
Abstract
High-grade gliomas are rapidly progressing tumors of the central nervous system (CNS) with a very poor prognosis despite extensive resection combined with radiation and/or chemotherapy. Histopathological and flow cytometry analyses of human and rodent experimental gliomas revealed heterogeneity of a tumor and its niche, composed of reactive astrocytes, endothelial cells, and numerous immune cells. Infiltrating immune cells consist of CNS resident (microglia) and peripheral macrophages, granulocytes, myeloid-derived suppressor cells (MDSCs), and T lymphocytes. Intratumoral density of glioma-associated microglia/macrophages (GAMs) and MDSCs is the highest in malignant gliomas and inversely correlates with patient survival. Although GAMs have a few innate immune functions intact, their ability to be stimulated via TLRs, secrete cytokines, and upregulate co-stimulatory molecules is not sufficient to initiate antitumor immune responses. Moreover, tumor-reprogrammed GAMs release immunosuppressive cytokines and chemokines shaping antitumor responses. Both GAMs and MDSCs have ability to attract T regulatory lymphocytes to the tumor, but MDSCs inhibit cytotoxic responses mediated by natural killer cells, and block the activation of tumor-reactive CD4+ T helper cells and cytotoxic CD8+ T cells. The presence of regulatory T cells may further contribute to the lack of effective immune activation against malignant gliomas. We review the immunological aspects of glioma microenvironment, in particular composition and various roles of the immune cells infiltrating malignant human gliomas and experimental rodent gliomas. We describe tumor-derived signals and mechanisms driving myeloid cell accumulation and reprogramming. Although, understanding the complexity of cell–cell interactions in glioma microenvironment is far from being achieved, recent studies demonstrated several glioma-derived factors that trigger migration, accumulation, and reprogramming of immune cells. Identification of these factors may facilitate development of immunotherapy for gliomas as immunomodulatory and immune evasion mechanisms employed by malignant gliomas pose an appalling challenge to brain tumor immunotherapy.
Similar content being viewed by others


INTRODUCTION
Gliomas are tumors of the central nervous system (CNS), originating from transformed neural stem or progenitor glial cells.1 On the basis of histopathological characteristics World Health Organization (WHO) divided gliomas into groups: low-grade gliomas (LGG, grades I and II) are well-differentiated, slow-growing tumors, whereas high-grade gliomas (HGG, grades III and IV) are less differentiated or anaplastic, and strongly infiltrate brain parenchyma. The most common and deadliest primary brain tumor is glioblastoma (GBM, grade IV). Histological classification is currently assisted by molecular genetic studies that provide diagnostic, prognostic, and predictive values.2, 3 The emerging molecular profile of gliomas is based on the studies of gene expression and DNA methylation, and recent discoveries of new biomarkers facilitate patient stratification, prognosis, and prediction of treatment responses.4 Gliomas that harbor mutations in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), and display the CpG island hypermethylator phenotype constitute a subtype of gliomas with improved survival.4 GBM is the most aggressive and difficult to treat malignancy, due to the frequent dysfunctions of tumor suppressors or oncogenes and highly diffusive growth, which prevents a successful resection of the tumor.5 Primary GBMs show alterations in EGFR, PDGFRA, PTEN, TP53, NF1, and CDKN2A/B, as well as TERT promoter mutations, but not IDH1/2 mutations.6, 7 On the basis of gene expression profiles, GBMs can be further divided into four subtypes: classical, proneural, neural, and mesenchymal, that differ in tumor aggressiveness, progression, and/or prognosis. The golden standard of GBM treatment is surgery combined with chemo- and radiotherapy, however, it remains only palliative and with median survival time of adult patients with GBM 15 months after diagnosis.8
Glioma cells secrete numerous chemokines, cytokines and growth factors that promote infiltration of various cells: astrocytes, pericytes, endothelial cells, circulating progenitor cells, and a range of immune cells such as microglia, peripheral macrophages, myeloid-derived suppressor cells (MDSC), leukocytes, CD4+ T cells, and Treg into the tumor.9, 10, 11, 12 These non-neoplastic cells create a specific niche called a tumor microenvironment, which has a crucial role in cancer growth, metastasis, and response to treatment. Locally produced cytokines and chemokines, and their crosstalk with components of the extracellular matrix, reprogram infiltrating immune cells to acquire distinct functional phenotypes, thereby directing the immune system into inflammatory or anti-inflammatory responses. Like many other non-CNS malignant cancers, GBMs developed multiple strategies to inhibit host antitumor responses (for a review see ref. 13). Several recent transcriptomic studies of GBM tissues have identified different signatures, based on immune and myeloid/macrophage genes expression, associated with GBM pathology, overall survival (OS), or response to treatment.14, 15, 16 These data strongly support a link between a type of the immune system response and glioma progression.
In this review, we discuss heterogeneity of the immune microenvironment of human gliomas and experimental rodent gliomas, and contribution of specific immune subpopulations to glioma pathology. We appraise recent discoveries of tumor-derived factors that stimulate migration and reprogramming of various immune cells infiltrating gliomas, and are promising novel targets for therapeutic treatments.
ORGANIZATION OF THE IMMUNE MICROENVIRONMENT OF GLIOMAS
Glioma-Associated Microglia/Macrophages in Human Gliomas
Microglia are myeloid cells residing in the CNS, which account for 10–20% of the non-neuronal cell population. Resident microglia and extra-parenchymal macrophages such as: perivascular, choroid plexus-associated, and meningeal macrophages, described collectively as ‘brain macrophages’, are involved in the maintenance of brain homeostasis and immunological responses.17 Detection of the CD11b antigen (integrin alpha M, Mac-1, or complement receptor CR3A) was used as a common marker for microglia in most studies on human tissues. However, bone marrow (BM) macrophages and MDSCs also express CD11b. Generally, markers used for the identification of microglia/macrophages are CD163, CD200, CD204, CD68, and Iba-1 (a lectin binding protein). Until now, no microglia-specific marker has been identified that does not also label extra-parenchymal or peripheral macrophages. The question remains unresolved whether CD11b+ cells detected under pathological conditions are CNS-resident microglia or come in part from other sources, such as blood or BM (for a review see ref. 18).
Clinical studies showed extensive infiltration of gliomas with microglia and peripheral macrophages, collectively termed glioma-associated microglia/macrophages (GAMs). Immunohistochemical studies using anti-Mac387, KP1 antibodies, and the lectin RCA-1 demonstrated the higher abundance of GAMs in HGGs than in LGGs.19 Immunohistochemical studies of forty gliomas (grades II–IV) with an antibody against CD45RO and lectin RCA-1 showed the high number of ramified GAMs in gemistocytic astrocytomas (grade II), both the ramified and ameboid GAMs in the protoplasmic and fibrillary astrocytomas (grade II), and the greatest number of ameboid GAMs with very rare ramified GAMs in anaplastic astrocytomas (grade III) and GBM (grade IV).20 Immunostaining for three microglial markers: CR3/43, Ki-M1P, and Iba1 detected the high number of GAMs in gemistocytic astrocytomas (grade II) that behave more aggressively than other grade II astrocytomas21 and are considered by some authors as grade III astrocytomas. Re-evaluation of GAM infiltration by staining of glioma sections with anti-HLA-DP,DQ,DR antibody (CR3/43) revealed morphological heterogeneity of immunopositive cells (more ramified GAMs were detected in grade I and II astrocytomas vs more ameboid GAMs in GBM), and the increased number of GAMs in GBM. Double staining with anti-HLA-CR3/43 and anti-CD11b antibodies demonstrated co-localization of both markers in a vast majority of positive cells.22 Interestingly, GAMs accumulation was not observed in pediatric high-grade tumors such as primitive neuroectodermal tumors (PNETs).
Flow cytometry was used to identify microglial cells within the CNS. A flow cytometry phenotype for ramified (non-activated) microglia isolated from adult CNS was defined as CD45low CD11b/c+ in Lewis and Brown Norway rats, and clearly distinguished these cells from all blood-derived leukocytes, the latter being CD45high. Isolated microglia were mostly MHC class II positive.23, 24 The extent of microglia (CD11bhigh, CD45low), macrophage (CD11bhigh, CD45high), and lymphocyte (CD11bnegative, CD45high) infiltration into tumors, tumor periphery, and contralateral tumor-free hemispheres was measured.25 Flow cytometry studies demonstrated accumulation of both microglia and macrophages in human gliomas. The number of infiltrating GAMs ranged from 1.8 to 3.7 × 107 cells per gram of tumor mass in GBM, whereas in normal brain it ranges from 4.6 to 3 × 105 cells per gram of tissue.26, 27 Intratumoural microglia/macrophage density increases during glioma progression and correlates with the grade of malignancy.28 Interestingly, glioma cells expressing a marker CD133+, considered to be glioma-initiating cells, are more effective in recruiting GAMs to the tumor, as evidenced by enhanced Iba1 and CD68 staining around them. Glioma-initiating cells that grow in vitro as 3-dimentional, floating spheres, express high levels of chemoattractants (2- to 3-fold higher level of CCL2, CCL5, and CCL7 mRNA, 7-fold higher level of VEGF-A mRNA, and nearly 50-fold higher level of NTS mRNA) as compared to the adherent glioma cells.28 Collectively those results point to a link between GAM accumulation and tumor grade. A lack of systematic studies on the abundance of ameboid GAMs in a tumor core/periphery prevents making definite statement about its link to patient survival.
Functional Polarization of Glioma-Associated Microglia/Macrophages in Human Gliomas
Macrophages are ‘plastic’ and their phenotype can adapt to specific environmental cues. These cells may change their effector mechanisms along a spectrum between a ‘M1’ pro-inflammatory phenotype, characterized by inflammatory, antitumor responses, and a ‘M2’ cytoprotective, immunosuppressive phenotype, displayed by macrophages involved in tissue repair and inflammation resolution.29 Cultured M2 macrophages display discrete functional states (M2a, b, c) depending on the cocktail of cytokines/factors used for stimulation, enhanced phagocytic, and cytoprotective properties, and are immunosuppressive.30 M2 macrophages produce high levels of IL-10, transforming growth factor (TGF)-β and low levels of IL-12. Several markers have been proposed to distinguish between M1- and M2-like states, eg, the expression of Arg1, Mrc1, Chi3l3, Socs2, CD163, Fizz-1, and Ccl2 mRNAs marks M2 activation, whereas M1 macrophages express Nos2, IL12b, and Ciita.29 Despite simplification of such distinctions representing the extremes of a spectrum of discrete states, it is useful to define such states using a set of functional features and gene expression profiles. Recent data on the functions of non-CNS tumor-associated macrophages (TAMs) suggest that their activities should be better described as pro- or antitumor, rather than ‘M1’ or ‘M2’ phenotypes.31, 32 A comprehensive analysis of gene expression profiles in TAMs isolated from various tumor models led to a surprising conclusion that TAMs from cancers associated with high amounts of endogenous commensal- and pathogen-derived molecules (such as colon cancer) will tend to the M1 phenotype, whereas tumors developing in sites with relative low inflammation such breast, head and neck tumors, developmental pediatric tumors, will have higher numbers of M2 macrophages.32
Microglia exhibit a remarkable degree of plasticity. Upon different stimulation mimicking pathological conditions, microglia can also polarize into functional states classified as the M1 pro-inflammatory activation or the M2 cytoprotective, immunosuppressive activation.33 M1 activation was induced by GM-CSF treatment followed by IFNγ/LPS in microglia isolated from adult brains and cultured ex vivo, whereas the M2 activation was induced by M-CSF treatment followed by IL-4/IL-13. High levels of Arginase-1 (Arg1), CCL17, CCL22, mannose and scavenger receptors, and production of anti-inflammatory cytokines such as: IL-4, IL-10, and TGF-β and low amount of IL-12 and NO were found in M2 activated microglia and macrophages. Myelin phagocytosis was augmented in microglia in comparison to macrophages and was increased in M2 cells.34 During an acute phase of brain injury microglia adopt mostly M1 phenotype and release chemical mediators such as nitric oxide (NO), reactive oxygen species (ROS), excitatory amino acids, and pro-inflammatory cytokines: interleukin-1β (IL-1β), IL-12, tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) (reviewed in refs 35, 36).
Despite many efforts there is no consensus regarding the activation status of GAMs. The preliminary analysis of the immune functions of GAMs (CD11b/c+CD45+ cells) from post-operatives samples indicated that these cells were phagocytic, expressed substantial levels of Toll-like receptors (TLRs) but did not produce pro-inflammatory cytokines: IL-1β, IL-6, TNF-α upon TLR stimulation.26 In addition, despite the expression of the MHC class II on CD11b/c+CD45+ cells, the expression of T-cell activation factors and co-stimulatory molecules such as CD86, CD80, and CD40 was downregulated, and these cells were less capable to mediate tumor cytotoxicity compared with the microglia isolated from normal brain.26
Several studies detected putative markers of M2 activation in gliomas. The expression of macrophage scavenger receptors CD163 and CD204 was increased in human gliomas, and the number of CD163 and CD204 positive cells among the GAM population was correlated with the grade of glioma and malignancy.37 Combination of in situ hybridization with CD68 immunohistochemistry showed that microglia are a main source of IL-10 expression in malignant gliomas.38, 39 Moreover, IL-10 mRNA levels were positively correlated with malignancy of the gliomas (87% of grade III and IV, and only 4% of grade II expressed IL-10 mRNA). Elevated levels of IL-10 were found in sera of 11% of low-grade and 63.6% of high-grade human gliomas.38
Applications of immunomagnetic beads or FACS immunosorting of CD11b+ cells allowed to isolate GAMs from gliomas, and make reasonable comparison to their counterparts in normal brain. Szulzewsky et al40 determined the expression of the M1 (Il1rn and Isg20), M2a (Clec7a), M2b (Tgfb1I), and M2c-specific (Cxcr4) genes in CD11b+ and CD11b− cells isolated from human 11 GBMs, 5 non-tumor brains, and 2 samples of blood monocytes. High expression of tested genes was detected in GAMs isolated from GBM samples, however only IL1RN, ISG20 (both M1-specific), and TGFBI (M2b-specific) levels were significantly higher in CD11b+ cells from GBMs than in CD11b+ cells isolated from control brains. The expression of CLEC7A was significantly lower in CD11b+ cells isolated from GBMs. The authors attributed a lack of differences to the fact that the control brain specimens were not taken from healthy patients, but patients suffering from epilepsy or and trauma injury.
