
Abstract
Nonalcoholic steatohepatitis is considered as a precancerous condition. However, hepatic carcinogenesis from NASH is poorly understood. This study aims to investigate the activation of pluripotent genes (c-Myc, Oct-4, KLF-4, and Nanog) and morphogenic gene (Gli-1) in hepatic progenitor cells from patient specimens and in an animal model to determine the possibility of normal stem/progenitor cells becoming the origin of NASH-HCC. In this study, expression of pluripotent and morphogenic genes in human NASH-HCC tissues was significantly upregulated compared to adjacent non-tumor liver tissues. After feeding high-fat/calorie diet plus high fructose/glucose in drinking water (HFC diet plus HF/G) for up to 12 months, mice developed obesity, insulin resistance, and steatohepatitis with significant necroptotic inflammation and fibrotic progression, as well as occurrence of hyperplastic nodules with dysplasia; and this model represents pathohistologically as a transition from NASH to NASH-HCC in a pre-carcinomatous stage. High expression of pluripotent and morphogenic genes was immunohistochemically visualized in the dysplasia areas of mouse liver, where there were many OV-6-positive cells, indicating proliferation of HOCs in NASH with fibrotic progression. Moreover, oncogenic transcription factors (c-Myc, KLF-4, and Nanog) were co-localized in these hepatic progenitor cells. In conclusion, pluripotent and morphogenic genes may contribute to the reprogramming of hepatic progenitor cells in driving these cells to be the origin of NASH-HCC in a steatotic and inflamed microenvironment.
Similar content being viewed by others


Main
Hepatocellular carcinoma (HCC) is one of the most common malignancies with increasing incidence in many countries. Various risk factors of HCC have been identified, including hepatitis B or C virus (HBV or HCV) infection and alcohol consumption. As nonalcoholic fatty liver disease (NAFLD) becomes epidemic, its progressive form, nonalcoholic steatohepatitis (NASH), may progress to fibrosis/cirrhosis or end-stage liver disease (ESLD). As one of the fatal consequences or complications of ESLD, NASH-associated HCC (NASH-HCC) has significantly contributed to an increased incidence of HCC in the United States in the latest few decades.1, 2 NASH-associated ESLD and HCC have become the second etiology for liver transplantation in the United States.3 It is not surprising that HCC develops in cirrhotic livers; however, NASH-HCC does occur in NASH without cirrhosis although the incidence remains relatively low.4 How HCC develops in a steatotic microenvironment and what factors facilitate the initiation of NASH and progression to fibrosis, and promote the development, progression, and metastasis of NASH-HCC are intriguing in revealing an oncogenic process in persistent steatohepatitis and progressive fibrosis in NASH patients, and of great value in the development of clinical monitoring guidelines in NASH individuals with high risk.
Cancer stem cells (CSCs) or tumor-initiating cells (T-ICs) are thought to be the origin for oncogenic development, progression, metastasis or relapse after resection, or adjuvant interventions. The origin and existence of CSCs or T-ICs in HCC development face great challenges and are under an active debate. One assumption postulates that CSCs or T-ICs may derive from steatotic hepatocytes.5, 6 Others hypothesize the transformation of liver progenitor cells, such as hepatic oval cells (HOCs) or ductular epithelial cells that normally are responsible for replacement of injured or dead hepatocytes or cholangiocytes, under an inflamed microenvironment with epigenetic or genetic modification.6, 7 However, what factors and how these factors (such as a microenvironment) affect the transformation from normal repairing or regenerative cells to malignant CSCs or T-ICs is elusive, and demands a great effort in clinical observation and laboratory investigation using a proper animal model.
Pluripotent genes are transcription factors (eg, c-Myc, KLF-4, Nanog, Oct-4, and Sox-2) that could reprogram somatic cells into induced pluripotent stem cells (iPSCs), and have been of the revolution in terms of their capability in reprogramming a cell fate, and are fundamental in regenerative medicine. However, these pluripotent transcription factors did induce tumors during the process of iPSC reprogramming in a rather high rate.8 At the same time, the involvement of hedgehog signaling in NASH and fibrotic progression may add an additional morphogenic influence toward malignant transformation.9 Therefore, we hypothesized that the occurrence of oncogenic pluripotent and morphogenic factors in steatotic liver might facilitate the oncogenic transformation of steatotic hepatocytes or liver progenitor cells, which may provide valuable insights into existing questions regarding how HCC develops during NASH and fibrotic progression.
In the present study, we examined pluripotent genes (c-Myc, Oct-4, KLF-4, and Nanog) and morphogenic gene (Gli-1) in NASH-HCC patient specimens and an animal model of neoplastic transition from NASH to hyperplastic nodules of pre-carcinomatous lesions. Our clinical and experimental findings conferred supporting evidence in demonstrating that activation of pluripotent and morphogenic genes in NASH might be crucial for the oncogenic transformation of steatotic hepatocytes or liver progenitor cells during NASH-HCC development.
Materials and methods
Procurement of Tissue Specimens from Fudan University Liver Cancer Institute Tissue Repository
The use of pathology-confirmed and surgically-resected liver cancer specimens from the Liver Cancer Institute Tissue Repository was approved by the Ethic Committee of Fudan University School of Basic Medical Sciences. From >2000 cases of resected paraffin-embedded HCC specimens, 14 pairs of HCC and peri-carcinoma liver tissues were identified after excluding HBV, HCV, alcoholic, and any other genetic or non-genetic liver diseases. Steatosis or steatohepatitis without cirrhosis in peri-carcinoma tissues was histologically confirmed.
Mouse Model of NASH-HCC
C57BL/6J male mice aged about 6 weeks were obtained from Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China. Mice were randomly separated into two groups: HFC diet+HF/G group (high-fat/calorie diet plus high fructose/glucose in drinking water, n=20) and control diet group (n=10). Mice were maintained at 22 °C in a specific pathogen-free facility of Fudan University Experimental Animal Center on a 12 h light/dark cycle. Mice in HFC diet+HF/G (high-fat/calorie diet plus high fructose and glucose) group were fed a high-fat/calorie diet with 60% kcal from fat (cat. no. D12492, Research Diets, New Brunswick, NJ, USA) similar in composition to an American fast-food diet, supplemented with a final concentration of 42 g/l of fructose/glucose in drinking water (23.1 g/l fructose (w/w: 55%)+18.9 g/l glucose (w/w: 45%) (Sigma-Aldrich, St Louis, MO, USA)).10 Control mice were fed a chow pellet diet containing 12% kCal from fat with normal drinking water. Food and drinking water were provided ad libitum. The consumption of diet was monitored by calculating difference between provided and remaining diet on each cage weekly. Body weight of each mouse was weighed weekly. Animal care and procedures were approved by the Institutional Animal Care and Use Committee, Fudan University School of Basic Medical Sciences, and all procedures were conducted following the NIH Guidelines of Experimental Animal Handling and Use.
