving over 50 days and 4 mice surviving to day 67, at which point the animals were harvested for tissue analysis (Figure 3b). We confirmed Col7a1 disruption in surviving mice by sequencing (data shown in Supplementary Figure S3). Importantly, none of the long-term surviving mice were positive for C7 protein, suggesting the therapeutic effect and enhanced survival was mediated by a mechanism other than restoration of the missing C7 protein (Figure 3c). Although all the long-term surviving mice were generally in good health, their coats had a scruffier appearance compared to wild-type littermates, and there was evidence of pseudosyndactyly (for images of these mice, see Supplementary Figure S4). Thus, although the therapeutic effect was substantial, it was not complete. Relevant to this, infused ABCB5+ cells were not detected in the tissue of long-term survivors, either by immunofluorescence microscopy or by quantitative PCR for human-specific DNA sequences (data not shown). We hypothesized that the transplanted ABCB5+ cells may be exerting their therapeutic effect via an alternate, inflammation-based mechanism due to their well-documented immunosuppressive capacity.
To explore this possibility, we used immunofluorescence microscopy to assess infiltration of CD68+ macrophages (which are intact in the NSG mice) into the dermis of mice treated with ABCB5+ cells and observed a significant reduction as soon as 48 h after transplant when compared to untreated neonates (Figures 3d and e). The fact that there was significant decrease as early as 48 h after transplantation suggests that early suppression of inflammatory cell infiltration is sufficient to prevent the severe phenotype associated with RDEB. Our observation that engrafted ABCB5+ cells may exist only transiently in skin offers further insight into RDEB pathology and provides evidence that there appears to be an early therapeutic window for intervention, during which the suppression of monocyte-mediated inflammation allows the mice to survive past a ‘crisis phase,’ after which they can survive for an extended period even in the absence of C7 protein.
Discussion
The pathogenesis of RDEB in humans is complex. The COL7A1 gene is transcribed in fibroblast and keratinocyte nuclei whereby 118 exons encode a polypeptide that gives rise to the C7 protein that forms anchoring fibrils that attach the lamina densa to underlying papillary dermal collagen fibrils. Hundreds of mutations in COL7A1 have been reported, and a wide variety of clinical manifestations may result from structurally and functionally altered or absent C7. Because even monozygotic twins with RDEB may show striking differences in disease severity,27 factors additional to C7 deficiency appear to define clinical phenotype. These include variability in production of pro-fibrotic and pro-inflammatory cytokines, as well as in pathways that may provide inhibitory signals.27 However, models have not until now existed to evaluate such co-factors and to test therapeutic interventions that would target and ameliorate such exacerbating influences that affect blister formation, lesion healing, and fibrotic sequellae.
In this report, we have generated with high efficiency Col7a1 knockout mice on the NSG background using the CRISPR/Cas9 system. When coupled with the scalability of embryo injection, this strategy allowed for rapid generation of large numbers of Col7a1−/− immunodeficient animals for downstream studies. The less severe phenotype of RDEB lesions in the NSG mouse, as compared to those with more intact immune status, is consistent with a role for local inflammatory pathways in lesion generation and persistence. The NSG model is also permissive to the administration of human cells with properties that may further suppress inflammatory and immune activation, as well as potentially restore deficient C7. To this end, we administered ABCB5+ dermal mesenchymal stem cells to RDEB/NSG mice we had generated. Although this maneuver had a significant effect in ameliorating disease severity, the mechanism did not appear to involve replenishment of C7 function. Rather, suppression of innate immune responses in NSG mice mediated by macrophages appeared to potentially play a role in ameliorating disease activity.