In the subsequent study, they analyzed the expression profile of human GBM GAMs and CD11b+ cells from non-tumor samples (epilepsy and control postmortem brains), and identified 334 significantly regulated genes in human GBM GAMs (292 upregulated and 42 downregulated).41 The upregulated genes were associated with mitotic cell cycle, cell migration, cell adhesion, response to wounding, and extracellular matrix organization. Chemokines upregulated in human GAMs include CCL2, 3, 4, 5, 8, and CXCL9. Interestingly, there was no overlap of the genes upregulated in human GAMs with the genes upregulated in macrophages stimulated with IL-4. In contrast to murine GAMs that upregulated Il-1β and Tnf-α, human GAMs did not upregulate genes associated with immune activation (ie, IFNG, NOS2, TNFα were barely expressed). RNA sequencing of GAMs from murine GL261 gliomas showed a fraction of upregulated similar transcripts as in human GAMs. The similar genes were mostly related to mitotic cell cycle. Among significantly upregulated genes in murine GAMs were those related to ‘interferon signaling’, ‘complement system’, and ‘pattern recognition receptors’.41 The study shows that a GL261 glioma model does not recapitulate well innate and adaptive immune responses occurring in human GBMs.
We have optimized a protocol for GAM isolation based on gentle mechanical/enzymatic tumor tissue digestion, and CD11b+ magnetic beads to capture all microglia, monocytes, and macrophages in gliomas. Using qRT-PCR we showed higher expression of ARG1, CXCL14, NOS2 mRNAs in GAMs from high-grade gliomas (mostly GBMs) than in low-grade gliomas.22 Interestingly, the IKBKB mRNA and its protein IKKβ were downregulated in GBM tissues, and lower IKBKB expression was detected in CD11b+ sorted from HGG than LGG.22 The IKBKB gene encodes a kinase IKKβ, which phosphorylates inhibitory IκB proteins and represents a convergence point for most signaling pathways leading to NFκB activation. The transcription factor NFκB is a downstream mediator of stimulation by the TLR or TNFα/IL-1β receptors, and its activation depends on the phosphorylation of an inhibitor IκB by IKKβ resulting in release of NFκB, translocation to the nucleus and activation of inflammatory genes. NFκB activation, which was prominent in GAMs infiltrating pilocytic astrocytomas, was reduced in GAMs infiltrating GBMs due to reduced IKKβ expression in GBMs.22 The failure of activating the IKKβ-NFκB signaling pathway could be responsible for reduced expression of immune/inflammatory genes in GAMs, and shaping the immunosuppressive microenvironment in GBMs. We performed the analysis of transcriptomes of GAMs (CD11b+ cells) isolated from WHO grade I pilocytic astrocytomas and GBMs. The upregulated genes in GAMs from GBM patients were associated with mitotic cell cycle, cell migration, cell adhesion, response to wounding, and extracellular matrix organization. Functional annotations showed overrepresentation of gene classes related to cell adhesion, chemotaxis, and cell proliferation. Dominating transcriptional responses in GBM CD11b+ cells are consistent with the protumorigenic, non-immune activation of GAMs (unpublished) which is in agreement with published studies.41 Table 1 summarizes data on phenotypic and functional markers of microglia/macrophages in human gliomas.
In a recent study, microarray analyses and microRNA expression profiling of different myeloid populations (microglia, macrophages, and MDSCs) from GBM with matched blood monocytes, normal brain microglia, non-polarized M0, and polarized M1, M2a, M2c macrophages were performed. GBM patients have the augmented number of monocytes relative to healthy donors. Among CD11b+ cells, microglia and MDSCs were more abundant than macrophages. Flow cytometry showed higher levels of MHC II and CD86 (B7-2) expression on MDSCs than on microglia and macrophages.42 Also CD80 (B7-1) was more highly expressed on MDSCs and macrophages than on microglia. B7-1 and B7-2, ligands for CD28, are prototypic co-stimulatory molecules expressed primarily on antigen-presenting cells. Optimal activation of T cells relies upon antigen presentation to the T-cell receptor (TCR), and is enhanced by co-stimulation via CD28/B7 ligation.43 Expression of CD163 and CD206 (M2 activation markers) was higher in the MDSCs and macrophages than in the microglia. The transcription factor pSTAT1 (involved in macrophage polarization) and its target a pro-inflammatory TNF-α was most commonly expressed in MDSCs. Immunosuppressive cytokines, TGF-β1 and IL-10, were expressed by all types of cells: MDSCs, microglia, and macrophages, but the highest expression was in macrophages.42 The mesenchymal subtype of GBM had the lowest expression of TNF-α. The highest expression of CD163 and CD206 was detected in MDSCs within classical GBMs, it was highly variable in MDSCs and macrophages in proneural GBMs. Nanostring expression profiling of 88 genes characteristic for M0, M1, M2a, and M2c macrophages in CD14+ blood cells, and tumor-infiltrating CD14+ cells from GBM patient demonstrated separation of GBM patient CD14+ blood cells from other groups, and displayed similarity of the GBM-infiltrating CD14+ cells to non-polarized M0 macrophages. The authors concluded that GAMs exhibit distinct immunological functions than peripheral macrophages and their expression profiles are similar to non-polarized M0 macrophages.42
One of the difficulties to refine definition of human GAMs phenotype lies in heterogeneity of isolated CD11b+ cells, which includes also CD11b+Gr1+ MDSCs bearing own transcriptomic characteristics. Altogether, gene expression profiling of human CD11b+ infiltrates points to a lack of the classical ‘immune response’ activation in GBMs, and indicates that GAM and MDSC populations assume discrete phenotypes along the M1–M2 continuum, therefore, more studies are required to ascertain their functions.
Myeloid-Derived Suppressor Cells in Gliomas
Myeloid-derived suppressor cells are a heterogeneous population of early myeloid progenitors and precursors at different stages of differentiation into granulocytes, macrophages, and dendritic cells. In mice, MDSCs are defined as CD11b+Gr-1/Ly-6G+IL4Rα+CD11c−F4/80+/−cells, but based on the relative expression levels of Ly-6G and Ly-6C two specific subsets: granulocytic (G-MDSCs) CD11b+Gr-1hiLy-6ClowLy-6G+CD49d− and monocytic (Mo-MDSCs) CD11b+Gr-1intLy-6ChiLy-6G−CD49d+ MDSCs were identified. A minor population of ‘early stage’ e-MDSC, defined as Lin− (including CD3−, CD14−, CD15−, CD19−, CD56) HLA-DR−CD33+ cells, contains more immature progenitors.44, 45 Gr-1 antigen is a cell-surface protein that belongs to the Ly-6 family of proteins. In rats MDSCs could be identified as CD11b+Gr-1+ cells.
Human MDSCs are identified as HLA-DR−CD11b+ CD14−CD33+ cells that express the common myeloid marker CD33 and lack the expression of markers of mature myeloid and lymphoid cells. The ortholog to Ly6C or Ly6G has not been described in humans. In humans, precise identification of G- and Mo-MDSC subsets is more difficult, and these cells have been characterized by combination of overlapping myeloid markers. Some studies discriminated six human MDSC phenotypes: MDSC1 (CD14+IL-4Rα+), MDSC2 (CD15+IL-4Rα+), MDSC3 (Lineage−HLA-DR−CD33+), MDSC4 (CD14+HLA-DRlow/−), MDSC5 (CD11b+CD14−CD15+), and MDSC6 (CD15+FSClowSSChigh) using multicolor staining.44, 45 The prior lack of standardization led to the establishment of a human MDSC proficiency panel under the guidance of the Association of Cancer Immunotherapy (CIMT) Immunoguiding Program. The currently accepted phenotypic definitions of human MDSCs are now CD11b+CD14+CD33+HLA-DR−/lowCo-receptor−/low for macrophage-MDSCs and CD11b+CD15+CD33+Lin−HLA−DR−/low for polymononuclear-MDSCs, present in the mononuclear fraction.45 A more immature subset of human early stage e-MDSCs was characterized by the absence of staining for the lineage markers (CD3−, CD14−, CD15−, CD19−, CD56−), HLA-DR− and expression of the common myeloid markers CD33 and CD11b.45
Normal BM contains 20–30% of cells with the MDSC phenotype, a small proportion (2–4%) is present in the spleen. Immature myeloid cells generated in BM quickly differentiate into mature granulocytes, macrophages or dendritic cells, therefore, in healthy individuals, immature myeloid cells comprise ∼0.5% of peripheral blood mononuclear cells. Under pathological conditions such as cancer, infectious diseases, sepsis, trauma, BM transplantation, or autoimmune disorders, the emergency myelopoiesis and/or blockade in cell differentiation leads to an expansion of this population.45 MDSCs have ability to suppress the cytotoxic activities of natural killer (NK) cells, the adaptive immune response mediated by CD4+ or CD8+ T cells in antigen dependent or non-dependent manner and induce apoptosis of T-cell subsets.45 The two MDSC subsets inhibit immune responses through different mechanisms: PMN-MDSCs suppress antigen-specific CD8+ T cells mainly by producing reactive oxygen species, whereas M-MDSCs express nitric oxide synthase 2 (NOS2) and arginase (ARG1), that metabolize l-arginine and block translation of the T cell CD3 zeta chain, generate reactive nitrogen species, inhibit T-cell proliferation, and promote T-cell apoptosis. Moreover, MDSCs secrete immunosuppressive cytokines and induce regulatory T-cell progression.45
Numbers of MDSCs are frequently increased in blood, spleen, and tumor mass, and correlate with cancer stage, metastasis, and chemotherapy response.45 Information regarding their presence and roles in gliomas is scarce.44, 46 Gielen et al reported increased percentages of both M- and PMN-MDSCs in the blood of GBM patients when compared with healthy donors. The myeloid activation markers B7-1/CD80 and PD-L1 were not detected. The MDSC population in tumor cell suspensions consisted almost exclusively of CD15+ PMN-MDSC cells. Immunohistochemistry confirmed infiltration of glioma tissues with CD15+/HLAII− cells.44 Raychaudhuri et al reported that patients with GBM have elevated levels of MDSCs compared with age-matched healthy donors and other cancer patients. The majority of the MDSCs in patients with GBM were CD15+ CD14− PMN-MDSCs (82%), followed by Lyn− eMSDcs (15%) and M-MDSCs (3%).46 Application of multicolor flow cytometry for the analysis of peripheral blood and tumor samples of 52 GBM patients revealed the significantly higher frequency of CD14+ M-MDSCs and CD15+ PMN-MDSCs in blood of GBM patients when compared with healthy controls. Moreover, correlation between the number of PMN-MDSCs and CD4+ effector memory T-cells (CD4+ Tem) within the tumors was detected. Tumor-derived CD4+Tem expressed high levels of an inhibitory molecule PD-1 and were functionally exhausted. The expression of PD-L1 was significantly upregulated on tumor-derived MDSCs.47 These findings show accumulation of different MDSC subsets in GBM patients and indicate that PMN-MDSCs in peripheral blood and at the tumor site may participate in GBM-induced T-cell suppression.
Growing evidence shows that the phenotype and mechanisms of action of MDSCs are tumor-dependent, therefore, it is important to determine the presence of all MDSC subsets in each cancer type and even individual patient. There is the lack of systematic studies on the abundance of specific MDSC subpopulations and their impact on numbers and subtypes of adaptive immunity cells. Furthermore, transcriptomic analyses should be carried out on MDSCs separately from microglia/macrophages to establish their functional phenotype. Molecular mechanisms responsible for MDSC migration to a tumor and inhibition of their differentiation need to be elucidated. It has to be clarified whether T-cell suppression occurs in an antigen-specific manner and whether targeting these cells in cancer patients will be of clinical significance.