Animal Killing and Tissue Collection
Mice were anesthetized with sodium pentobarbital (60 mg/kg, i.p.). Blood was collected from the eye orbit, and mice were killed by cervical dislocation under anesthesia. Serum was separated by centrifugation (1000 g for 10 min at room temperature). Fresh liver tissue from each mouse was divided into three parts: the first was for formalin fixation and paraffin embedding; the second was transferred to 1.5 ml RNase-free microcentrifuge tubes containing 1 ml TRIZOL reagent for total RNA extraction; and the third was snap-frozen in liquid nitrogen and stored at −80 °C.
Intraperitoneal Glucose Tolerance Test
To assess glucose tolerance, intraperitoneal glucose tolerance test was performed as follows: after being fasted for 15 h, mice were injected intraperitoneally with glucose (2.5 g/kg body weight), and glucose level in blood from tail vein incision was measured at 0, 30, 60, 90, and 120 min after glucose administration by a portable glucometer (cat. no. HGM-112, Omron Company, Dalian, China).
Biochemical Analysis
Serum levels of alanine transaminase (ALT), aspartate transaminase (AST), total bilirubin (TBIL), and total cholesterol (TCHO) were determined by an automated biochemical analyzer (Olympus AU1000; Olympus Corporation, Tokyo, Japan). Liver triglyceride (TG) levels were measured with a TG quantification kit according to a protocol provided by the manufacturer (cat. no. E1013, Applygen Technologies, Beijing, China). Total hepatic hydroxyproline content was measured with a commercial detection kit following the manufacturer’s instructions (cat. no. A030, Jiancheng Biotechnology Company, Nanjing, China).11 Fasting serum insulin (FINS) was measured using an Ultrasensitive Mouse Insulin ELISA kit (cat. no. EZRMI-13K, Millipore, Billerica, MA, USA). Homeostasis model assessment-insulin resistance (HOMA-IR) index was calculated based on the following formula:12 HOMA-IR=(FINS (mU/l) × fasting serum glucose (mmol/l))/22.5.
RNA Extraction and Real-Time Reverse Transcription PCR
RNA was extracted from two different types of specimens: paraffin-embedded human liver tissue and fresh mouse liver tissue. RNA from paraffin-embedded human tissue was extracted from thick (10 mm) paraffin-embedded HCC and peri-carcinoma sections by using the Recover-All Total Nucleic Acid Isolation Kit (cat. no. AM1975, Ambion, Austin, TX, USA). Total RNA of fresh liver tissue was extracted using TRIZOL reagent. The amount and quality of RNA were determined by a Nanodrop spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Total RNA (2 μg) was reversely transcribed into cDNA templates using a PrimeScript RT reagent kit (cat. no. RR037A, Takara Bio, Dalian, China). Quantitative RT-PCR amplification was performed with a Power SYBR green PCR Master Mix (cat. no. 4368708, Applied Biosystems, Foster City, CA, USA) in triplicate using an Eppendorf realplex cycler (Eppendorf, Hamburg, Germany). Relative gene expression was calculated using 2−ΔΔCt method13, 14 and normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) house-keeping gene control. Primers utilized in quantitative RT-PCR are listed in Supplementary Table S1.
Histopathological Analysis
Formalin-fixed and paraffin-embedded liver tissues were sectioned routinely and stained with hematoxylin and eosin (H&E). All liver sections were examined, and extent of steatosis, cell necrosis, inflammatory infiltration, and fibrosis were semi-quantitatively scored according to the criteria of NAFLD activity by a board-certified pathologist blind to the animal experimental protocol. In the scoring system proposed by Kleiner et al,15 the NAFLD activity score (NAS) is defined as the unweighted sum of steatosis (0–3), lobular inflammation (0–3), and hepatocellular ballooning degeneration (0–2). ‘NAS ≥5’ means ‘definitive NASH,’ ‘NAS=3 or 4’ stands for ‘borderline NASH,’ whereas ‘NAS ≤2’ represents ‘non-NASH.’ After Masson’s trichrome staining (cat. no. HT-15, Sigma-Aldrich), hepatic fibrosis was semi-quantitatively scored using the following criteria:15 4=cirrhosis; 3=bridging fibrosis; 2=zone 3 and portal/periportal fibrosis; 1=zone 3 fibrosis only; and 0=no fibrosis. Ten randomly chosen fields of Masson’s stained sections were photographed under × 200 magnification. Percentage of the positive staining area was analyzed with ImageJ software (the National Institutes of Health, Bethesda, MD, USA). Liver apoptosis was assessed after TUNEL staining (cat. no. 11684817910, Roche, Indianapolis, IN, USA). In addition, frozen liver sections from the different groups of mice were stained oil Red-O (cat. no. O0625, Sigma-Aldrich) to clearly differentiate lipid accumulation in the hepatocytes. The oil Red-O-positive areas were semi-quantitatively analyzed with ImageJ software.
ELISA Determination of TNF-α and IL-1β Concentration in the Mouse Liver
To determine the concentrations of TNF-α and IL-1β, mouse livers in 50 mg wet weight were cut into small pieces on dry ice and then homogenated in 0.65 ml of tissue homogenization buffer containing 20 mM Tris, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, and 0.1% SDS (pH 7.5) with protease inhibitors cocktail (cat. no. P8340, Sigma-Aldrich). The resulting homogente was centrifuged for 30 min at 16 000 g at 4 °C. Supernatant was collected and determined for protein content using the BCA protein assay kit (cat. no. 23225, Pierce, Rockford, IL, USA). TNF-α and IL-1β concentration was then determined by commercially available ELISA kits (cat. no. 88-7324, 88-7013 eBioscience, San Diego, CA, USA) according to manufacturer’s instructions. TNF-α and IL-1β levels were expressed as pg/mg total protein.
Immunofluorescent and Immunohistochemical Staining
Immunofluorescent and immunohistochemical staining was performed according to the standard protocols. Briefly, tissue sections were deparaffinized, rehydrated, and subjected to antigen retrieval by immersing sections in citrate buffer (10 mM sodium citrate; pH 6.0) at 95 °C for 30 min, and allowed to cool for 30 min. Slices were washed in PBS for 5 min for three times, treated with 3% H2O2 for 30 min to block endogenous peroxidase activity, and rinsed in PBS for 5 min. Sections were blocked with 3% bovine serum albumin, followed by incubation with primary antibody at 4 °C overnight, and rinsed in PBS for 5 min for three times. For immunohistochemical staining, sections were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature, and color was developed with a 3,3′-diaminobenzidine chromogenic reagent (cat. no. ZLI-9017, Beijing Zhongshan Golden Bridge Biotechnology, Beijing, China) and counterstained with hematoxylin. For immunofluorescent staining, sections were incubated for 2 h at room temperature with secondary antibody, and nucleus was counterstained with 4′6-diamidino-2-phenylindole (DAPI) (cat. no. C1005, Beyotime Biotechnology, Haimen, China). Sources and dilutions of antibodies used in this study are specified in Supplementary Table S2. Ten randomly chosen fields of immunohistochemical or immunofluorescent staining sections were photographed under × 400 magnification. Percentage of the positive cell count was analyzed with ImageJ software (the National Institutes of Health). In addition, OV-6 was co-stained with Nanog, c-Myc, or KLF-4, and nucleus was counterstained with DAPI.