ABCB5+ dermal mesenchymal stem cells are known to have the following immunosuppressive properties:24 (i) expression of PD-1; (ii) ability to suppress T-cell proliferation; (iii) capacity to evade immune rejection; (iv) capability to induce regulatory T cells; and (v) homing potential to localize to skin after systemic administration. Although T-cell immunosuppressive capacity is not germane to the NSG model, evidence indicates that PD-1 engagement with PD-L1 displayed by macrophages (and present in NSG animals) induces a regulatory profile characterized by activation of TLR4 downstream MAPK signaling pathways and results in a decrease in inflammatory mediators and production of anti-inflammatory cytokines.28 Moreover, fibroblasts in RDEB have long been known to be abnormally sensitive to macrophage-derived factors with regard to collagenase production involved in lesion evolution and severity.29 Although these pathogenic observations and related speculations are preliminary, and require confirmation and mechanistic validation, they provide proof-of-principle for further studies deploying human immunomodulatory cells in this novel NSG RDEB model system. In particular, although murine ABCB5+ cells have been demonstrated to home to skin,24 we were unable to document the presence of human ABCB5+ cells at the timepoints examined in this study. This is consistent with the transient nature of transplanted MSCs, and follow-on mechanistic studies examining human ABCB5+ cell homing and localization post-delivery will be necessary to maximize therapeutic outcome. Furthermore, it will be of interest to explore the therapeutic capacity of ABCB5+ cells in an immunocompetent setting. The ability of ABCB5+ cells to evade immune rejection and suppress T-cell-based inflammation suggests a strong potential for therapeutic benefit, not only in an immunocompetent setting but also potentially in allogeneic settings as an off-the-shelf cellular intervention.
In sum, we have shown that the CRISPR/Cas9 system is a highly efficient method for generating disease model mice on the immunodeficient NSG background conducive to experiments using human cell xenotransplantation. We show that, in the case of Col7a1, a lower dose of CRISPR/Cas9 is preferable to obtain viable animals harboring mono-allelic frameshift mutations for subsequent breeding, while the increased frequency of pups presenting with RDEB at higher doses allows for reproducible, temporally controlled production of large numbers of biallelic knockout animals suitable for transplantation experiments. Importantly, we show the utility of CRISPR/Cas9-mediated knockout in studying strain-specific differences in disease pathology. This is particularly relevant in RDEB and may explain discrepancies in the results obtained across independent studies in different research labs. Finally, the capability to rapidly produce large numbers of knockout animals allowed us to identify a novel therapeutic modality based on the immunomodulatory ABCB5+ cell population, where transient modulation of the underlying inflammatory response, with particular reference to macrophage inhibition, could be a complementary approach to reducing RDEB pathology. Considering the ease with which the CRISPR/Cas9 system can be programmed to disrupt discrete genomic loci, we envision that this strategy can be rapidly deployed to produce virtually any disease model on the NSG background.
References
South AP, O'Toole EA . Understanding the pathogenesis of recessive dystrophic epidermolysis bullosa squamous cell carcinoma. Dermatol Clin 2010;28:171–178.
Pourreyron C, Cox G, Mao X et al. Patients with recessive dystrophic epidermolysis bullosa develop squamous-cell carcinoma regardless of type VII collagen expression. J Invest Dermatol 2007;127:2438–2444.
Uitto J, Christiano AM, McLean WH et al. Novel molecular therapies for heritable skin disorders. J Invest Dermatol 2012;132 (3 Pt 2):820–828.
Uitto J, Has C, Vahidnezhad H et al. Molecular pathology of the basement membrane zone in heritable blistering diseases: the paradigm of epidermolysis bullosa. Matrix Biol 2017; 57–58: 76-85.
Tolar J, Wagner JE . Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. Lancet 2013;382:1214–1223.
Nystrom A, Thriene K, Mittapalli V et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med 2015;7:1211–1228.
Heinonen S, Mannikko M, Klement JF et al. Targeted inactivation of the type VII collagen gene (Col7a1 in mice results in severe blistering phenotype: a model for recessive dystrophic epidermolysis bullosa. J Cell Sci 1999;112 (Pt 21):3641–3648.
Fritsch A, Loeckermann S, Kern JS et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Invest 2008;118:1669–1679.
Tolar J, Ishida-Yamamoto A, Riddle M et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood 2009;113:1167–1174.
Wagner JE, Ishida-Yamamoto A, McGrath JA et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 2010;363:629–639.
Carbery ID, Ji D, Harrington A et al. Targeted genome modification in mice using zinc-finger nucleases. Genetics 2010;186:451–459.
Sung YH, Baek IJ, Kim DH et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 2013;31:23–24.
Wang H, Yang H, Shivalila CS et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013;153:910–918.
Ito M, Hiramatsu H, Kobayashi K et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002;100:3175–3182.
Landel CP, Dunlap J, Patton JB et al. A germline-competent embryonic stem cell line from NOD.Cg-Prkdc (scid) Il2rg (tm1Wjl)/SzJ (NSG) mice. Transgenic Res 2013;22:179–185.