LEUKOCYTE INFILTRATES IN GLIOMA MICROENVIRONMENT
Glioma Infiltrating T Helper (Th), T Cytotoxic (Tc), and Treg Cells
The glioma microenvironment is infiltrated with leukocytes, mostly CD4+ T helper (Th), CD8+ T cytotoxic (Tc), and CD4+CD25+FoxP3+ Treg.9, 10, 11, 26, 48 CD8+ T cells that express the transcription factors Eomes and T-bet are destined to develop into cytotoxic effector cells that produce IFNγ, granzyme B, and perforin. CD8+ T cells sorted from GBM tissue were phenotypically CD8+CD25−, which indicates a lack of activation.26 It is believed that CD8+ T cells with a constitutive CD25 (the IL-2Rα chain) expression and expressing CD45RO, CD28, and the lymph node-homing markers CD62L and CCR7 are memory T cells. CD4+ T cells were more numerous than CD8+ T cells in a GBM tissue (20 vs 11% gated lymphocytes). The CD4+ population increased with tumor grade, starting at 39% in WHO grade II to 73% in WHO grade III, and 98% in grade IV. Percentages of both CD4+ and CD8+ tumor-infiltrating T cells increased with tumor grade.49 FACS analysis of freshly dissociated 93 glioma samples (II–IV grades), revealed that the CD8− T-cell population consisted >93% of CD3+CD8− cells. Infiltration of T helper cells (Th, CD3+CD8−) and T cytotoxic cells (Tc, CD3+CD8+) was observed in all samples. A significant correlation between increased intratumoral numbers of CD3+ and CD8+ cells and prolonged patient survival was observed, whereas no association was found with CD4+ cells.49 A large neuropathological study evaluated infiltrating CD8+ T cells histologically in patients with newly diagnosed GBM in relation to long-term clinical survival (>403 days). GBM patients with intermediate or extensive CD8+ T-cell infiltrate at the time of diagnosis were more likely to have long-term survival than patients with rare or focal CD8+ T-cell infiltrates.11
Immunocytochemical studies demonstrate that infiltrating T cells were frequently observed in WHO IV fibrinogen-positive GBM areas, which supports a hypothesis that leaky vessels, which typically occur in GBMs, facilitate T-cell transmigration.10 On the other hand, augmented infiltration of effector CD3+Foxp3− T cells was associated with the expression of a cell adhesion molecule ICAM-1 on the vessels, and transmigration of T cells in vitro was markedly reduced in the presence of CAM-blocking antibodies.10
The lack of proper T-cell activation in tumor microenvironment is attributed to the fact that antitumor T-cell responses are suppressed by cytokines TGF-β and IL-10 secreted by glioma cells (for a review ref. 13). Moreover, glioma cells lack B7.1/2 (CD80/86) co-stimulatory molecules and overexpress B7-H1 mRNA and protein. B7-H1 expressed in glioma cells is a strong inhibitor of CD4+ as well as CD8+ T-cell activation as assessed by cytokine production (IFN-γ, interleukin-2, interleukin-10) and the expression of CD69, the T-cell activation marker.50 GBM-infiltrating T cells had decreased INF-γ production and the increased PD-1 mRNA level.47 Apart from directly blocking effective activation of naive T cells, glioma cells promote the accumulation of immunosuppressive cells including Treg and regulatory dendritic cells in the tumor environment. These cells mediate inhibition of T cells, whereas MDSCs and Treg maintain inhibition of NK cells.
Treg (CD4+CD25+FoxP3+) cells are known as potent suppressors of the adaptive immune response, because of the ability of this subset to inhibit the proliferation of any cytokine-secreting effector T cells. Accumulation of Treg subsets was observed in different types of cancer and correlated with worse prognosis.12 Treg are necessary to restrict and resolve activation of the immune system. In one of the first reports evaluating Tregs using flow cytometry, it has been reported that gliomas were infiltrated predominantly with CD8+CD25− T cells and CD4+CD25+FOXP3+ regulatory T cells.26 Relatively high content of Tregs (CD4+FOXP3+CD25highCD127low) accounting for 14% of total CD4+ T-cell population was further reported.51 Immunocytochemical studies indicated the presence of up to 10% Treg in human GBMs and contribution of mostly thymus-derived, Helios expressing Tregs.48 In contrast to these reports, Lohr et al10 observed a very limited infiltration of Treg cells (detected as FOXP3+CD3+ cells in flow cytometry) that accounted for <1% of total T cells identified in GBMs. In another study, CD8+CD28−FOXP3+ were identified in 3/5 of patients, making up 2.08±0.99% of all T cells.52 These contrasting results likely occurred due to the use of different methods and markers for cell type detection. Gene expression profiling of human GBMs indicated overexpression of Treg markers such as: nuclear transcription factor (FOXP3), membrane-resident interleukin-2 receptor alpha (IL-2Rα/CD25), cytotoxic T lymphocyte antigen-4 (CTLA-4), glucocorticoid-induced tumor necrosis factor (TNF) receptor (GITR).12 Despite reported differences, the FOXP3+ cells likely represent 10–14% of the total CD4+ population.13 The prognostic implication of Tregs accumulation in patients with GBM is not clear. Although two groups: Heimberger et al51 and Lohr et al10 found no association between Tregs and prognosis in patients with GBM, Yue et al52 reported a significant association between the density of Tregs infiltrating GBM and poor prognosis. Further analysis of Tregs, other T-cell subsets and their activation state in GBM tissue and patient blood is required to draw conclusions. Dissecting heterogeneity and specific roles of intratumoral T cells in gliomas may be of critical importance for the design of future immunotherapies.
Natural Killer Cells in Gliomas
Natural killer cells (NK; characterized as CD3−CD56+CD16+) are effective cytotoxic lymphocytes that contain both perforin-rich and granzyme-rich granules, and are capable of killing cancer cells and virally infected cells. Human NK cells can be subdivided into different populations based on the relative expression of the surface markers: CD16 and CD56. Activation of NK and effector T cells is inhibited by interactions of HLA class I antigens with certain receptors originally identified as NK cell receptors but also variably expressed on αβ and γδ T cells. These inhibitory NK cell receptors include isoforms of the killer cell immunoglobulin-like receptors (KIR) and immunoglobulin-like transcript/leukocyte immunoglobulin-like receptors (ILT/LIR) that interact with HLA-A, -B, -C, or -G antigens, and a heterodimer formed by the C-type lectin CD94/NKG2A that interact with HLA-E.53 Binding of inhibitory KIRs, expressed by NK cells, to the HLA class I molecules protects non-pathologic cells from being killed and maintains NK cells tolerance. After disruption of the homeostasis, HLA class I expression is reduced and this impairs the tolerance of NK cells.54 A major barrier to effective NK cell-mediated killing of malignant gliomas is high HLA class I expression. The expression of ectopic HLA-G, a non-classical MHC molecule, was described as a different, tumor-dependent mechanism to paralyze the immune system. Primary GBMs and established glioma cell lines express ectopic HLA-G, which may protect the tumor from T and NK-mediated killing.55 NKG2D (natural killer group 2, member D) is a C-type, lectin-like homodimeric receptor expressed by human NK, γδ T, and CD8+ αβ T cells. Ligation of specific NKG2D results in the production of pro-inflammatory cytokines such as IFN-γ and the release of cytotoxic granules that cause tumor cell lysis. NKG2D interacts with ligands that are not constitutively expressed but are inducible, such as human MHC class I chain-related A (MICA) and MICB, and UL16-binding proteins.56 Escape from the NKG2D-mediated immune surveillance of malignant gliomas may be promoted by the inhibition of MICA and ULBP2 expression by glioma-derived TGF-β and by metalloproteinase-dependent shedding from the cell surface.57
Moreover, percentages of NK cells among peripheral blood mononuclear cells isolated from GBM patients were reduced.58 Poli et al reported that NK cells account for a minor part of infiltrating CD45+ cell population in GBMs. Infiltrating NK cells were non-functional, possibly due to the contact with immunosuppressive cells, such as GAMs, MDSCs, and Tregs.59 These cells inhibit activities of NK cells by suppressing NKG2D expression and production of INF-γ.60 Kmiecik et al identified NK cells (2.11±0.54%) and rare B cells (0.66±0.27%) in eight GBM biopsies. NK cells from GBM were characterized by the CD56dimCD16− phenotype and 57.45±12.05% expressed NKG2D.49 B cells (HLA-DR+CD19+) were infrequent in both GBM and normal CNS tissue.26
LESSONS FROM GLIOMA EXPERIMENTAL MODELS
Glioma Experimental Models
The use of in vivo models of malignant glioma shed a light on composition of tumor microenvironment, its influence on disease progression and outcome, as well identified new therapeutic targets for treatment. Animal models attempt to mimic molecular profiles of gliomas as well as underlying biology. Molecular analysis of human gliomas have identified a number of mutations and alterations within the human glioma genome.61 Retroviral or transgenic glioma models allowed for incorporation of most common mutations found in GBM, creating molecular profiles as seen in the clinic.62 Another models are xenografts and transplantation of established glioma cell lines. Because of the availability of the reviews describing glioma animal models,63 here we only briefly outline most commonly used models underlying their usefulness for defining immune microenvironment.
The method widely used in biomedical research is intracranial or subcutaneous injection of tumor cells into the mouse or rat (Figure 1). Established cell lines (eg, C6, 9 L, GL261) that originated from tumors chemically induced by methylnitrosourea or methylcholanthrene, are used to study biology of glioma or new therapeutic agents. The C6 and 9 L gliomas were created using the same method but show distinct characteristics. The first one, resembles more the molecular and histopathological profile of the human GBM.64, 65 The diffuse nature of human GBM is well represented when C6 cells are implanted into the Wistar rats, but not to Sprague–Dawley or Long–Evans rats.63

Most common glioma experimental models. Xenografts of human U87-MG and LN17 cell lines. U87-MG carries mutation in PTEN,226 wild-type p53 (p53 WT),227 wild-type IDH1,228 and overexpression of MHC I and MHC II.229 Human LN18 glioma cell line carries mutation in p53,230 has wild-type PTEN,231 overexpression of TNFR1,232 and downregulation of Kras. Mice allograft commonly used employ implantation of GL261 that carries mutation in p53 with overexpression of MHC I and c-myc.233 C6 rat glioma show downregulation of MHC I and MHC II,234 and carries wild-type p53.235
Orthotopic xenografts retain human GBM features and are considered to be a useful model for therapeutic studies.66 Gene expression analysis of mouse xenografts growing from transplanted human U87-MG, LN18, or U251 glioma cells revealed similar pattern to that of human GBM samples, suggesting that the behavior of human U87-MG or U251 glioma cells is representative of that seen in the clinical settings.67 Nevertheless, this was only shown for one subset of the heterogeneous human GBM. Xenograft models also lack the proper immune environment due to the use of immunocompromised mice. The BALB/c-nude strain has an innate immune system but no acquired immunity, whereas both the innate and acquired immune systems are impaired in the NOD-scid IL2Rgammanull (NSG) mice. This raises an issue for the tumor immunology research in which such models should not be employed.
The advances in molecular biology and gene editing made it possible to generate models that spontaneously form tumors in immunocompetent mice. Disruption of the p53, Rb, Pten, and Nf1 pathways, and others lead to formation of tumors resembling human tumors.68 Mice harboring heterozygous loss of function mutations for the Nf1 and p53 tumor suppressor genes develop glioma spontaneously, however, with different tumor incidence depending on mouse strain genetic background. A second generation of NF1-based glioma models relies on the use of conditional knockout (Cre/LoxP) system that can specify the cell type in which tumor suppressor gene inactivation is induced. These mice developed glioma with manifestation of pathological features of grades II, III, and IV tumors. An S100b-v-erbB transgenic mice and rats develop low-grade and malignant anaplastic oligodendroglioma with over 80% of tumors localized near or within the cerebellum. The use of retrovirus-mediated expression of oncogene allowed for more precise initiation oftumor formation, thus allowing to study a cell origin of GBM. This was shown by triggering GBM formation in genetically engineered mice that expressed a retroviral receptor under nestin promoter (neuronal stem cells—NSCs and early progenitors) or GFAP (primarily in glia, but also NSCs). Introduction of activated K-ras and Akt to neuronal progenitors led to formation of GBM. This was not observed when introduced to more differentiated astrocytes/glia.69 Using a similar system two mouse models of PDGF-driven glioma were generated. First model employs a Moloney murine leukemia virus containing the human PDGF B-chain gene for infecting proliferating cells in the neonatal brain, which leads to c.a. 40% of mice developing tumors within 14-29 weeks with mixed histology (majority exhibit characteristics of GBM or PNET). The second strategy uses the RCAS/tv-a system that employs genetically engineered mice and a viral vector RCAS that is introduced only to the cells expressing tv-a receptor.70 This system was used to model a human adult glioma driven by the PDGF oncogene in mice expressing tv-a. Newborn tv-a pups with PDGFB overexpression in different locations such as cortex, brain stem, and cerebellum have been generated to identify the role of anatomical location in tumorigenesis or were combined with a loss of various tumor suppressors such as Ink4a-arf, p53, and Pten (for a review see ref. 71). It is believed that these transgenic murine tumors recapitulate the three main GBM subtypes: proneural-PDGF, classical-EGFR and mesenchymal-NF1.72 The information about genetic background of frequently used animal glioma models is presented in the Figure 1.