Statistical Analysis
Numerical data were presented as means±s.e.m. Unpaired student t-test was used for comparison between two groups. Fisher test was used for analysis of more than two groups followed by q-test for comparison between given two groups. The Wilcoxon rank sum test was used for analysis of semi-quantitative histopathological scores. P-value <0.05 is considered to be statistically significant.
Results
Histopathological Features of NASH-HCC
In the present study, 14 cases of non-cirrhotic NASH-HCC tissues without HBV or HCV infection, alcohol abuse, and other liver diseases were identified from >2000 pathologically-confirmed resecting specimens. NASH-HCC represents great heterogeneity in histopathology, which was classified morphologically into six subtypes (Supplementary Figure S1). In well-differentiated HCC (Supplementary Figure S1A), tumor cells are ranged in a trabecular but irregular pattern of hepatocellular plates. The sinusoids between hepatocellular plates are underlined with sinusoidal endothelial cells. Malignant cells are large with cholestasis in the bile canaliculi. In poorly differentiated HCC (Supplementary Figure S1B), neoplastic cells are occupied with large nuclei in an irregular shape ranging from small or spindle to giant in size. Nuclear division with a loose arrangement of chromatin is frequently visualized. In a pseudoglandular subtype (Supplementary Figure S1C), malignant cells are ranged in a glandular pattern surrounding extended cholangio ducts. Effusion positive in eosin staining is observed in the central glandular space. In a clear-cell subtype (Supplementary Figure S1D), large and irregular malignant cells with transparent cytoplasm appear to be moderately differentiated. In an acinar subtype of NASH-HCC (Supplementary Figure S1E), small malignant cells are situated in an acinar pattern surrounding cholangio ducts. Eosinophilic secretion is often seen in the acinus antrum. The scirrhous subtype is characterized with thick fibril bundles surrounding small and malignant cell clusters with sinusoid-like space between clusters (Supplementary Figure S1F).
Expression of Pluripotent and Morphogenic Genes (c-Myc, Oct-4, KLF-4, Nanog, and Gli-1) in Human NASH-HCC Specimens
As shown in Figure 1, expression of c-Myc (Figure 1a), Oct-4 (Figure 1b), KLF-4 (Figure 1c), Nanog (Figure 1d), and Gli-1 (Figure 1e) in human NASH-HCC tissues was remarkably positive compared to adjacent non-tumor liver tissues in fluorescent immunohistochemical staining sections. Distinct positivity of c-Myc, Oct-4, KLF-4, Nanog, or Gli-1 was clearly visualized in a neoplastic area, but was not seen in peri-tumor tissues with clear separation of fibril septa (Figure 1). There existed a sharp difference in the positivity of c-Myc, Oct-4, KLF-4, Nanog, and Gli-1 staining between NASH-HCC areas and non-tumor tissues. These nuclear transcription factors are overlapped with DAPI staining in albumin-positive NASH-HCC (Supplementary Figure S2A–E). These findings of immunohistochemical staining were confirmed by quantitative RT-PCR as shown in bar graft panels next to corresponding micrographic images (Figure 1), in which paired NASH-HCC and non-tumor specimens from same patients were shown in red.

Representative micrographs of fluorescent immunohistochemical staining of pluripotent and morphogenic genes: c-Myc, Oct-4, KLF-4, Nanog, and Gli-1 in human NASH-HCC specimens. (a) c-Myc. (b) Oct-4. (c) KLF-4. (d) Nanog. (e) Gli-1. For each row, the left three images were the representative immunofluorescent images of c-Myc (red), Oct-4 (red), KLF-4 (red), Nanog (green), and Gli-1 (red) staining on adjacent non-tumor liver specimens of nonalcoholic steatohepatitis (NASH, first column), separation of fibrous septa (second column), and NASH-HCC area (third column). DAPI was used for nuclear staining (blue). Images were taken at original magnification (× 400). Scale bars=50 μm. On the right column are panels of the quantitative RT-PCR data of the corresponding pluripotent morphogenic gene expression in paraffin-embedded human liver tissue. *P<0.05, tumor compared to peri-tumor liver tissue. Number of tumor specimen=14; number of peri-tumor specimen=5 (only 5 in 14 peri-tumor specimens had adequate RNA from formalin-fixed and paraffin-embedded tissues for quantitative RT-PCR assay). In each scatter diagram, red dots with same shape represent the specimens from the same patient.
Diet-Induced Obesity, Glucose Intolerance, and Insulin Resistance
Mice fed HFC diet plus HF/G exhibited a gradual gain in body weight and obesity over time, and reached approximately more than twofold in body weight of control mice after 12 months of feeding (Supplementary Figure S3A). The gain in body weight was accompanied with an increase in liver weight (Supplementary Figure S3E). In contrast, mice receiving the control diet displayed a normal growth curve of body weight (Supplementary Figure S3A). The volume of brown, subcutaneous, and visceral adipose tissue in mice fed HFC diet plus HF/G was remarkably greater than that in control mice at 9 months (Supplementary Figure S5). To determine the extent of glucose intolerance, glucose (2.5 g/kg body weight) was injected intraperitoneally. It was evident that fasting blood glucose concentration in mice fed HFC diet plus HF/G was strikingly elevated when compared to control mice (161.6±10.7 vs 73.8±3.7 mg/dl at 12th month, P<0.05), and plasma blood glucose levels sustained at much higher levels 2 h after an intraperitoneal glucose load (Supplementary Figure S3B–D). In accordance, a two to threefold increase in fasting insulin levels and striking elevation in HOMA-IR values were notable in mice fed HFC diet plus HF/G compared to the controls (Supplementary Figure S3F–G). Thus, these results verified that feeding HFC diet plus HF/G resulted in severe obesity, glucose intolerance, and insulin resistance.