Li F, Cowley DO, Banner D et al. Efficient genetic manipulation of the NOD-Rag1−/−IL2RgammaC-null mouse by combining in vitro fertilization and CRISPR/Cas9 technology. Sci Rep 2014;4:5290.
Sweeney CL, Choi U, Liu C et al. CRISPR-mediated knockout of Cybb in NSG mice establishes a model of chronic granulomatous disease for human stem-cell gene therapy transplants. Hum Gene Ther 2017;28:565–575.
Behringer R . Manipulating the Mouse Embryo: a Laboratory Manual. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2014.
Jiang D, Muschhammer J, Qi Y et al. Suppression of neutrophil-mediated tissue damage—a novel skill of mesenchymal stem cells. Stem Cells 2016;34:2393–2406.
Ksander BR, Kolovou PE, Wilson BJ et al. ABCB5 is a limbal stem cell gene required for corneal development and repair. Nature 2014;511:353–357.
Mandal PK, Ferreira LM, Collins R et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014;15:643–652.
Osborn MJ, Webber BR, Knipping F et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther 2016;24:570–581.
Shen B, Zhang W, Zhang J et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 2014;11:399–402.
Schatton T, Yang J, Kleffel S et al. ABCB5 identifies immunoregulatory dermal cells. Cell Rep 2015;12:1564–1574.
Galipeau J, Krampera M, Barrett J et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016;18:151–159.
Tolar J, Le Blanc K, Keating A et al. Concise review: hitting the right spot with mesenchymal stromal cells. Stem Cells 2010;28:1446–1455.
Odorisio T, Di Salvio M, Orecchia A et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-beta signalling in modifying disease severity. Hum Mol Genet 2014;23:3907–3922.
Lee Y-J, Moon Y-H, Hyung KE et al. Macrophage PD-L1 strikes back: PD-1/PD-L1 interaction drives macrophages toward regulatory subsets. Adv Biosci Biotechnol 2013;4:19–29.
Nomura K, Imaizumi T, Sawamura D et al. Response of epidermolysis bullosa fibroblasts to factors derived from macrophages and polymorphonuclear leukocytes in terms of collagenase production. J Invest Dermatol 1988;90:170–174.
Acknowledgements
We thank Dr. George Murphy, Brigham and Women's Hospital, Harvard Medical School, for critical reading of the manuscript. This work was supported in part by NIH Grant R01EY025794 (to MHF and NYF), the Department of Veterans Affairs VA Merit Review Award VA RR&D 1I01RX000989 (to NYF), and a Harvard Stem Cell Institute Seed Grant (to MHF). JT is supported in part by NIH R01AR063070 and P01CA065493. MJO is supported by NIH 8UL1TR000114-02. BRW is supported by NIH T32HL007062.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MHF and NYF are inventors of issued or pending ABCB5-related US patents assigned to Boston Children’s Hospital and/or Brigham and Women’s Hospital, Boston, MA, USA and licensed to Ticeba (Heidelberg, Germany) and Rheacell (Heidelberg, Germany). MHF serves as a scientific advisor to Ticeba and Rheacell. CG is CEO and MAK is CSO of Rheacell and Ticeba. This work was supported in part by a grant from Rheacell to MHF. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
T-immunodeficient mouse models of genetic disorders that are able to accept human cell grafts would facilitate development of novel cellular therapies. The authors describe use of the CRISPR/Cas9 system via pronuclear microinjection into immunodeficient mice to rapidly create a model of a genetic disorder with F0 biallelic knockout efficiencies exceeding 80%.
Rights and permissions
About this article

Cite this article
Webber, B., O'Connor, K., McElmurry, R. et al. Rapid generation of Col7a1−/− mouse model of recessive dystrophic epidermolysis bullosa and partial rescue via immunosuppressive dermal mesenchymal stem cells. Lab Invest 97, 1218–1224 (2017). https://doi.org/10.1038/labinvest.2017.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.85
This article is cited by
-
A Review of CRISPR-Based Advances in Dermatological Diseases
Molecular Diagnosis & Therapy (2023)
-
Process data of allogeneic ex vivo-expanded ABCB5+ mesenchymal stromal cells for human use: off-the-shelf GMP-manufactured donor-independent ATMP
Stem Cell Research & Therapy (2020)
-
Human skin-derived ABCB5+ stem cell injection improves liver disease parameters in Mdr2KO mice
Archives of Toxicology (2019)
-
Stammzell-basierter biologischer Gefäßersatz
Gefässchirurgie (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}