IMMUNE ENVIRONMENT OF EXPERIMENTAL GLIOMAS
Microglia/Macrophages in Rodent Glioma Models
Animal models have been used to study the microenvironment of glioma and its influence on the surrounding cells.73 Using flow cytometry to determine the proportion of microglia (CD11b/chigh, CD45low), macrophages (CD11b/chigh, CD45high), and lymphocytes (CD11b/c−, CD45high) in single cell suspensions of rat C6, 9 L, and RG-2 gliomas, Badie and Schartner25 demonstrated that microglia infiltration is dependent on the glioma cell line and does not correlate with the tumor size. Microglia accounted for the highest proportion of the mass (13–34%) within the tumor periphery. The highest infiltration was seen in C6 gliomas. Macrophages accounted for 5–12% of cells and showed no significant difference between C6, 9 L, and RG-2 gliomas.25 Gliomas generated by an intracranial transplantation of GL261 cells to the CSF-1 R-GFP(+) macrophage Fas-induced apoptosis (MAFIA) transgenic mice showed accumulation of both cell populations, with macrophages accumulating preferentially within the tumor center, rather than periphery.74
The RCAS-PDGFb tumor model, in which primary RCAS-PDGFb tumors were re-transplanted into Cx3cr1GFP/wt Ccr2RFP/wt mice, was used to evaluate GAM infiltration. The system allows to isolate RFP−/GFP+ microglia and RFP+/GFPlow macrophages/monocytes from tumors. Flow cytometry demonstrated 14% microglia and 8.5% macrophages/monocytes in the RCAS-PDGFb gliomas and similar proportion of GAMs in GL261 tumors (16% microglia and 6.5% macrophages/monocytes).40
The intriguing question is a phenotype of GAMs in different glioma models. Microglia stimulated in vitro to glioma-conditioned medium or lipopolysaccharide exhibit responses similar to M1 and M2 activation.75 First studies showed that GAMs infiltrating GL261 glioma express M2 phenotype genes: Arg1, Mmp14 (Mt1-mmp), Cxcl14, Chi3l3 (Ym1).76, 77 Similar pattern of gene expression was detected in CD11b+ cells isolated from rat C6 gliomas and human GBM (unpublished). Mmp-12 and Mmp-13 mRNA levels were upregulated in GAMs isolated from GL216 gliomas indicating their pro-invasive function.78 Transcriptomic profiles of GAMs from GL261 tumors showed gene overlaps with transcriptomes of M1- and M2b-polarized macrophages.40 The analysis of genes characteristic for M1 (Isg20, Il1rn) or M2 (Tgfbi, Clec7a, and Cxcr4) activation showed a significantly higher expression of selected genes in glioma-associated macrophages, and increased Il1rn, Clec7a, and Cxcr4 mRNA levels in glioma-associated microglia. The expression of the selected genes (in particular Isg20, Il1rn, and Clec7a) was lower in GAMs from the RCAS-PDGFb gliomas when compared with the GL261 gliomas.40 Gpnmb and Spp1 genes were upregulated in GAMs in both GL261 and RCAS-PDGFb gliomas, with invading macrophages as the major source for Gpnmb in the RCAS-PDGFb model, whereas resident microglia were the main source for Spp1 in both models.40
It is apparent that GAMs from rodent models of gliomas (C6, GL261, and less extent RCAS-PDGFb gliomas) show a pro-invasive, immunosuppressive phenotype reminiscent of the M2 phenotype observed in non-CNS tumors. However, intersection of the marker genes of discrete M-type responses points to a distinct phenotype of GAMs, only partially overlapping with markers of M1/M2 phenotype.40 Data on molecular profiles of GAMs from different animal models are still inconclusive and more studies are required to reach a consensus. Interpretation of human GAMs data, which should take into account genomic heterogeneity of tumors and subtypes, is even more complicated.
Lymphocytes in Glioma Models
Comparative analysis of infiltrating lymphocytes into the glioma in patients and RCAS-PDGFB gliomas, have shown that infiltrating CD3+CD4+ cells comprise only around 3%, and CD3+CD8+ only 2% of the tumor cell suspension.79 Infiltrating lymphocytes when compared with GAMs were of very low frequency. In addition, there is a correlation between infiltrating microglia/macrophages and functional impairment of T cells.80 The extent of lymphocyte infiltration differs in experimental gliomas. CD8+ cells represented around 22% of infiltrating immune cells in C6 and 9 L gliomas, but only 5% in RG-2 glioma.25 This suggests that two first glioma cell lines possess more immunogenic nature than RG-2 cells, thus the C6 and 9 L models are not very representative for GBM lymphocyte accumulation.81 An increased frequency of CD11b+Gr-1+ immature myeloid cells (e-MDSCs) was associated with diminished CD8+ T-cell infiltration. In mice with a knockout of the histidine decarboxylase, the enzyme responsible for histamine production, more profound suppression of CD8+ T-cell proliferation and functions, associated with increased prostaglandin PGE2 expression levels, was demonstrated.82 Maes et al83 have shown that depletion of Tregs within the tumor allows infiltration of non-immunosuppressive myeloid cells in mice with GL261 gliomas. This shows that, even though the infiltration of immune cells is very low, it has a big impact on immunosuppressive environment and glioma growth. Owing to a scarce number of studies, there is no sufficient data to compare immune infiltrates between experimental glioma models, in particular transgenic ones. Tumor immunology should be considered when choosing the animal model for preclinical or drug screening studies, as it can influence the therapeutic outcome.
Plasticity and Identity of Microglia and Infiltrating Macrophages within the Brain
Microglia originate from hematopoietic progenitor cells during the early embryonic development and share some of the common markers such as CD11b and CD14 with the other peripheral immune cells. However, microglia populate the CNS early in development and have the exclusive functions, such as synaptic pruning, interactions with neurons and other glial cells, maintenance of proper functioning of the neural circuitry.84, 85, 86 Microglia secrete neurotropic factors that recruits neuronal progenitor cells to the site of injury contributing to regeneration and tissue repair. Even though microglia are capable of self-renewing and maintain their steady state population, peripheral macrophages do not contribute to the turn-over of microglia under physiological conditions. Experiments with the use of BM transplantation after irradiation or parabiosis (in which two mice are surgically joined) did not support the notion that BM-macrophages enter CNS under normal conditions.87, 88
In most of neurologic diseases and CNS cancers, the infiltration of peripheral immune cells into the brain has a very important role in the etiopathology of the disease (for reviews see refs 89, 90). In mouse disease models, labeling of cells and the use of chimeras facilitated neuroscientists to distinguish resident cells (CD45low) from the infiltrating ones (CD45high). Various studies on animal models of cerebral ischemia and multiple sclerosis using mouse chimeras showed that microglia maintain their CD45low status even under inflammatory conditions and do not contribute significantly to the CD45high pool of BM-macrophages.91, 92
Recent studies comparing gene expression and chromatin landscape of myeloid cells from seven tissues (including CNS) revealed distinct enhancer/promoter landscapes, which reflects the ontogeny and function of an individual type of macrophages. Experiments with transplantation of BM cells from an adult donor into the lethally irradiated recipients have demonstrated that donor cells replace the endogenous embryo-derived macrophages and at the same time acquire a unique epigenetic landscape in accordance with their tissue of migration. Moreover, transplantation of differentiated macrophages from a place of residence into a different microenvironment induced the genes that are specific to the transplanted microenvironment, whereas downregulate genes constituting the identity of its original source.93, 94 These compelling evidence shows how plastic these cells are and uncovers their capability of reshaping their epigenetic landscape according to the microenvironment. Although all myeloid cells, including microglia, depend on a master transcription factor Pu.1, co-binding of other tissue specific transcription factors mediates the expression of specific gene pattern. For example, the function of macrophages largely depends on the activity of transcription factors Myb and FLT3, whereas microglial functions depends on CSF-1 and FLT3. 87, 88
Recent transcriptomics studies using microarray and direct RNA sequencing have revealed unique patterns of gene expression in microglia and BM-macrophages. Among all transcripts 40% were shared by both microglia and peripheral macrophages, which indicates their lineage relationship and functional similarities. More than 30% of the transcripts were expressed only in microglia or macrophages. Genes such as Hexb, Trem2, Gpr34, Tmem119, Cx3cr1, Siglech, and P2ry12, P2ry13 were shown to be expressed exclusively in microglia, whereas genes such as Cxcr7, Ifitm3, Pilra, Itgb2 were enriched on peritoneal macrophages.95, 96 Functions of those putative discriminating markers are summarized in the Table 2.
The question whether the circulating monocytes entering the CNS acquire microglial signature was addressed by Butovsky et al97 in an EAE model using Cx3cr1GFP/+ chimeric mice. Trancriptomic analysis demonstrated that microglia and macrophages maintain their unique molecular signature even under inflammatory conditions. However, some degree of plasticity exists, eg, Trem2 is expressed only on microglia under normal conditions, however IL-4 stimulation induced Trem2 in peripheral and infiltrating BM-macrophages. We found that primary microglial cultures exposed to glioma-conditioned medium upregulate Trem2 mRNA levels, whereas LPS stimulation sharply downregulates its expression (unpublished). This is in agreement with the previous reports depicting Trem2 as a M2 phenotype marker induced by IL-4 stimulation, whereas IFN-γ/LPS rapidly downregulated its level.98, 99 These results suggest a considerable functional plasticity of microglia and its adaptation to environmental cues.
It has been suggested that in BM transplantation studies immune infiltration to the CNS was exaggerated due to irradiation affecting the blood-brain barrier integrity. In a study using head-protected irradiation chimeras, in which an unspecific influx of donor-derived myeloid cells was reduced, the reduction in the tumor volume was demonstrated. A minor subset of microglia upregulated the CD45 expression, which could contribute to detection of the CD45high population in the tumor.100 Altogether, recent evidence points out that microglia and macrophages exhibit a unique gene expression pattern under normal conditions, but pathological microenvironment may alter transcriptomic signatures and cell functions. Understanding cellular heterogeneity of microglia and infiltrating macrophages, and dissecting their roles would help in exploiting the peripheral macrophages to manipulate the disease microenvironment.101
FACTORS INVOLVED IN RECRUITMENT AND POLARIZATION OF IMMUNE CELLS IN GLIOMAS
Chemotactic Signals for Myeloid Cells
In malignant gliomas, as in many other cancers, various immune cells accumulate and are polarized within the tumor to acquire new properties, further supporting tumor growth by a variety of growth factors and pro-angiogenic cytokines. Here, we would like to briefly discuss chemoattractants and cytokines that have been shown to stimulate migration and polarization of microglia/macrophages, as well as other types of immune cells, into the tumor site.
Hepatocyte growth factor (HGF)/scatter factor (SF) is a soluble ligand for transmembrane tyrosine kinase receptor c-Met,102, 103 required for wound healing and development.104 In malignant gliomas, expression of HGF/SF and its receptor c-Met was found both in GAMs and glioma cells isolated from GBM tissues,102, 103 and positively correlated with a grade of malignancy103, 105, 106 and inversely with patient mortality.107 In vitro studies revealed that HGF/SF secreted by glioma cells induces migration of microglial cells and BM-macrophages.102 Moreover, HGF/SF-c-Met signaling stimulates proliferation, migration, and invasiveness of human glioma cells in vitro,105, 108 as well as glioma growth and angiogenesis in vivo.109, 110 The expression of c-Met and HGF/SF secretion are induced by hypoxia111 and radiation,112 pointing to their role in tumor angiogenesis and glioma radio-resistance.