Mice Fed HFC diet plus HF/G Developed Steatohepatitis and Disordered Lipid Metabolism
Feeding mice HFC diet plus HF/G led to an at least eightfold increase in hepatic TG content (Figure 2d), which was in accordance with the increase of liver weight (Supplementary Figure S3E). The hepatic TG content remained at around 82.9±5.8 and 79.5±3.7 μg/mg fresh tissue at 5 months and 9 months, with a slight decline at 12 months (73.7±2.9 μg/mg fresh tissue, P>0.05). Moreover, steatosis extent was semi-quantitatively assessed according to histopathological criteria and oil red-O staining-positive areas (Figure 2c). Mice fed HFC diet plus HF/G showed a much extensive microsteatosis in an early stage and macrosteatosis or mixed type of microsteatosis and macrosteatosis at a late stage (Figure 2aB, D and F and Supplementary Figure S6A, P<0.01). Semi-quantitative scores of hepatocellular ballooning degeneration (Supplementary Figure S6D) reached a peak 12 months after feeding HFC diet plus HF/G. These findings indicated that HFC diet plus HF/G caused hepatic steatosis with marked necroptotic activity (spotty or focal cell death) in mice by 5 months (Figure 2aB). An at least threefold increase in serum ALT and AST levels was seen in mice receiving HFC diet plus HF/G feeding (Supplementary Figure S4A and B) compared to the controls. In mice fed HFC diet plus HF/G, both ALT and AST levels reached a peak at 9 months (ALT: 555.0±45.9 IU/l; AST: 487.5±49.9 IU/l) and then declined at 12 months (ALT: 270.3±50.3 IU/l; AST: 381.7±54.4 IU/l). Mice fed HFC diet plus HF/G also showed an increase in total serum TBIL (approximately onefold increase at 12th month) (Supplementary Figure S4C) and serum TCHO (more than twofold) (Supplementary Figure S4D). Notably, the difference of serum TBIL between these two groups was remarkably increased overtime (Supplementary Figure S4C), suggesting the progression and persistence of steatohepatitis in mice fed HFC diet plus HF/G. Inflammatory infiltration (Figure 2aF) was semi-quantitatively assessed, and the score seemed to be increasing after feeding HFC diet plus HF/G for 5, 9, and 12 months (Supplementary Figure S6B).

Liver histopathologic features of mice fed HFC diet plus HF/G for 12 months. C57BL/6J mice were fed a control diet or high-fat/-calorie diet plus high fructose/glucose in drinking water (HF/C diet plus HF/G) for 12 months. (a) Representative micrographs of hematoxylin and eosin (H&E) staining of liver tissues at 5, 9, and 12 months of the control group and the HF/C diet plus HF/G feeding group. Focal necrosis (B) and cells with double nuclei (D, F) are indicated by yellow and black arrow symbols, respectively. Images were taken at original magnification (× 200). Scale bars=100 μm. (b) Representative micrographs of liver lipid accumulation shown by Oil Red-O staining. Images were taken at original magnification (× 400). Scale bars=50 μm. (c) Quantitative analysis of Oil Red-O staining. (d) Liver triglyceride content at 5, 9, and 12 months. Data are expressed as the means±s.e.m. Number of mice in control group at 5 and 9 months=3; number of mice in control group at 12 months=4; number of mice in HF/C diet plus HF/G group at 5 and 9 months=5; and number of mice in HF/C diet plus HF/G group at 12 months=10. *P<0.05, **P<0.01 compared to mice fed control diet at corresponding time points.
Disordered Cytokines and Adipokines in Mice Fed HFC Diet Plus HF/G
There was an increase in liver TNF-α and IL-1β mRNA levels (Figure 3a and b) and protein content (Figure 3c and d) in mice receiving HFC diet plus HF/G feeding, especially at late stages. In consistence with increased TNF-α and IL-1β levels, CD163-positive Kupffer cells were progressively more than control livers, especially in the late stage (Figure 3e and f). Expression of liver leptin was suppressed in the late stage of HFC diet plus HF/G feeding, while, a low expression level of liver adiponectin was observed at all time points during HFC diet plus HF/G feeding (Supplementary Figure S4E and F).

Enhanced inflammation in the liver of mice fed HFC diet plus HF/G. (a–d) Upregulation of TNF-α and IL-1β at mRNA and protein levels in mice fed HFC diet plus HF/G over time. (e) CD163 immunofluorescent staining for activated Kupffer cells in the liver of mice fed HFC diet plus HF/G overtime. Images were taken at original magnification (× 400). Scale bars=50 μm. (f) Quantitative count of CD163-positive cells in the liver of mice fed HFC diet plus HF/G overtime. (g) α-SMA immunohistochemical staining for activated hepatic stellate cells (HSCs) in mice. Images were taken at original magnification (× 400). Scale bars=50 μm. (h) Semi-quantitative analysis of α-SMA-positive cells in the liver mice fed HFC diet plus HF/G overtime. *P<0.05, **P<0.01 compared to mice fed control diet at corresponding time points.
Mice Receiving HFC Diet Plus HF/G Feeding Developed Marked Hepatic Fibrosis
Liver mRNA levels of smooth muscle α-actin (α-SMA) and procollagen type I (COL1A1 and COL1A2) in mice fed HFC diet plus HF/G were significantly higher than the controls (Figure 4e–g), which was verified by quantitation of liver hydroxyproline content (Figure 4h), collagen type I staining (Figure 4a and b), and morphometric analysis of fibrotic area (Figure 4b). There was a progressing and more than twofold increase in liver hydroxyproline content (Figure 4h) and threefold increase in fibrotic area semi-quantitation (Figure 4d) at 12th month compared to these at 5 months of HFC diet plus HF/G feeding. As shown in Masson’s trichrome staining and collagen type I staining, collagenous fiber deposited in the hepatic lobule (5, 9, and 12 months), the portal tract (9 and 12 months), and the perisinusoidal and peri-hepatocellular space (12 months) (Figure 4aB, D and F and cB, D and F). On the basis of α-SMA staining (Figure 3g and h), mRNA levels (Figure 4e), collagen type I staining (Figure 4a and b), and hydroxyproline quantitation (Figure 4h), it appeared that hepatic fibrosis was progressing with much more collagen fibril deposition at a late stage of HFC diet plus HF/G feeding. Semi-quantitative fibrotic score further confirmed the persistence and progression of hepatic fibrosis. Whereas, proliferation of bile duct epithelial cells (BDECs) was not evidently enhanced with a long-term of HFC diet plus HF/G feeding (data from counter-staining of CK-19 and Ki67 not shown). From the sum of semi-quantitative scores of steatosis (Supplementary Figure S6A), inflammation (Supplementary Figure S6B), and hepatocellular ballooning degeneration (Supplementary Figure S6D), overall NAFLD activity appeared to keep increasing overtime (Supplementary Figure S6E). By 12 months of feeding, extent of steatosis and necroptotic activity actually became moderate; however, hepatic fibrosis appeared to be at an advanced stage without obvious disruption of liver architecture (Figure 4a, c, d and h).

Mice fed HFC diet plus HF/G developed hepatic fibrosis. C57BL/6J mice were fed a control diet or high-fat/-calorie diet plus high fructose/glucose in drinking water (HF/C diet plus HF/G) for 12 months. (a) Representative micrographs and semi-quantification (b) of immunohistochemical staining of collagen I in mouse liver at 5, 9, and 12 months. The positive signal was developed with DAB (in brown color indicated by arrow symbols). Representative images were recorded at original magnification (× 400), and scale bars=50 μm. (c) Representative micrographs of Masson’s trichrome staining of liver tissues at 5, 9, and 12 months. Hepatic fibrosis is indicated by arrow symbols. Images were recorded at original magnification (× 200). Scale bars=100 μm. The increase in hydroxyproline content overtime is consistent with progressive fibrotic deposition in the parenchyma and the portal triads. (d) The quantitative analysis of Masson’s trichrome staining. (e) mRNA level of α-SMA in mouse liver at 5, 9, and 12 months. (f, g) mRNA levels of procollagen type I (COL1A1 and COL1A2) in mouse liver at 5, 9, and 12 months. (h) Liver hydroxyproline content (ng/mg tissue) at 5, 9, and 12 months. Data are expressed as the means±s.e.m. Number of mice in control group at 5 and 9 months=3; number of mice in control group at 12 months=4; number of mice in HF/C diet plus HF/G group at 5 and 9 months=5; and number of mice in HF/C diet plus HF/G group at 12 months=10. *P<0.05 and **P<0.01 compared to mice fed control diet at corresponding time points. Figure legends are the same in panel e, f, g and h.