The monocyte chemotactic proteins (MCPs) belong to the MCP subgroup of the C-C chemokines family. CCL2 (MCP-1) by binding to its receptor CCR2 promotes chemotaxis and stimulates migration of immune cells, such as macrophages, monocytes, NK and T cells to the site of inflammation or injury. CCL2 is overexpressed in many cancers, including GBM and astrocytoma.113, 114, 115, 116 Levels of MCP-1 were correlated to numbers of GAMs in tumor tissues,114, 115 grade of malignancy and poor clinical prognosis.117 CCL2 supports proliferation and migration of GAMs to the tumor site and activates inflammatory responses in these cells.118 Moreover, gliomas expressing higher levels of CCL2 have more blood vessels.56 The role of distinct MCPs in glioma pathology is not yet clear, as it was shown that several tested glioma lines predominantly express MCP-3, but not MCP-1 mRNAs, and infiltration of GAMs in human gliomas was correlated with MCP-3, but not MCP-1 expression.119 MCP-3 promotes chemotaxis of immune cells by binding to CCR1, CCR2, and CCR3 receptors (while MCP-1 binds exclusively to CCR2).120 Consequently, MCP-3 is able to attract not only monocytes, T cells and NK as MCP-1 does, but it attracts also dendritic cells.121 Human monocytes express both CCR1 and CCR2 receptors on their surface,122 thus only MCP-3 can activate both of them. Knockdown of Mcp-1 expression in murine Gl261 gliomas did not decrease of GAM accumulation.123 Interestingly, CCR2A is frequently overexpressed in GBM cells and reduction of CCL2 level suppressed migration of these cells.124
Glial-derived neurotrophic factor (GDNF) contains seven conserved cysteine residues typical for all members of the TGF-β superfamily, but shares no more than 20% homology with family members. GDNF binds to GDNF receptor-α1 (GFR-α1), which leads to activation of tyrosine kinase RET signaling.125, 126 GDNF acts as a trophic factor promoting survival and differentiation of a various neuronal cell populations in the CNS.127, 128 In both human and rat, GDNF expression was detected not only in neurons,129, 130 but also in other types of cells such as astrocytes,131, 132 glioma cell lines,133, 134, 135 and activated microglia/macrophages after injury.136 GDNF expression was highly upregulated in glioma cell lines and human high-grade gliomas.137 GDNF is involved in chemoattraction of different cells,138, 139 including microglia.140 Overexpression of GDNF in fibroblasts favored microglia attraction, whereas silencing of GDNF expression in GL261 glioma cells decreased accumulation of GAMs and reduced tumor growth in vivo. GDNF promotes glioma cell migration in an autocrine manner.141, 142
GAM POLARIZING FACTORS
The macrophage colony-stimulating factor (M-CSF), also known as colony-stimulating factor 1 (CSF-1), is a secreted cytokine expressed by a variety of cells, such as macrophages, endothelial cells, fibroblasts, osteoblasts, and smooth muscles.143 CSF-1 is involved in the differentiation, proliferation and survival of monocytes, macrophages, and BM progenitor cells. In osteopetrotic mice (op/op), carrying a single mutation in the Csf-1 gene, a lack of CSF-1 prevents growth of macrophages and osteoclasts, thus leads to decrease in a number of macrophages and monocytes, both in blood and the tissues.144, 145, 146 CSF-1 R, a receptor for CSF-1, is expressed on monocytes, tissue macrophages, and monocyte-derived dendritic cells, and can bind not only CSF-1, but also IL-34.147 Binding a ligand to CSF-1 R induces phosphorylation of receptor followed by recruitment and activation of Src, PI-3 K, and Cbl kinases along with downstream signaling pathways.148, 149
CSF-1 is expressed in GBM cells, normal human astrocytes, and in operative specimens of human gliomas. The CSF-1 receptor, encoded by the c-Fms gene, is also expressed in human gliomas, which suggests autocrine/paracrine interactions.150 Murine GL261 glioma cells secrete CSF-1, which stimulates microglia invasion, and PLX3397, an inhibitor of CSF-1 R signaling, reduces the number of GAMs and GL261 glioma invasion in vivo.151 Treatment of mice bearing RCAS-PDGFb gliomas with PLX3397 increased survival and regressed established tumors.152 CSF-1 R blockade reduced intracranial growth of patient-derived glioma xenografts. However, GAMs were not depleted in treated mice, which suggests a negligible role of CSF-1 in glioma-microglia communication. Stimulation with granulocyte-macrophage CSF (GM-CSF/CSF-2) and IFN-γ facilitated GAM survival in animals with CSF-1 R inhibition. Expression of M2 activation markers (Adm, Mrc1, Arg1, F13a1) decreased in surviving GAMs, which is consistent with impairment of tumor-promoting functions.152 In both studies, Csf-1 levels were not compared with those in non-transformed astrocytes. In a MAFIA-based glioma model, temporal ablation of the CSF-1 R+ population affects the myeloid population in gliomas and results in switching to M1 phenotype, which was associated with decreased tumor cell proliferation, angiogenesis, and growth.74
We found the low expression and secretion of Csf-1 in murine GL261 glioma cells and human glioma cells (at similar levels as in non-transformed astrocytes).76 CSF-1 mRNA levels were not significantly upregulated in GBMs in comparison to normal brains and WHO grade I juvenile pilocytic astrocytomas.77 Growth of GL261 gliomas and accumulation of GAMs were not affected Csf-1 deficient mice.77 These conflicting results may stem from differences in applied animal models, selection of GBM cell lines and lack of specificity of a pharmacological inhibitor.
Granulocyte-macrophage colony-stimulating factor (GM-CSF)/colony-stimulating factor 2 (CSF-2), is a cytokine secreted by macrophages, T cells, NK cells, mast cells, endothelial cells, and fibroblasts. CSF-2 stimulates survival, proliferation and differentiation of hematopoietic myeloid cells, and in combination with other factors also hematopoietic multipotent precursors and differentiated cells of other lineages.153 CSF-2 binds to a dimeric receptor (CSF-2 R), which contains a ligand-specific a-chain subunit binding CSF-2 with a low affinity, and a b-chain subunit, which is able to bind CSF-2, IL-3, and IL-5.154, 155 The structure of CSF-2 R allows its sequential activation,146, 156 thus CSF-2 can have distinct effect on survival or proliferation of myeloid cells, depending on concentration. Activation of CSF-2 R stimulates PI-3 K, JAK-STAT, and MAPK signaling pathways.157
Currently data regarding expression of CSF-2 and its receptor in gliomas are conflicting. CSF-2 mRNA was found to be upregulated in human and rodent glioma tissues, and GBM cell lines76, 158, 159, 160, 161 and high expression of CSF-2 R was observed in those samples.161 Sawamura and colleague reported that CSF-2 was produced by 10 glioma cells lines in vitro but CSF-2 receptor was not expressed. No CSF-2 protein was detected in the cerebrospinal fluid of three malignant glioma patients.162 Further studies demonstrated that CSF-2 and CSF-2 R expression is highly upregulated in GBMs in comparison to low-grade gliomas and normal brain tissue.161 We demonstrated highly upregulated CSF-2 expression (but not CSF-1) in GBMs. Kaplan–Meier survival curves acquired from the Rembrandt depository showed correlation between CSF-2 expression and poor survival of glioma patients.77 Csf-2 expression was high in GL261 glioma cells and its knockdown reduced accumulation of GAMs, impaired GL261 glioma growth in mice, and prolonged their survival.77 We corroborated these results by the analysis of TCGA data set and found upregulation of CSF-2 expression in a subset of GBMs (unpublished). Upregulated CSF-2 expression in human GBMs and the autocrine or paracrine CSF-2 effects on glioma proliferation in vitro have been reproducibly demonstrated161, 163 CSF-2 was upregulated in both human and mouse glioma microenvironment compared with normal brain or peripheral blood samples, and stimulated the IL-4Rα expression on MDSCs.164 In conclusion, most data show the increased expression of CSF-2 in GBMs and several studies point to a critical role of this cytokine in accumulation and polarization of GAMs and MDSCs.
Transforming growth factor-beta (TGF-β) belongs to the TGF-β superfamily consisting three isoforms: TGF-β1, TGF-β2, and TGF-β3 in humans.114 TGF-β is a pleiotropic cytokine able to bind to a TGF-βR complex composed of type I and type II receptors. Upon activation, transcription factors Smad2 and Smad3 are phosphorylated and form homo- and heterotrimeric complexes with a common mediator Smad4. These complexes translocate to the nucleus, where they regulate transcription (for a review see ref. 165). Activity of TGF-β/SMAD pathway in human gliomas was negatively correlated with clinical prognosis.166
Glioma-derived TGF-β is a soluble cytokine which regulates cell proliferation, differentiation, invasiveness (by inducing MMP expression and endothelial–mesenchymal transition), and angiogenesis.167, 168 TGF-β1, TGF-β2, and TGF-β3 are differentially expressed in glioma cells in vitro and glioma tissues,169, 170, 171 with TGF- β 2 expression highly upregulated in malignant gliomas.170, 171 TGF-β2 has been shown to induce immunosuppressive polarization of immune cells,170 especially GAMs.172 TGF-β, together with prostaglandin E2 (PGE2), diminishes activation of GAMs through an inhibition of MHC I and MHC II expression on glioma and microglial cells.173 Glioma-conditioned medium induced production of PGE2 in rat microglial cultures.174 TGF-β treatment reduced a phagocytic activity of GAMs and IFN-γ-induced production of inflammatory cytokines Il-6, Il-1, and TNF-α.175 TGF-β was also described as an inhibitor of Tc and NK cells activation and differentiation. 176 Downregulation of TGF-β expression in glioma cells led to increased susceptibility to lysis by both NK cell and Tc cells,177 which could be explained by the fact that TGF-β downregulates NKG2D receptor on the NK and Tc cells.178 Microglial cells produce TGF-β1 when exposed to glioma.179, 180 Microglia-derived TGF-β1 strongly increased human and rat glioma cell invasion in vitro and in immunocompromised mice.180 TGF-β secreted by glioma cells is implicated into a functional shift of GAMs to the immunosuppressive, pro-invasive phenotype, which supports tumor growth. A dual function of TGF-β in gliomas has been suggested: TGF-β secreted by glioma exerts immunosuppression by shifting GAMs to the immunosuppressive phenotype, whereas GAMs-derived TGF-β promotes tumor progression by upregulation of its own receptors: TBRI and TBRII on glioma cells.181
Osteopontin (OPN/SPP1) is a chemokine-like, secreted phosphoglycoprotein, which belongs to a small integrin-binding ligand N-linked glycoproteins (SIBLINGs) family. OPN has distinct roles intra- and extracellularly, and is expressed by many cells: macrophages, neutrophils, dendritic cells, natural killer cells, epithelial cells, smooth muscle cells, osteoblasts, Purkinje cells, and neoplastic cells. Originally, OPN was described as a protein involved in osteoclast adherence and bone remodeling, however its role in pathophysiologic processes, including cancer progression, and immune responses was also shown.182, 183, 184 Overexpression of SPP1 (a gene encoding osteopontin)185 and elevated protein levels were found in GBMs,186 and correlated with poor patient prognosis.187
Multi-functionality of osteopontin is related to numerous post-translational modifications, including phosphorylation, glycosylation, proteolytic cleavage by thrombin and matrix metalloproteinases 3 and 7, as well as alternative splicing of Spp1 transcripts. Three different isoforms of human osteopontin were detected: OPN-a (a full-length protein), OPN-b (without exon 5), and OPN-c (without exon 4).188 In glioma cells OPN-c stronger induced MMP-2, MMP-9, and uPA (urokinase plasminogen activator) expression, than OPN-a and OPN-b.189 Expression pattern of different isoforms and a type of post-translational modification is cell-specific and may affect the potential role of osteopontin in cancer.190
Osteopontin contains an arginine-glycine-aspartate (RGD) motif, which binds to integrin receptors α5β1, α8β1, αvβ1, αvβ3, αvβ5, and αvβ6,191, 192 whereas OPN cleaved by thrombin exposes C-terminal SVVYGLR motif, which binds to α4β1, α9β1, α4β7, and α9β4 integrins.193, 194, 195 Such integrins are present on variety of cells, including endothelial cells, macrophages, neutrophils, and lymphocytes.196, 197, 198, 199 In human gliomas, osteopontin enhanced attraction of leukocytes and GAMs to the tumor site in the RGD-dependent manner and protein itself was co-localized with neutrophils and macrophages.200 Thrombin-cleaved fragments of osteopontin were abundant in malignant glial tumors.201 Giachelli et al202 reported that both a full-length and thrombin-cleaved osteopontin induce chemotactic migration of monocytes, macrophages, neutrophils and lymphocytes. OPN can also act in an autocrine manner and interacts with CD44v6, a specific variant of CD44 receptor expressed on neoplastic cells. Recent data show that osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem-cell phenotype and promotes aggressive tumor growth.203, 204 High levels of Spp1 mRNA were detected in dendritic cells and GAMs infiltrating murine GL261 gliomas,40 but there is no information regarding protein form and processing.
We demonstrated that C6 rat glioma cells overexpress Spp1 and secrete the processed protein, which participates in the pro-invasive, immunosuppressive activation of microglia. Pre-treatment with the RGD peptide, blocking interaction between a ligand and a integrin receptor, reduced the expression of genes characteristic for the protumorigenic microglia phenotype and abolished activation of focal adhesion kinase (FAK) as well as downstream kinases, such as Akt and ERK in cultured microglial cells. We found that osteopontin, produced by non-neoplastic cells, is a primarily pro-inflammatory factor, but sequential processing by thrombin and MMP-3 and -7 in glioma cells, generates a microglia-activating form devoid of the pro-inflammatory activity. In rat experimental gliomas, knockdown of osteopontin in glioma cells did not affect accumulation of GAMs, but blocked their polarization into the protumorigenic phenotype and tumor growth.205 Our findings provide the evidence for the role of glioma-derived osteopontin in polarization of infiltrating myeloid cells to the immunosuppressive, tumor supportive phenotype.
Periostin (POSTN), also known as osteoblast-specific factor 2 (OSF-2), is a multifunctional, evolutionarily conserved extracellular matrix (ECM) protein.206 POSTN also contains cysteine-rich domain, four internal repeats, fascilin domain, and hydrophilic C-terminal domain.207 Owing to the splice alterations at the C terminus, at least eight isoforms of periostin can be found in humans.208 POSTN interacts with cells through various integrin receptors and is an essential downstream effector of TGF-β superfamily signaling. The protein is involved in many processes in cancer development and progression, such as growth, invasiveness, and metastasis.209, 210, 211 Tumorigenic properties of periostin are associated with acting through Akt/PI3K and Wnt signaling pathways.210, 212 A recent study demonstrated that POSTN is one of the key cytokines involved in attraction and activation of GAMs in malignant gliomas.213 POSTN was abundantly expressed in glioma stem-like cells (GSCs) and its silencing led to significant reduction of GAMs infiltration, which indicated that POSTN is involved in attraction of GAMs by less differentiated glioma cells. Knockdown of POSTN inhibited tumor growth in vivo and increased survival of mouse xenografts. Moreover, knockdown of POSTN significantly reduced a number of M2-like GAMs (reduced expression of Fizz-1, Cd163, and Arg1) favoring GAMs expressing M1-like markers (MHC II and CD11c). Overexpression of POSTN in GSCs promoted recruitment of M2-like GAMs in vivo. The exact mechanism is yet to be understood, however, the proposed mode of action is through integrin-dependent activation of phosphatidylinositol 3-kinase signaling, which supports activation of macrophages. POSTN binds to αvβ3 integrin on GAMs and the use of neutralizing αvβ3 integrin antibody or a synthetic RGD peptide blocked this activation. The authors concluded that a majority of GAMs were derived from circulating monocytes, rather than from microglia as they were abundant around blood vessels, both in case of murine xenografts and human malignant brain tumors.213 POSTN secreted by glioma may also act in an autocrine manner by binding to the αv integrins on their surface and increasing tumor invasiveness.214 In human GBM tissues, GAMs density was positively correlated with POSTN protein expression.213 TCGA database analysis revealed that the POSTN level is higher in GBM than in low-grade gliomas, and high POSTN expression is associated with poor patients survival.213
Findings of two glioma-derived ligands that polarize GAMs in the glioma microenvironment via integrins, open a new perspective in glioma therapy. Factors secreted by glioma such as OPN and POSTN that bind to the integrins on immune cells are potential candidates for therapeutic interventions. Preclinical and clinical research on integrins in cancer, particularly on αvβ3 and αvβ5, revealed their substantial potential as therapeutic targets for the treatment of different cancer types.215 Cilengitide is a cyclic peptide (cyclo(-RGDfV-)), that selectively binds to the target integrins αvβ3 and αvβ5 present on the surface of endothelial cells, macrophages, and neoplastic cells. Cilengitide has been shown to act by inhibiting the FAK/Src/AKT pathway,216 and preclinical studies in mice revealed efficacious tumor regression. Initial clinical trials have shown promising results of cilengitide treatment in GBM therapy, but when combined with radiation and temozolomide in the phase III CENTRIC clinical trial, the drug failed to provide additive benefits.217 Integrins αvβ3, αvβ5 and αvβ8 are differentially expressed in GBM. In the CORE phase II trial, cilengitide was more effective in GBM patients with higher αvβ3 levels in tumor cells.218
Versican is a chondroitin-sulfate proteoglycan, which forms complexes with hyaluronan and is cleaved at specific peptide bonds by ADAMTS (a disintegrin-like and metalloprotease domain with thrombospondin type 1 motifs) proteases. ADAMTS proteases are expressed in coordinate manner and generate a bioactive versican fragment containing the N-terminal G1 domain—versikine.219 It has been shown that microglia promote glioma expansion through upregulation of membrane type 1 matrix metalloproteinase (MT1-MMP. MMP14), which activates glioma-derived MMP-2.220 This upregulation is mediated by the Toll-like receptor (TLR)2 signaling.221 TLRs belong to the superfamily of pattern recognition receptors that classically activate and mediate pro-inflammatory responses in innate immune cells by recognizing invading pathogens. TLR2 is highly expressed in all gliomas compared with non-tumor tissue, and patients with high TLR2 expression had reduced survival compared with patients having lower TLR2 expression.222
In glioma cells, synthesis of versican is primarily stimulated by TGF-β1.220 TGF-β2 has been shown to interact with versican V1 and support glioma proliferation and invasion.221 In the ECM, versican interacts with various partners, such as tenascin-R, fibulin-1, fibrillin-1, fibronectin, P- and L-selectin, and various chemokines via the globular domains or GAG chains.223 Versican can also bind to the epidermal growth factor receptor, the cell-surface proteins CD44 and integrin β1. Versican has been shown to act on macrophages through toll-like receptors (TLR2 and TLR6), and promote inflammatory cytokine production and Lewis lung carcinoma cell metastasis.224 The splice variants V0/V1 of versican were expressed in mouse and human glioma cell lines and tissues, and versican released from glioma promoted tumor invasion through TLR2 signaling in GAMs.225
CONCLUSIONS
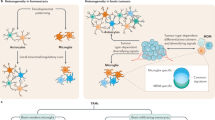
Combined work from a number of laboratories worldwide supports the thrilling concept that gliomas rely on tumor infiltrating microglia/macrophages during development and progression. GAMs produce a large number of cytokines, interleukins, and growth factors that directly stimulate glioma cell growth and invasion, or create a more permissive tumor microenvironment (Figure 2). These changes establish a supportive environment, rich in extracellular substrates, and facilitating glioma growth or invasion. Moreover, glioblastomas produce a large number of cytokines (IL-6, TGFβ) and small molecules (osteopontin, periostin, versican) that act on the glioma cells to increase glioma stem-cell self-renewal, and support cell proliferation, survival, and/or invasion of more differentiated glioma cells. Identifying the cellular and extracellular matrix-dependent relationships unique to the glioma microenvironment may provide exceptional opportunities to develop effective treatments to target these symbiotic associations that support glioma progression.

Summary of glioblastoma-immune system interactions. The expression of all presented factors/cytokines is upregulated in GBMs and inversely correlates with patient survival.
References
Ilkanizadeh S, Lau J, Huang M et al. Glial progenitors as targets for transformation in glioma. Adv Cancer Res 2014;121:1–65.
Weller M, Stupp R, Hegi ME et al. Personalized care in neuro-oncology coming of age: Why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol 2012;14:iv100–iv108.
Weller M, Pfister SM, Wick W et al. Molecular neuro-oncology in clinical practice: a new horizon. Lancet Oncol 2013;14:e370–e379.
Ceccarelli M, Barthel FP, Malta TM et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016;164:550–563.
Sarin H . Recent progress towards development of effective systemic chemotherapy for the treatment of malignant brain tumors. J Transl Med. 2009;7:77.
Tabatabai G, Stupp R, Van Den Bent MJ et al. Molecular diagnostics of gliomas: the clinical perspective. Acta Neuropathol 2010;120:585–592.
Vigneswaran K, Neill S, Hadjipanayis CG . Beyond the World Health Organization grading of infiltrating gliomas: advances in the molecular genetics of glioma classification. Ann Transl Med 2015;3:95.
Van Meir EG, Hadjipanayis CG, Norden AD et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignantglioma. CA Cancer J Clin 2010;60:166–193.
Fecci PE, Mitchell DA, Whitesides JF et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res 2006;66:3294–3302.
Lohr J, Ratliff T, Huppertz A et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin Cancer Res 2011;17:4296–4308.
Alexiou GA, Vartholomatos G, Karamoutsios A et al. Circulating progenitor cells: a comparison of patients with glioblastoma or meningioma. Acta Neurol Belg 2013;113:7–11.
Wainwright DA, Dey M, Chang A, et al. Targeting tregs in malignant brain cancer: overcoming IDO. Front Immunol 2013;4:116.
Perng P, Lim M . Immunosuppressive mechanisms of malignant gliomas: parallels at non-CNS sites. Front Oncol 2015;5:153.
Godard S, Getz G, Delorenzi M et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res 2003;63:6613–6625.
Shirahata M, Iwao-Koizumi K, Saito S et al. Gene expression-based molecular diagnostic system for malignant gliomas is superior to histological diagnosis. Clin Cancer Res 2007;13:7341–7356.
Vauleon E, Tony A, Hamlat A et al. Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genomics 2012;5:41.
Guillemin GJ, Brew BJ . Microglia macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol 2004;75:388–397.
Kaminska B . Microglia in gliomas: friend or foe? In: Sedo A, Mentlein R (eds). Glioma Cell Biology. Vienna: Springer, 2014, pp 241–270.
Esiri MM, Morris CS . Immunocytochemical study of macrophages and microglial cells and extracellular matrix components in human CNS disease. 2. Non-neoplastic diseases. J Neurol Sci 1991;101:59–72.
Wierzba-Bobrowicz T, Kuchna I, Matyja E . Reaction of microglial cells in human astrocytomas (preliminary report). Folia Neuropathol 1994;32:251–252.
Geranmayeh F, Scheithauer BW, Spitzer C, et al. Microglia in gemistocytic astrocytomas. Neurosurgery 2007;60:159–166.
Mieczkowski J, Kocyk M, Nauman P et al. Down-regulation of IKKβ expression in glioma-infiltrating microglia/macrophages is associated with defective inflammatory/immune gene responses in glioblastoma. Oncotarget 2015;6:33077–33090.
Sedgwick JD, Schwender S, Imrich H, et al. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA 1991;88:7438–7442.
Sedgwick JD . Resident macrophages (ramified microglia) of the adult brown Norway rat central nervous system are constitutively major histocompatibility complex class II positive. J Exp Med 1993;177:1145–1152.
Badie B, Schartner JM . Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 2000;46:957–962.
Hussain SF, Yang D, Suki D et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol 2006;8:261–279.
Badie B, Schartner J . Role of microglia in glioma biology. Microsc Res Tech 2001;54:106–113.
Yi L, Xiao H, Xu M et al. Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J Neuroimmunol 2011;232:75–82.
Murray PJ, Allen JE, Biswas SK et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14–20.
Murray PJ, Wynn T . A. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol 2011;89:557–563.
Ostuni R, Kratochvill F, Murray PJ, et al. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 2015;36:229–239.
Kratochvill F, Neale G, Haverkamp JM et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep 2015;12:1902–1914.
Ellert-miklaszewska A, Ciechomska I, Kaminska B . Glioma signaling. Adv Exp Med Biol 2013;986:209–220.
Durafourt BA, Moore CS, Zammit DA et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012;60:717–727.
Walker DG, Lue L . Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 2015;7:56.
Kaminska B, Mota M, Pizzi M . Signal transduction and epigenetic mechanisms in the control of microglia activation during neuroinflammation. Biochim Biophys Acta-Mol Basis Dis 2016;1862:339–351.
Komohara Y, Ohnishi K, Kuratsu J et al. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 2008;216:15–24.
Huettner C, Czub S, Kerkau S, et al. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res 1997;17:3217–3224.
Wagner S, Czub S, Greif M et al. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int J Cancer 1999;82:12–16.
Szulzewsky F, Pelz A, Feng X et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015;10:e0116644.
Szulzewsky F, Arora S, de Witte L et al. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016;64:1416–1436.
Gabrusiewicz K, Rodriguez B, Wei J et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI insight 2016;1:1–32.
Chen L . Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol 2004;4:336–347.
Gielen PR, Schulte BM, Kers-Rebel ED et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol 2015;74:390–400.
Marvel D, Gabrilovich DI . Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Investig 2015;125:3356–3364.
Raychaudhuri B, Rayman P, Ireland J et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol 2011;13:591–599.
Dubinski D, Wölfer J, Hasselblatt M et al. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol 2015;18:807–818.
Wainwright DA, Sengupta S, Han Y, et al. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol 2011;13:1308–1323.
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res 2008;14:5166–5172.
Wintterle S, Schreiner B, Mitsdoerffer M et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res 2003;63:7462–7467.
Jacobs JFM, Idema AJ, Bol KF et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol 2010;225:195–199.
Kmiecik J, Poli A, Brons NHC et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemiclevel. J Neuroimmunol 2013;264:71–83.
Borrego F, Kabat J, Kim D-KK et al. Structure and function of major histocompatibility complex (MHC) class I specific receptors expressed on human natural killer (NK) cells. Mol Immunol 2002;38:637–660.
Ryan JC, Naper C, Hayashi S et al. Physiologic functions of activating natural killer (NK) complex-encoded receptors on NK cells. Immunol Rev 2001;181:126–137.
Wiendl H, Mitsdoerffer M, Hofmeister V et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol 2002;168:4772–4780.
Friese MA, Platten M, Lutz SZ et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res 2003;63:8996–9006.
Eisele G, Wischhusen J, Mittelbronn M et al. TGF-β and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 2006;129:2416–2425.
Fadul CE, Fisher JL, Gui J, et al. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol 2011;13:393–400.
Poli A, Wang J, Domingues O et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget 2013;4:1527–1546.
Ogbomo H, Cinatl J, Mody CH, et al. Immunotherapy in gliomas: limitations and potential of natural killer (NK) cell therapy. Trends Mol Med 2017;17:433–441.
Vitucci M, Hayes DN, Miller CR . Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. Br J Cancer 2011;104:545–553.
Fomchenko EI, Holland EC . Mouse models of brain tumors and their applications in preclinical trials. Clin Cancer Res 2006;12:5288–5297.
Stylli SS, Luwor RB, Ware TMB et al. Mouse models of glioma. J Clin Neurosci 2015;22:619–626.
Auer RN, Del Maestro RF, Anderson R . A simple and reproducible experimental in vivo glioma model. Can J Neurol Sci 1981;8:325–331.
Sibenaller Za, Etame AB, Ali MM et al. Genetic characterization of commonly used glioma cell lines in the rat animal model system. Neurosurg Focus 2005;19:E1.
Richmond A, Su Y . Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech 2008;1:78–82.
Shankavaram UT, Bredel M, Burgan WE et al. Molecular profiling indicates orthotopic xenograft of glioma cell lines simulate a subclass of human glioblastoma. J Cell Mol Med 2012;16:545–554.
Huse JT, Holland EC . Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol 2009;19:132–143.
Holland EC, Celestino J, Dai C et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 2000;25:55–57.
McKinney AJ, Holmen SL . Animal models of melanoma: a somatic cell gene delivery mouse model allows rapid evaluation of genes implicated in human melanoma. Chin J Cancer 2011;30:153–162.
Sukhdeo K, Hambardzumyan D, Rich JN . Glioma development: where did it all go wrong? Cell 2011;146:187–188.
Hambardzumyan D, Amankulor NM, Helmy KY, et al. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol 2009;2:89–95.
Badie B, Bartley B, Schartner J . Differential expression of MHC class II and B7 costimulatory molecules by microglia in rodent gliomas. J Neuroimmunol 2002;133:39–45.
Gabrusiewicz K, Hossain MB, Cortes-Santiago N et al. Macrophage ablation reduces M2-Like populations and jeopardizes tumor growth in a MAFIA-based glioma model. Neoplasia 2015;17:374–384.
Ellert-Miklaszewska A, Dabrowski M, Lipko M, et al. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013;61:1178–1190.
Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, et al. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011;6:e23902.