Mice Fed HFC Diet Plus HF/G Developed Hyperplastic Nodules with Dysplasia
No visible nodule was observed in mouse liver after HFC diet plus HF/G feeding for 5 or 9 months. However, 3 of 10 mice developed hyperplastic nodules with dysplasia: two visible nodules (4 × 5 mm and 2 × 3 mm) occurred in the liver of two mice after feeding for 12 months (Figure 5aB), and the whole liver of another mouse appeared to be in a hyperplastic stage (Figure 5aC). In contrast to the liver from mice fed control diet (Figure 5aA), the livers from mice fed HFC diet plus HF/G were generally larger, with a faint yellow and greasy surface. In tissue from the hyperplastic liver, CD31 staining (an indicator of angiogenesis, Figure 5aD and E), CD34 staining (Figure 5aF and G), reticular fiber (Figure 5bA and B), Ki67 staining (Figure 5bC and D), and TUNEL staining (Figure 5bE and F) confirmed that the tissue was hyperplastic with significant dysplasia, a pre-carcinomatous stage which occurred in the liver of mice fed HFC diet plus HF/G by 12 months. Thus, we established a transition model from NASH to NASH-HCC in mice, and this model was then utilized to determine whether aberrant activation of pluripotent and morphogenic genes existed in the pre-carcinomatous lesion. CD31 staining (Figure 5aD and E),reticular fiber (Figure 5bA and B), Ki67 staining (Figure 5bC and D), and TUNEL staining (Figure 5bE and F), as well as their positive cell counts (Figure 5c–f) verified the morphological characteristics of dysplasia status.

Mice fed HF/C diet plus HF/G developed hyperplastic nodules with dysplasia. C57BL/6J mice were fed a control diet or high-fat/-calorie diet plus high fructose/glucose in drinking water (HF/C diet plus HF/G) for up to 12 months. (a) The appearance of the liver with hyperplastic nodules (B) and the whole liver of another mouse appeared to be in a hyperplastic stage (C) in mice fed HF/C diet plus HF/G and mice fed control diet, as well as representative micrographs of CD31 (D, E) and CD34 (F, G) staining at original magnification (× 400, scale bars=50 μm). Hyperplastic nodule (B) and positive staining of CD31 are indicated by arrow symbols. (b) Representative micrographs of reticular fiber, Ki67, and TUNEL staining in liver tissues at 12 months. For reticular fiber staining, immunohistochemistry images were taken at × 400 original magnification (scale bars=50 μm). For Ki67 and TUNEL (apoptosis) staining, immunohistochemistry images were taken at × 200 original magnification (scale bars=100 μm). Positive staining of reticular fiber, Ki67, and TUNEL is indicated by arrow symbols. Reduced CD31, negative CD34, much less reticular fiber, and increased Ki67 staining are in accordance with the morphologic criteria of hyperplastic nodules with dysplasia at the end stage of HF/C diet plus HF/G feeding. (c) The quantitative analysis of CD31 staining. (d) The quantitative analysis of reticular fiber staining. (e) The quantitative analysis of Ki67 staining. (f) The quantitative analysis of TUNEL staining.
Activation of Pluripotent and Morphogenic Genes (c-Myc, Oct-4, KLF-4, Nanog, and Gli-1) in Hyperplastic Lesion
As shown in the results of immunohistochemical staining, expression levels of c-Myc (Figure 6aF), Oct-4 (Figure 6bF), KLF-4 (Figure 6cF), Nanog (Figure 7aF), and Gli-1 (Figure 7bF) in the hyperplastic lesion from mice fed HFC diet plus HF/G were remarkably higher than that in early stages (Figure 6aB/D, bB/D and cB/D and Figure 7aB/D and bB/D) and control livers (negative for all these makers). After semi-quantification, number of gene-positive cells per field in the hyperplastic lesion from mice fed HFC diet plus HF/G was remarkably bigger than that in early stages and control livers (Figure 6d–f and Figure 7c and d). In tissue of the non-hyperplastic liver from mice fed HFC diet plus HF/G for 12 months, expression of c-Myc (Supplementary Figure S7A), Oct-4 (Supplementary Figure S7B), KLF-4 (Supplementary Figure S7C), Nanog (Supplementary Figure S7D), and Gli-1 (Supplementary Figure S7E) was also remarkably positive compared to that in early stages (Figure 6aB/D, bB/D and cB/D and Figure 7aB/D and bB/D) and control livers. However, number of gene-positive cells per field in the non-hyperplastic tissue (Supplementary Figure S7A–F) was remarkably less than that in hyperplastic lesions (Figure 6aF, bF, cF and dF and Figure 7aF and bF). These results were in close agreement with the immunofluorescent staining and quantitative RT-PCR results in human tumor and non-tumor tissues (Figure 1).

Enhanced expression of pluripotent c-Myc, Oct-4, and KLF-4 genes in hyperplastic dysplasia tissue. C57BL/6J mice were fed a control diet or high-fat/-calorie diet plus high fructose/glucose in drinking water (HF/C diet plus HF/G) for 12 months. (a–c) Representative micrographs and semi-quantification (d–f) of immunohistochemical staining of c-Myc, Oct-4, and KLF-4 in mouse liver at 5, 9, and 12 months. The positive signal was developed with DAB (in brown color indicated by arrow symbols). Representative images were recorded at original magnification (× 400), and scale bars=50 μm. *P<0.05 and **P<0.01 compared to mice fed control diet at corresponding time point.

Enhanced expression of pluripotent Nanog gene and morphogenic Gli-1 gene in hyperplastic dysplasia tissue. C57BL/6J mice were fed a control diet or high-fat/-calorie diet plus high fructose/glucose in drinking water (HF/C diet plus HF/G) for 12 months. (a, b) Representative micrographs and semi-quantitation (c, d) of immunohistochemical staining of Nanog and Gli-1 in mouse liver at 5, 9, and 12 months. The positive signal was developed with DAB (in brown color indicated by arrow symbols). Representative images were recorded at original magnification (× 400), and scale bars=50 μm. *P<0.05 and **P<0.01 compared to mice fed control diet at corresponding time point.