Sielska M, Przanowski P, Wylot B et al. Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J Pathol 2013;230:310–321.
Levy A, Blacher E, Vaknine H, et al. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro Oncol 2012;14:1037–1049.
Raychaudhuri B, Rayman P, Huang P et al. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol 2015;122:293–301.
Kennedy BC, Maier LM, D’Amico R et al. Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol 2009;10:11.
Parney IF, Waldron JS, Parsa AT . Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J Neurosurg 2009;110:572–582.
Ahn B, Kohanbash G, Ohkuri T et al. Histamine deficiency promotes accumulation of immunosuppressive immature myeloid cells and growth of murine gliomas. Oncoimmunology 2015;4:e1047581.
Maes W, Verschuere T, Van Hoylandt A, et al. Depletion of regulatory T cells in a mouse experimental glioma model through anti-CD25 treatment results in the infiltration of non-immunosuppressive myeloid cells in the brain. Clin Dev Immunol 2013;2013:952469.
Kettenmann H, Kirchhoff F, Verkhratsky A . Microglia: new roles for the synaptic stripper. Neuron 2013;77:10–18.
London A, Cohen M, Schwartz M . Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci 2013;7:34.
Zhan Y, Paolicelli RC, Sforazzini F et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 2014;17:400–406.
Prinz M, Mildner A . Microglia in the CNS: immigrants from another world. Glia 2011;59:177–187.
Ginhoux F, Greter M, Leboeuf M et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2014;330:841–845.
Hambardzumyan D, Gutmann DH, Kettenmann H . The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 2015;19:20–27.
Glass R, Synowitz M . CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol 2014;128:347–362.
Campanella M, Sciorati C, Tarozzo G, et al. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke 2002;33:586–592.
O’Koren EG, Mathew R, Saban DR . Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci Rep 2016;6:20636.
Lavin Y, Winter D, Blecher-Gonen R et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014;159:1312–1326.
Gosselin D, Link VM, Romanoski CE et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014;159:1327–1340.
Hickman SE, Kingery ND, Ohsumi TK et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013;1:1896–1905.
Satoh Jichi, Kino Y, Asahina N et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016;36:39–49.
Butovsky O, Jedrychowski MP, Moore CS et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci 2014;17:131–143.
Sieber MW, Jaenisch N, Brehm M et al. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS ONE 2013;8:e52982.
Takahashi K, Rochford CDP, Neumann H . Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005;201:647–657.
Müller A, Brandenburg S, Turkowski K et al. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer 2015;137:278–288.
Lahmar Q, Keirsse J, Laoui D et al. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. Biochim Biophys Acta-Rev Cancer 2016;1865:23–34.
Badie B, Schartner J, Klaver J et al In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery 1999;44:1077–1083.
Kunkel P, Müller S, Schirmacher P et al. Expression and localization of scatter factor/hepatocyte growth factor in human astrocytomas. Neuro Oncol 2001;3:82–88.
Sonnenberg E, Meyer D, Weidner KM et al. Scatter factor/hepatocyte growth factor and its receptor, the c-met tyrosine kinase, can mediate a signal exchange between mesenchyme and epithelia during mouse development. J Cell Biol 1993;123:223–235.
Koochekpour S, Jeffers M, Rulong S et al. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 1997;57:5391–5398.
Esencay M, Newcomb EW, Zagzag D . HGF upregulates CXCR4 expression in gliomas via NF-kappaB: implications for glioma cell migration. J Neurooncol 2010;99:33–40.
Arrieta O, Garcia E, Guevara P et al. Hepatocyte growth factor is associated with poor prognosis of malignant gliomas and is a predictor for recurrence of meningioma. Cancer 2002;94:3210–3218.
Tu H, Zhou Z, Liang Q et al. CXCR4 and SDF-1 production are stimulated by hepatocyte growth factor and promote glioma cell invasion. Onkologie 2009;32:331–336.
Laterra J, Rosen E, Nam M, et al. Scatter factor/hepatocyte growth factor expression enhances human glioblastoma tumorigenicity and growth. Biochem Biophys Res Commun 1997;235:743–747.
Moriyama T, Kataoka H, Hamasuna R et al. Up-regulation of vascular endothelial growth factor induced by hepatocyte growth factor/scatter factor stimulation in human glioma cells. Biochem Biophys Res Commun 1998;249:73–77.
Eckerich C, Zapf S, Fillbrandt R, et al. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int J Cancer 2007;121:276–283.
Sheng-hua C, Yan-bin M, Zhi-an Z et al. Radiation-enhanced hepatocyte growth factor secretion in malignant glioma cell lines. Surg Neurol 2007;68:610–613.
Prat E, Baron P, Meda L et al. The human astrocytoma cell line U373MG produces monocyte chemotactic protein (MCP)-1 upon stimulation with β-amyloid protein. Neurosci Lett 2000;283:177–180.
Platten M, Kretz A, Naumann U et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol 2003;54:388–392.
Leung SY, Wong MP, Chung LP, et al. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol 1997;93:518–527.
Desbaillets I, Tada M, De Tribolet N, et al. Human astrocytomas and glioblastomas express monocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro. Int J Cancer 1994;58:240–247.
Kuratsu J, Yoshizato K, Yoshimura T, et al. Quantitative study of monocyte chemoattractant protein-1 (MCP-1) in cerebrospinal fluid and cyst fluid from patients with malignant glioma. J Natl Cancer Inst 1993;85:1836–1839.
Hinojosa AE, Garcia-Bueno B, Leza JC et al. CCL2/MCP-1 modulation of microglial activation and proliferation. J Neuroinflammation 2011;8:77.
Okada M, Saio M, Kito Y et al. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol 2009;34:1621–1627.
Rot A, Von Andrian UH . Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 2004;22:891–928.
Menten P, Wuyts A, Van Damme J . Monocyte chemotactic protein-3. Eur Cytokine Netw 2001;12:554–560.
Serbina NV, Jia T, Hohl TM, et al. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol 2008;26:421–452.
Kirk EA, Sagawa ZK, McDonald TO, et al. Monocyte chemoattractant protein-1 deficiency fails to restrain macrophage infiltration into adipose tissue. Diabetes 2008;57:1254–1261.
Liang Y, Bollen AW, Gupta N . CC chemokine receptor-2A is frequently overexpressed in glioblastoma. J Neurooncol 2008;86:153–163.
Treanor JJ, Goodman L, de Sauvage F et al. Characterization of a multicomponent receptor for GDNF. Nature 1996;382:80–83.
Trupp M, Arenas E, Fainzilber M et al. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Natur 1996;381:785–789.
Lapchak PA, Jiao S, Collins F, et al. Glial cell line-derived neurotrophic factor: distribution and pharmacology in the rat following a bolus intraventricular injection. Brain Res 1997;747:92–102.
Unsicker K . GDNF: a cytokine at the interface of TGF-betas and neurotrophins. Cell Tissue Res 1996;286:175–178.
Hunot S, Bernard V, Faucheux B et al. Glial cell line-derived neurotrophic factor (GDNF) gene expression in the human brain: a post mortem in situ hybridization study with special reference to Parkinson’s disease. J Neural Transm 1996;103:1043–1052.
Pochon NAM, Menoud A, Tseng JL, et al. Neuronal GDNF expression in the adult rat nervous system identified by in situ hybridization. Eur J Neurosci 1997;9:463–471.
Moretto G, Walker DG, Lanteri P et al. Expression and regulation of glial-cell-line-derived neurotrophic factor (GDNF) mRNA in human astrocytes in vitro. Cell Tissue Res 1996;286:257–262.
Schaar DG, Sieber Ba, Dreyfus CF, et al. Regional and cell-specific expression of GDNF in rat brain. Expl Neurol 1993;124:368–371.
Lin L, Doherty D, Lile J, et al. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993;260:1130–1132.
Verity aN, Wyatt TL, Hajos B, et al. Regulation of glial cell line-derived neurotrophic factor release from rat C6 glioblastoma cells. J Neurochem 1998;70:531–539.
Verity AN, Wyatt TL, Lee W et al. Differential regulation of glial cell line-derived neurotrophic factor (GDNF) expression in human neuroblastoma and glioblastoma cell lines. J Neurosci Res 1999;55:187–197.
Batchelor PE, Liberatore GT, Wong JYF et al. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci 1999;19:1708–1716.
Wiesenhofer B, Stockhammer G, Kostron H et al. Glial cell line-derived neurotrophic factor (GDNF) and its receptor (GFR-alpha 1) are strongly expressed in human gliomas. Acta Neuropathol 2000;99:131–137.
Cornejo M, Nambi D, Walheim C et al. Effect of NRG1, GDNF, EGF and NGF in the migration of a Schwann cell precursor line. Neurochem Res 2010;35:1643–1651.
Dudanova I, Gatto G, Klein R . GDNF acts as a chemoattractant to support ephrina-induced repulsion of limb motor axons. Curr Biol 2010;20:2150–2156.
Ku MC, Wolf SA, Respondek D et al. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol 2013;125:609–620.
Lu D-Y, Leung Y-M, Cheung C-W, et al. Glial cell line-derived neurotrophic factor induces cell migration and matrix metalloproteinase-13 expression in glioma cells. Biochem Pharmacol 2010;80:1201–1209.
Wan G, Too H-P . A specific isoform of glial cell line-derived neurotrophic factor family receptor alpha 1 regulates RhoA expression and glioma cell migration. J Neurochem 2010;115:759–770.
Chitu V, Stanley ER . Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol 2006;18:39–48.
Yoshida H, Hayashi S, Kunisada T et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990;345:442–444.
Bergmann CE, Hoefer IE, Meder B et al. Arteriogenesis depends on circulating monocytes and macrophage accumulation and is severely depressed in op/op mice. J Leukoc Biol 2006;80:59–65.
Hamilton JA, Achuthan A . Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol 2013;34:81–89.
Wei J, Barr J, Kong L-Y et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther 2010;9:67–78.
Hamilton JA . CSF-1 signal transduction. J Leukoc Biol 1997;62:145–155.
Yu W, Chen J, Xiong Y et al. CSF-1 receptor structure/function in MacCsf1r−/− macrophages: regulation of proliferation, differentiation, and morphology. J Leukoc Biol 2008;84:852–863.
Alterman RL, Stanley ER . Colony stimulating factor-1 expression in human glioma. Mol Chem Neuropathol 1994;21:177–188.
Coniglio SJ, Eugenin E, Dobrenis K et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med 2012;18:519–527.
Pyonteck SM, Akkari L, Schuhmacher AJ et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013;19:1264–1272.
Metcalf D . The colony-stimulating factors and cancer. Cancer Immunol Res 2013;1:351–356.
Gasson JC, Kaufman SE, Weisbart RH et al. High-affinity binding of granulocyte-macrophage colony-stimulating factor to normal and leukemic human myeloid cells. Proc Natl Acad Sci USA 1986;83:669–673.
Hayashida K, Kitamura T, Gorman DM et al. Molecular cloning of a second subunit of the receptor for human granulocyte-macrophage colony-stimulating factor (GM-CSF): reconstitution of a high-affinity GM-CSF receptor. Proc Natl Acad Sci USA 1990;87:9655–9659.
Hercus TR, Thomas D, Guthridge MA et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood 2009;114:1289–1298.
Fleetwood AJ, Cook AD, Hamilton J . Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol 2005;25:405–428.
Frei K, Nohava K, Malipiero UV et al. Production of macrophage colony-stimulating factor by astrocytes and brain macrophages. J Neuroimmunol 1992;40:189–195.
Guillaume T, Sekhavat M, Rubinstein DB et al. Transcription of genes encoding granulocyte-macrophage colony-stimulating factor, interleukin 3, and interleukin 6 receptors and lack of proliferative response to exogenous cytokines in nonhematopoietic human malignant cell lines. Cancer Res 1993;53:3139–3144.
Mueller MM, Herold-Mende CC, Riede D et al. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. AmJPathol 1999;155:1557–1567.
Revoltella RP, Menicagli M, Campani D . Granulocyte-macrophage colony-stimulating factor as an autocrine survival-growth factor in human gliomas. Cytokine 2012;57:347–359.
Murata J, Sawamura Y, Tada M et al. Human glioblastoma cells produce granulocyte-macrophage colony-stimulating factor in vitro, but not in vivo, without expressing its receptor. Neurol Med Chir 1993;33:603–609.
Curran CS, Evans MD, Bertics PJ . GM-CSF production by glioblastoma cells has a functional role in eosinophil survival, activation, and growth factor production for enhanced tumor cell proliferation. J Immunol 2011;187:1254–1263.
Kohanbash G, McKaveney K, Sakaki M et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-α. Cancer Res 2013;73:6413–6423.
Kaminska B, Kocyk M, Kijewska M . TGF beta signaling and its role in glioma pathogenesis. Adv Exp Med Biol 2013;986:171–187.
Bruna A, Darken RS, Rojo F et al. High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007;11:147–160.
Verrecchia F, Mauviel A . Control of connective tissue gene expression by TGF beta: role of Smad proteins in fibrosis. Curr Rheumatol Rep 2002;4:143–149.
Leivonen S-K, Kähäri V-M . Transforming growth factor-beta signaling in cancer invasion and metastasis. Int J Cancer 2007;121:2119–2124.
Constam DB, Philipp J, Malipiero UV et al. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol 1992;148:1404–1410.
Bodmer S, Strommer K, Frei K et al. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol 1989;143:3222–3229.
Kjellman C, Olofsson SP, Hansson O et al. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int J Cancer 2000;89:251–258.
Wu A, Wei J, Kong LY et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 2010;12:1113–1125.
Taniguchi Y, Ono K, Yoshida S et al. Antigen-presenting capability of glial cells under glioma-harboring conditions and the effect of glioma-derived factors on antigen presentation. J Neuroimmunol 2000;111:177–185.
Nakano Y, Kuroda E, Kito T et al. Induction of prostaglandin E2 synthesis and microsomal prostaglandin E synthase-1 expression in murine microglia by glioma-derived soluble factors. Laboratory investigation. J Neurosurg 2008;108:311–319.
Suzumura A, Sawada M, Yamamoto H, et al. Transforming growth factor-beta suppresses activation and proliferation of microglia in vitro. J Immunol 1993;151:2150–2158.
Flavell RA, Sanjabi S, Wrzesinski SH, et al. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol 2010;10:554–567.
Friese MA, Wischhusen J, Wick W et al. RNA interference targeting transforming growth factor-β enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res 2004;64:7596–7603.
Crane CA, Han SJ, Barry JJ et al. TGF-β downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol 2010;12:7–13.
Kiefer R, Supler ML, Toyka KV et al In situ detection of transforming growth factor-beta mRNA in experimental rat glioma and reactive glial cells. Neurosci Lett 1994;166:161–164.
Wesolowska A, Kwiatkowska A, Slomnicki L et al. Microglia-derived TGF-β as an important regulator of glioblastoma invasion—an inhibition of TGF-β-dependent effects by shRNA against human TGF-β type II receptor. Oncogene 2007;27:918–930.
Li W, Graeber MB . The molecular profile of microglia under the influence of glioma. Neuro Oncol 2012;14:958–978.
Wai PY, Kuo PC . Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev 2008;27:103–118.
Wang KX, Denhardt DT . Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev 2008;19:333–345.
Kazanecki CC, Uzwiak DJ, Denhardt DT . Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J Cell Biochem 2007;102:912–924.
Saitoh Y, Kuratsu J, Takeshima H et al. Expression of osteopontin in human glioma. Its correlation with the malignancy. Lab Invest 1995;72:55–63.
Sreekanthreddy P, Srinivasan H, Kumar DM et al. Identification of potential serum biomarkers of glioblastoma: Serum osteopontin levels correlate with poor prognosis. Cancer Epidemiol Biomarkers Prev 2010;19:1409–1422.
Matusan-Ilijas K, Behrem S, Jonjic N, et al. Osteopontin expression correlates with angiogenesis and survival in malignant astrocytoma. Pathol Oncol Res 2008;14:293–298.
He B, Mirza M, Weber GF . An osteopontin splice variant induces anchorage independence in human breast cancer cells. Oncogene 2006;25:2192–2202.
Yan W, Qian C, Zhao P et al. Expression pattern of osteopontin splice variants and its functions on cell apoptosis and invasion in glioma cells. Neuro Oncol 2010;12:765–775.
Anborgh PH, Mutrie JC, Tuck AB, et al. Pre- and post-translational regulation of osteopontin in cancer. J Cell Commun Signal 2011;5:111–122.
Denhardt DT, Noda M, O’Regan AW, et al. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest 2001;107:1055–1061.
Yokosaki Y, Tanaka K, Higashikawa F, et al. Distinct structural requirements for binding of the integrins alphavbeta6, alphavbeta3, alphavbeta5, alpha5beta1 and alpha9beta1 to osteopontin. Matrix Biol 2005;24:418–427.
Sharif SA, Du X, Myles T et al. Thrombin-activatable carboxypeptidase B cleavage of osteopontin regulates neutrophil survival and synoviocyte binding in rheumatoid arthritis. Arthritis Rheum 2009;60:2902–2912.
Laffon A, Garcia-Vicuna R, Humbria A et al. Upregulated expression and function of VLA-4 fibronectin receptors on human activated T cells in rheumatoid arthritis. J Clin Invest 1991;88:546–552.
Yokosaki Y, Matsuura N, Sasaki T et al. The integrinalpha9beta1 binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J Biol Chem 1999;274:36328–36334.
Ito K, Kon S, Nakayama Y et al. The differential amino acid requirement within osteopontin inaplha4 andalpha9 integrin-mediated cell binding and migration. Matrix Biol 2009;28:11–19.
Nishimichi N, Higashikawa F, Kinoh HH, et al. Polymeric osteopontin employs integrinalpha9beta1 as a receptor and attracts neutrophils by presenting a de Novo binding site. J Biol Chem 2009;284:14769–14776.
Diao H, Iwabuchi K, Li L et al. Osteopontin regulates development and function of invariant natural killer T cells. Proc Natl Acad Sci USA 2008;105:15884–15889.
Fong YC, Liu SC, Huang CY et al. Osteopontin increases lung cancer cells migration via activation of the alphavbeta3 integrin/FAK/Akt and NF-kappaB-dependent pathway. Lung Cancer 2009;64:263–270.
Atai NA, Bansal M, Lo C et al. Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 2011;132:39–48.
Yamaguchi Y, Shao Z, Sharif S et al. Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J Biol Chem 2013;288:3097–3111.
Giachelli CM, Lombardi D, Johnson RJ, et al. Evidence for a role of osteopontin in macrophage infiltration in response to pathological stimuli in vivo. Am J Pathol 1998;152:353–358.
Pietras A, Katz AM, Ekström EJ et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014;14:357–369.
Lamour V, Henry A, Kroonen J et al. Targeting osteopontin suppresses glioblastoma stem-like cell character and tumorigenicity in vivo. Int J Cancer 2015;137:1047–1057.
Ellert-Miklaszewska A, Wisniewski P, Kijewska M et al. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene 2016;35:6366–6377.
Takeshita S, Kikuno R, Tezuka K et al. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J 1993;294:271–278.
Rani S, Barbe MF, Barr AE, et al. Induction of periostin-like factor and periostin in forearm muscle, tendon, and nerve in an animal model of work-related musculoskeletal disorder. J Histochem Cytochem 2009;57:1061–1073.
Morra L, Rechsteiner M, Casagrande S et al. Relevance of periostin splice variants in renal cell carcinoma. Am J Pathol 2011;179:1513–1521.
Bao S, Ouyang G, Bai X et al. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 2004;5:329–339.
Malanchi I, Santamaria-Martinez A, Susanto E et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012;481:85–89.
Michaylira CZ, Wong GS, Miller CG et al. Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Res 2010;70:5281–5292.
Baril P, Gangeswaran R, Mahon PC et al. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene 2007;26:2082–2094.
Zhou W, Ke SQ, Huang Z et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 2015;17:170–182.
Mikheev AM, Mikheeva SA, Trister AD et al. Periostin is a novel therapeutic target that predicts and regulates glioma malignancy. Neuro Oncol 2015;17:372–382.
Goodman SL, Picard M . Integrins as therapeutic targets. Trends Pharmacol Sci 2012;33:405–412.
Yamada S, Bu XY, Khankaldyyan V, et al. Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice. Neurosurgery 2006;59:1304–1312.
Stupp R, Hegi ME, Gorlia T et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:1100–1108.
Weller M, Nabors LB, Gorlia T et al. Cilengitide in newly diagnosed glioblastoma: biomarker expression and outcome. Oncotarget 2016;7:15018–15032.
Markovic DS, Vinnakota K, Chirasani S et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA 2009;106:12530–12535.
Arslan F, Bosserhoff A-K, Nickl-Jockschat T, et al. The role of versican isoforms V0/V1 in glioma migration mediated by transforming growth factor-beta2. Br J Cancer 2007;96:1560–1568.
Onken J, Moeckel S, Leukel P et al. Versican isoform V1 regulates proliferation and migration in high-grade gliomas. J Neurooncol 2014;120:73–83.
Vinnakota K, Hu F, Ku MC et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol 2013;15:1457–1468.
Wu YJ, La Pierre DP, Wu J, et al. The interaction of versican with its binding partners. Cell Res 2005;15:483–494.
Kim S, Takahashi H, Lin W-W et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009;457:102–106.
Hu F, Dzaye ODA, Hahn A et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol 2015;17:200–210.
Clark MJ, Homer N, O’Connor BD et al. U87MG decoded: the genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet 2010; 6.
Badie B, Goh CS, Klaver J et al. Combined radiation and p53 gene therapy of malignant glioma cells. Cancer Gene Ther 1999;6:155–162.
Lazovic J, Soto H, Piccioni D et al. Detection of 2-hydroxyglutaric acid in vivo by proton magnetic resonance spectroscopy in U87 glioma cells overexpressing isocitrate dehydrogenase-1 mutation. Neuro Oncol 2012;14:1465–1472.
Jung TY, Choi YD, Kim YH et al. Immunological characterization of glioblastoma cells for immunotherapy. Anticancer Res 2013;33:2525–2534.
Naumann U, Durka S, Weller M . Dexamethasone-mediated protection from drug cytotoxicity: association with p21WAF1/CIP1 protein accumulation? Oncogene 1998;17:1567–1575.
Zhang R, Banik NL, Ray SK . Differential sensitivity of human glioblastoma LN18 (PTEN-positive) and A172 (PTEN-negative) cells to Taxol for apoptosis. Brain Res 2008;1239:216–225.
Kumar PS, Shiras A, Das G, et al. Differential expression and role of p21cip/waf1 and p27kip1 in TNF-alpha-induced inhibition of proliferation in human glioma cells. Mol Cancer 2007;6:42.
Szatmári T, Lumniczky K, Désaknai S et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci 2006;97:546–553.
Zagzag D, Salnikow K, Chiriboga L et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest 2005;85:328–341.
Merkel CA, da Silva Soares RB, de Carvalho ACV et al. Activation of endogenous p53 by combined p19Arf gene transfer and nutlin-3 drug treatment modalities in the murine cell lines B16 and C6. BMC Cancer 2010;10:316.
Kyrkanides S, Miller AW, Miller JH et al. Peripheral blood mononuclear cell infiltration and neuroinflammation in the HexB−/− mouse model of neurodegeneration. J Neuroimmunol 2008;203:50–57.
Kronlage M, Song J, Sorokin L et al. Autocrine Purinergic Receptor Signaling Is Essential for Macrophage Chemotaxis. Sci Signal 2010;3:ra55–ra55.
Webster CM, Hokari M, McManus A et al. Microglial P2Y12 deficiency/inhibition protects against brain ischemia. PLoS One 2013;8:e70927.
Ford JW and McVicar DW . TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol 2009;21:38–46.
Neumann H, Takahashi K . Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 2007;184:92–99.
Bédard A, Tremblay P, Chernomoretz A et al. Identification of genes preferentially expressed by microglia and upregulated during cuprizone-induced inflammation. Glia 2007;55:777–789.
Fuhrmann M, Bittner T, Jung CKE et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci 2010;13:411–413.
Held-Feindt J, Hattermann K, Muerkoster SS et al. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp Cell Res 2010;316:1553–1566.
Kopatz J, Beutner C, Welle K et al. Siglec-h on activated microglia for recognition and engulfment of glioma cells. Glia 2013;61:1122–1133.
Walters MJ, Ebsworth K, Berahovich RD et al. Inhibition of CXCR7 extends survival following irradiation of brain tumours in mice and rats. Br J Cancer 2014;110:1179–1188.
Han JH, Lee S, Park YS et al. IFITM6 expression is increased in macrophages of tumor-bearing mice. Oncol Rep 2011;25:531–536.
Wang J, Shiratori I, Uehori J et al. Neutrophil infiltration during inflammation is regulated by PILRα via modulation of integrin activation. Nat Immunol 2013;14:34–40.
Yee NK, Hamerman JA . β(2) integrins inhibit TLR responses by regulating NF-κB pathway and p38 MAPK activation. Eur J Immunol 2013;43:779–792.
Acknowledgements
Studies supported by a Grant 2012/04/A/NZ3/00630 from the National Science Center and by the project DIMUNO: ‘Development of new cancer therapies based on selective antitumor immunomodulators’—co-financed by the National Centre for Research and Development in the framework of STRATEGMED-3 Prevention Practices and Treatment of Civilization Diseases. KAW is funded by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant Agreement No. 665735.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Malignant gliomas are aggressive brain tumors that are heavily infiltrated with myeloid cells deficient in antitumor activity but supportive of tumor invasion. This review summarizes data on immune cell heterogeneity and cell-cell interactions in the tumor microenvironment, as well as tumor-derived signals and mechanisms driving myeloid cell accumulation and reprogramming.
Rights and permissions
About this article

Cite this article
Gieryng, A., Pszczolkowska, D., Walentynowicz, K. et al. Immune microenvironment of gliomas. Lab Invest 97, 498–518 (2017). https://doi.org/10.1038/labinvest.2017.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.19
This article is cited by
-
Immune cells: potential carriers or agents for drug delivery to the central nervous system
Military Medical Research (2024)
-
MIF contribution to progressive brain diseases
Journal of Neuroinflammation (2024)
-
Myeloid-derived suppressor cells in cancer: therapeutic targets to overcome tumor immune evasion
Experimental Hematology & Oncology (2024)
-
Antibiotics treatment promotes vasculogenesis in the brain of glioma-bearing mice
Cell Death & Disease (2024)
-
Characterization of purinergic signaling in tumor-infiltrating lymphocytes from lower- and high-grade gliomas
Purinergic Signalling (2024)