Co-Localization of OV-6 with Activation of c-Myc, KLF-4, or Nanog in the Hyperplastic Lesion
OV-6 is a marker of HOCs that are the progenitor cells in the mouse liver. HOCs are proliferative and matured to be hepatocytes or cholangiocytes in chronic liver injury for a regenerative recovery. To examine whether pluripotent transcription factors are positive in these cells, liver sections were stained with OV-6 in combination with c-Myc, KLF-4, or Nanog, the transcription factors with oncogenic potential. As shown in the results of fluorescent immunohistochemical staining, OV-6 was positive in areas of the portal triads, and co-localized with positive staining of c-Myc (Figure 8a), KLF-4 (Figure 8b), or Nanog (Figure 8c) in hyperplastic lesions from mice fed HFC diet plus HF/G for 12 months. However, there was no OV-6-positive cell in the control and non-hyperplastic tissue.

Co-localization of c-Myc, KLF-4, and Nanog in hepatic oval cells in hyperplastic dysplasia tissue. C57BL/6J mice were fed a control diet or high-fat/-calorie diet plus high fructose/glucose in drinking water (HF/C diet plus HF/G) for 12 months. (a) c-Myc, (b) KLF-4, (c) Nanog. Representative micrographs of fluorescent immunohistochemical co-staining of OV-6 (red) with c-Myc (green), KLF-4 (green), or Nanog (green) in mouse liver after feeding for 12 months. DAPI was used for nucleus staining (blue). Yellow color represents the overlapping of oval-6 with c-Myc, KLF-4, or Nanog. Images were recorded at original magnification (× 400). Scale bars=50 μm. HV, hepatic vein.
Discussion
The pathogenesis of NASH-HCC development and progression is poorly understood, and we hypothesize that activation of pluripotent and morphogenic genes in steatotic liver accelerates oncogenic transformation of steatotic hepatocytes or liver progenitor cells. c-Myc, KLF-4, Nanog, and Oct-4 are pluripotent genes and they are so-called Yamanaka’s factors. They participate in the formation of stem/progenitor cells and control their self-renewal, proliferation, and differentiation. These factors (including c-Myc, KLF-4, Oct-4, Sox-2, or Lin28) are sufficient to reprogram somatic cells into few clones (<1.0%) of iPSCs and much more granular clones.8, 16, 17, 18 In fact, most of non-embryonic stem cell (ESC)-like colonies are tumor-like spheroids.17 It is known that both undifferentiated ESCs and iPSCs form teratoma in immunodeficient mice, and teratoma formation is a standard method to assess the pluripotency of stem cells.8, 19 The remaining question is what these pluripotent transcription factors will do when they are activated in vivo such as in hepatic progenitor cells or steatotic hepatocytes in an inflammatory environment, since both hepatic progenitor cells and steatotic hepatocytes are thought to be possible origins of NASH-HCC.20, 21 Growing evidence from hepatitis virus-positive specimens has shown that c-Myc, KLF-4, Nanog, and Oct-4, as well as a morphogenic gene Gli-1 are highly expressed in HCC with HBV infection, and may serve as an indication of poor prognosis.22, 23 However, no studies are seen whether these factors are activated in NASH-HCC, and the influences of these factors on hepatic progenitor cells in the transition from NASH to HCC remain unexplored.
CSCs or T-ICs are believed to be responsible for the oncogenesis, progression, metastases and relapse after surgical resection or adjuvant therapies. This hypothesis has been convincing for tumorigenesis, metastases and relapse of various types of cancers, such as breast, colon, pulmonary, prostate, and liver cancers. However, controversies do exist regarding the origins of CSCs or T-ICs, surface markers for identification, and their derivation and new occurrence or induction through progression and intervention.6 We have previously identified CD133+/ADH+ or EpCAM+ cells in HCC specimens from HBV and HCV patients.24 Although we believed that these markers are able to identify CSCs in cancer tissue, they may be less useful for the indication of malignancies. Because there was a higher positive rate of these markers in liver specimens with chronic viral hepatitis than HCC tissue due to the fact that these markers are positive in progenitor cells in the liver, and there is a profound activation of progenitor cells in the liver with chronic injury.24 In the present study, activation of pluripotent and morphogenic genes was confirmed in the liver from mice fed HFC diet plus HF/G, and the activation levels tended to be increased overtime (Figures 7 and 8). Therefore, we postulate that there were more progenitor cells activated during the late stage of HFC diet plus HF/G feeding along with persistent necroptotic, inflammatory activity, and fibrotic progression as observed in this transition model of NASH to malignancy.
In rodents, HOCs in the canal of Hering are believed to be the progenitor cells. HOCs are bipotent and able to further mature to hepatocytes or cholangiocytes when there is injury causing loss of hepatocytes or cholangiocytes, especially under a condition in which hepatocellular proliferation is inhibited,25 and so do small hepatocyte-like cells in a scenario of pre-treatment with retrorsine before partial hepatectomy.26 One study indicated that bone morrow cells may migrate into injured liver to become the source of oval cells.27 In human liver, ductular epithelial cells,28 or primitive hepatic stem cells29 and EpCAM-positive cells are believed in a progenitor cell compartment. The activation of hepatic progenitor cells in human specimens with NASH have been reported in the area with significant hepatic fibrosis in pediatric patients.30 Proliferation of BDECs is often seen in cholestatic disorders, such as primary biliary cholangitis or primary sclerosing cholangitis (PSC), and NASH patient specimens.31 The counter-staining of CK-19 with Ki67 did not show significant overlapping in the livers from mice fed HFC diet plus HF/G for 12 months in the present study, indicating BDEC proliferation was not a major event in the progression of NASH to fibrosis and dysplasia in these mice. It is known that aberrant or continuing proliferation of HOCs may lead to HCC or cholangio-carcinoma in a choline-deficient plus ethionine model or by 2-acetylaminoflourene treatment followed by partial hepatectomy with or without initiation by diethylnitrosamine.29, 32, 33 However, how these cells are activated and whether they are the origin of oncogenic development in the transition from NASH to HCC have not been investigated. One supporting notion is that targeting precancerous CD133+/CD44+ of HOC subpopulation with acyclic retinoid treatment reduces HCC development both in vitro and in vivo.34 This study indirectly implies the possibility of HOCs as the origin of HCC.
On the basis of these understandings, we first examined the expression of pluripotent and morphogenic genes (c-Myc, Oct-4, KLF-4, Nanog, and Gli-1) in NASH-HCC tissues, and found that they were much more positive than adjacent non-tumor tissues with NASH evidence. These findings are in concordance with the previous studies on HCC.22, 23 Although evidence had shown the downregulation of KLF-4 in HCC,35 we found its high expression in NASH-HCC. This deviation might be caused by HCC heterogeneity or the base diseases (most of cases were HBV positive in the previous study). Therefore, c-Myc, Oct-4, KLF-4, Nanog, and Gli-1 were activated in NASH-HCC tissue; however, their significance was unclear.
To further delineate whether the activation of pluripotent and morphogenic genes would affect responses or cell fate of HOCs, we developed a transition model of NASH-HCC in mice. After feeding HFC diet plus HF/G for up to 12 months, mice developed obesity, glucose intolerance, steatohepatitis with progressive fibrosis, insulin resistance, and disturbed lipid metabolism. In this model, there was significant activation of Kupffer cells with increased TNF-α and IL-1β at both mRNA and protein levels. Progressive fibrosis was evident at late stages of HFC diet plus HF/G feeding as indicated by more activated HSCs (α-SMA staining and mRNA levels), increased collagen type I staining, semi-quantitative score of fibrosis, as well as hydroxproline quantitation. However, both steatotic extent and necroptotic activity (ALT and AST levels), became less severe at 12 months compared to those parameters at 9 months in this model. Slightly decreased steatotic extent at later stages may be resulted from negative feedback in fatty acid synthesis as indicated by less enhanced gene expression of enzymes (acetyl CoA carboxylase and fatty acid synthase) in a similar model of NASH-HCC for 1 year duration.36 The discrepancy between less necroptotic activity and progressive fibrosis at the late stage may result from fibrotic influence and is seen in other models of severe fibrosis/cirrhosis, such as carbon tetrachloride (CCl4) or thioacetamide (TAA) intoxication.11
Hyperplastic nodules are indicative of aberrant proliferation of hepatocytes, and were seen with dysplasia in some of mice. Thus, these hyperplastic nodules are in an intermediate stage of transformation to malignancy, and pluripotent genes (c-Myc, Oct-4, KLF-4, and Nanog) and a morphogene (Gli-1) were positive in the liver with significant dysplasia (Figures 6 and 7) as evidenced by specific staining (CD34, CD31, reticular fiber, and Ki67 in Figure 5b and c). Furthermore, in the transition model with pre-carcinomatous lesion, oval cell antigen 6 (OV-6) was extensively expressed in the portal triads of hyperplastic dysplasia tissue. As a specific marker of HOCs, OV-6 co-localization with c-Myc, Nanog, and KLF-4 is therefore indicative of malignant transformation in cell fate under a persistent steatotic and inflamed microenvironment. These HOCs with malignant tendency may be well reprogrammed to the candidates of CSCs/T-ICs with co-expression of pluripotent and oncogenic transcription factors in combination with a disordered profile of inflammatory cytokines (TNF-α, IL-1β) or/and adipokines (leptin and adiponectin). Thus, presumably, with an extended duration, these hyperplastic nodules with dysplasia will develop to hepatic malignancy of either HCC or cholangio-carcinoma.
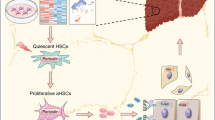
c-Myc is a well-known oncogenic factor, which regulates cell growth, proliferation and differentiation, and it is a driving force for oncogenic transformation in various conditions.37, 38, 39, 40 KLF-4 possesses a potent oncogenic function in mammary tumorigenesis via maintaining stem cell-like features, and is critical for the development, stemness, and tumorigenesis of HCC.41 Elevated expression of KLF-4 in HCC is significantly correlated with poor tumor differentiation, vascular invasion, overall survival, and recurrence-free survival.22 As a major pluripotent factor, Oct-4 is highly expressed in liver CSCs42, 43, 44 and may contribute to the reprogramming of CSCs. Gli-1 is the main transcription factor in hedgehog signaling pathway, which is activated in various chronic liver diseases. It is believed to be responsible for fibrogenesis and oncogenic transformation of steatotic hepatocytes,9 as well as drug resistance and metastasis of HCC as demonstrated in our previous studies.13, 14 Nanog is also an oncogenic transcription factor with participation in the reprogramming process of cell fate changes. Its involvement in hepatic carcinogenesis was evidenced by the fact that silencing Nanog attenuated tumor-initiating capability of liver CSCs.45 Nanog also participates in the self-renewal of hepatic CSCs that express CD24.46 Therefore, we hypothesize that proliferation of HOCs in NASH participates in repairing liver damage, and some of them may transform into CSCs/T-ICs under the steatotic and inflamed microenvironment although further evidence is needed to functionally prove this with technologies, such as lineage tracing. Thus, hypothetically, enhanced expression of pluripotent and morphogenic genes (c-Myc, Oct-4, KLF-4, Nanog, and Gli-1) may reprogram these progenitor cells to change their fate toward origins of malignancy in an inflamed and steatotic microenvironment as illustrated in a schematic depiction (Figure 9), although more rigorous investigations are needed to approve this in vivo.

A schematic illustration of possible transformation of hepatic progenitor cells toward the origin of cancer cells in an inflamed and steatotic microenvironment. In chronic injury liver progenitor cells, such as hepatic oval cells may be activated and differentiated into hepatocytes or bile duct epithelial cells for repair of damage and loss of these two cell types. In a steatotic microenvironment, hepatic inflammatory infiltration, fibrotic deposition, altered cytokine or adipokine profile, and oxidant stress may lead to aberrant activation of pluripotent transcription factors, such as c-myc, nanog, KLF-4, oct-4, and morphogenic factor, Gli-1, which may facilitate trans-differentiation of hepatic progenitor cells toward a malignant phenotype, and become one of cell origins of hepatocellular carcinoma or cholangio-carcinoma in NASH.
In conclusion, the findings of the present study demonstrate that pluripotent and morphogenic genes, such as c-Myc, Oct-4, KLF-4, Nanog, and Gli-1 are highly expressed in NASH-HCC tissue, and expression of stemness genes in hepatic progenitor cells may contribute to the reprogramming of HOCs to transform toward cell origins of malignancy in a steatotic and inflamed microenvironment. This initial observation highlights the possibility that hepatic progenitor cell compartment may become one of origins in NASH-HCC development.
References
Lazo M, Hernaez R, Eberhardt MS et al. Prevalence of nonalcoholic fatty liver disease in the United States: the third National Health and Nutrition Examination survey, 1988-1994. Am J Epidemiol 2013;178:38–45.
Michelotti GA, Machado MV, Diehl AM . NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656–665.
Wong RJ, Aguilar M, Cheung R et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547–555.
Wu J . Utilization of animal models to investigate nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Oncotarget 2016;7:42762–42776.
Chong LW, Chou RH, Liao CC et al. Saturated fatty acid induces cancer stem cell-like properties in human hepatoma cells. Cell Molecular Biol 2015;61:85–91.
Ding J, Wu J . Epigenetic regulation of hepatic tumor-initiating cells. Front Biosci 2015;20:946–963.
Yang S, Koteish A, Lin H et al. Oval cells compensate for damage and replicative senescence of mature hepatocytes in mice with fatty liver disease. Hepatology 2004;39:403–411.
Kooreman NG, Wu JC . Tumorigenicity of pluripotent stem cells: biological insights from molecular imaging. J R Soc Interface 2010;7:S753–S763.
Chan IS, Guy CD, Machado MV et al. Alcohol activates the hedgehog pathway and induces related procarcinogenic processes in the alcohol-preferring rat model of hepatocarcinogenesis. Alcohol Clin Exp Res 2014;38:787–800.
Asgharpour A, Cazanave SC, Pacana T et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016;65:579–588.
Li F, Song Z, Li Q et al. Molecular imaging of hepatic stellate cell activity by visualization of hepatic integrin alphavbeta3 expression with SPECT in rat. Hepatology 2011;54:1020–1030.
Adkins Y, Schie IW, Fedor D et al. A novel mouse model of nonalcoholic steatohepatitis with significant insulin resistance. Lab Invest 2013;93:1313–1322.
Fan YH, Ding J, Nguyen S et al. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene 2016;35:116–124.
Chen X, Lingala S, Khoobyari S et al. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol 2011;55:838–845.
Kleiner DE, Brunt EM, Van Natta M et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313–1321.
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663–676.
Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872.
Yu J, Vodyanik MA, Smuga-Otto K et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007;318:1917–1920.
Shirahashi H, Wu J, Yamamoto N et al. Differentiation of human and mouse embryonic stem cells along a hepatocyte lineage. Cell Transplant 2004;13:197–211.
Sia D, Villanueva A, Friedman SL et al. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017;152:745–761.
Oikawa T . Cancer stem cells and their cellular origins in primary liver and biliary tract cancers. Hepatology 2016;64:645–651.
Yin X, Li YW, Jin JJ et al. The clinical and prognostic implications of pluripotent stem cell gene expression in hepatocellular carcinoma. Oncol Lett 2013;5:1155–1162.
Zhang J, Tu K, Yang W et al. Evaluation of Jagged2 and Gli1 expression and their correlation with prognosis in human hepatocellular carcinoma. Mol Med Rep 2014;10:749–754.
Lingala S, Cui YY, Chen X et al. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp Mol Pathol 2010;89:27–35.
Apte U, Thompson MD, Cui S et al. Wnt/beta-catenin signaling mediates oval cell response in rodents. Hepatology 2008;47:288–295.
Carpino G, Renzi A, Franchitto A et al. Stem/progenitor cell niches involved in hepatic and biliary regeneration. Stem Cells Int 2016;2016:3658013.
Petersen BE, Bowen WC, Patrene KD et al. Bone marrow as a potential source of hepatic oval cells. Science 1999;284:1168–1170.
Shin S, Kaestner KH . The origin, biology, and therapeutic potential of facultative adult hepatic progenitor cells. Curr Top Dev Biol 2014;107:269–292.
Sanders JA, Brilliant KE, Clift D et al. The inhibitory effect of rapamycin on the oval cell response and development of preneoplastic foci in the rat. Exp Mol Pathol 2012;93:40–49.
Sobaniec-Lotowska ME, Lebensztejn DM, Lotowska JM et al. Ultrastructure of liver progenitor/oval cells in children with nonalcoholic steatohepatitis. Adv Med Sci 2011;56:172–179.
Ganz M, Bukong TN, Csak T et al. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J Transl Med 2015;13:193.
Fan L, Xu C, Wang C et al. Bmi1 is required for hepatic progenitor cell expansion and liver tumor development. PLoS ONE 2012;7:e46472.
Guest I, Ilic Z, Sell S . Age dependence of oval cell responses and bile duct carcinomas in male fischer 344 rats fed a cyclic choline-deficient, ethionine-supplemented diet. Hepatology 2010;52:1750–1757.
Zheng YW, Tsuchida T, Shimao T et al. The CD133+CD44+ precancerous subpopulation of oval cells is a therapeutic target for hepatocellular carcinoma. Stem Cells Dev 2014;23:2237–2249.
Lin ZS, Chu HC, Yen YC et al. Kruppel-like factor 4, a tumor suppressor in hepatocellular carcinoma cells reverts epithelial mesenchymal transition by suppressing slug expression. PLoS ONE 2012;7:e43593.
Dowman JK, Hopkins LJ, Reynolds GM et al. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am J Pathol 2014;184:1550–1561.
Wilson A, Murphy MJ, Oskarsson T et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 2004;18:2747–2763.
Nesbit CE, Tersak JM, Prochownik EV . MYC oncogenes and human neoplastic disease. Oncogene 1999;18:3004–3016.
Dang CV, Resar LM, Emison E et al. Function of the c-Myc oncogenic transcription factor. Exp Cell Res 1999;253:63–77.
Akita H, Marquardt JU, Durkin ME et al. MYC activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer Res 2014;74:5903–5913.
Lu XJ, Shi Y, Chen JL et al. Kruppel-like factors in hepatocellular carcinoma. Tumour Biol 2015;36:533–541.
Gidekel S, Pizov G, Bergman Y et al. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell 2003;4:361–370.
Zhu P, Wang Y, He L et al. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J Clin Invest 2015;125:3795–3808.
Kuo KK, Lee KT, Chen KK et al. Positive feedback loop of OCT4 and c-JUN expedites cancer stemness in liver cancer. Stem Cells 2016;34:2613–2624.
Machida K, Chen CL, Liu JC et al. Cancer stem cells generated by alcohol, diabetes, and hepatitis C virus. J Gastroenterol Hepatol 2012;27:19–22.
Lee TK, Castilho A, Cheung VC et al. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011;9:50–63.
Acknowledgements
We are grateful to Yuhong Li in the Joint Live Imaging Laboratory of Fudan University Shanghai Medical College-PerkinElmer Company, for her technical support in the use of high-resolution X-ray microCT scanning. We thank Ke Qiao in the Department of Medical Microbiology and Parasitology, Fudan University School of Basic Medical Sciences for her technical support in the use of confocal microscope. This work is supported by the National Natural Science Foundation of China (NSFC #81272436 and 81572356 to JW), (NSFC #81272725 and 81530077 to JF), and (NSFC #81470857 to X-PL), as well as Shanghai Commission of Sciences and Technologies (#16140903700), and the Ministry of Science and Technology of China (#2016YFE0107400) to JW.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Activation of pluripotent and morphogenic genes was examined in nonalcoholic steatohepatitis-associated hepatocellular carcinoma (NASH-HCC) specimens and an animal model of hyperplastic nodules of pre-carcinomatous lesions. The authors demonstrate that activation of pluripotent and morphogenic genes in NASH might be crucial for the oncogenic transformation of steatotic hepatocytes or liver progenitor cells during NASH-HCC development.
Supplementary information
Rights and permissions
About this article

Cite this article
Xu, G., Ye, J., Liu, XJ. et al. Activation of pluripotent genes in hepatic progenitor cells in the transition of nonalcoholic steatohepatitis to pre-malignant lesions. Lab Invest 97, 1201–1217 (2017). https://doi.org/10.1038/labinvest.2017.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.84
This article is cited by
-
GPM6A expression is suppressed in hepatocellular carcinoma through miRNA-96 production
Laboratory Investigation (2022)
-
Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis
Laboratory Investigation (2019)
-
Characterization of a murine nonalcoholic steatohepatitis model induced by high fat high calorie diet plus fructose and glucose in drinking water
Laboratory Investigation (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}