
Abstract
Digital image analysis (DIA) is becoming central to the quantitative evaluation of tissue biomarkers for discovery, diagnosis and therapeutic selection for the delivery of precision medicine. In this study, automated DIA using a new purpose-built software platform (QuPath) is applied to a cohort of 293 breast cancer patients to score five biomarkers in tissue microarrays (TMAs): ER, PR, HER2, Ki67 and p53. This software is able to measure IHC expression following fully automated tumor recognition in the same immunohistochemical (IHC)-stained tissue section, as part of a rapid workflow to ensure objectivity and accelerate biomarker analysis. The digital scores produced by QuPath were compared with manual scores by a pathologist and shown to have a good level of concordance in all cases (Cohen’s κ>0.6), and almost perfect agreement for the clinically relevant biomarkers ER, PR and HER2 (κ>0.86). To assess prognostic value, cutoff thresholds could be applied to both manual and automated scores using the QuPath software, and survival analysis performed for 5-year overall survival. DIA was shown to be capable of replicating the statistically significant stratification of patients achieved using manual scoring across all biomarkers (P<0.01, log-rank test). Furthermore, the image analysis scores were shown to consistently lead to statistical significance across a wide range of potential cutoff thresholds, indicating the robustness of the method, and identify sub-populations of cases exhibiting different expression patterns within the p53 and Ki67 data sets that warrant further investigation. These findings have demonstrated QuPath’s suitability for fast, reproducible, high-throughput TMA analysis across a range of important biomarkers. This was achieved using our tumor recognition algorithms for IHC-stained sections, trained interactively without the need for any additional tumor recognition markers, for example, cytokeratin, to obtain greater insight into the relationship between biomarker expression and clinical outcome applicable to a range of cancer types.
Similar content being viewed by others


Main
The microscopic analysis of tissue samples continues to be the mainstay of diagnosis, grading and therapeutic decision making in solid cancers, also for precision medicine and targeted therapy. In this context, the diagnostic test therefore acts as a reliable companion to the selection of therapy for an individual patient. Indeed, in solid cancers, tissue or cellular samples are the core of predictive pathology or personalized medicine.1
Tissue samples can be used in multiple manners to achieve a diagnostic goal.2 They serve to identify mutations or other genomic anomalies from extracted nucleic acids by low-throughput PCR or next-generation sequencing, by labeling sequences in vitro using FISH or DISH, RNA in situ detection or immunohistochemical (IHC) detection of proteins. The latter probably presents the most widely used, cost-effective and practical solution for biomarker evaluation in primary diagnostics, and is used extensively in research for the rapid screening of candidate biomarkers in multiplex platforms such as tissue microarrays (TMAs).
Despite the existence of robust guidelines for optimization,3, 4 on its own IHC is limited by the subjectivity associated with (a) the characterization of the appropriate tumor areas to analyze within a complex tissue section; and (b) the visual interpretation of the expression patterns and intensities in tumor cells and surrounding tissues. Numerous studies have shown poor inter-laboratory, inter-observer and intra-observer reproducibility in the assessment of IHC expression patterns of tissue.5, 6 This variability is slowing the progress of biomarker discovery and delivery of precision medicine.
Automated digital image analysis (DIA) has the potential to provide the objectivity, reliability and speed required to radically transform tissue biomarker research, discovery and routine delivery. It is only recently, however, that the scanning technology, image processing and image understanding algorithms are aligning to allow solutions to be developed with genuine impact.7
This article demonstrates the application and evaluation of a new, comprehensive digital pathology image analysis platform developed in-house at Queen’s University Belfast: QuPath. The platform allows (a) the accurate recognition of tumor and non-tumor cells within whole or TMA sections using automated digital algorithms, and (b) reproducible IHC scoring of relevant biomarkers, which provides an alternative to traditional, subjective manual scoring. Hereby we demonstrate QuPath’s ability to achieve robust biomarker scoring to support high-throughput studies, and to provide additional, quantitative data that can offer new insights into biomarker interpretation. The QuPath software presented here represents an advanced platform for algorithm development, with the ability to accelerate tissue research and provide the tools to link discovery with diagnostics.
Materials and methods
Study Design
Image analysis was applied to score the clinically important biomarkers estrogen receptor (ER), progesterone receptor (PR) and HER2, in addition to Ki67 and p53, in TMAs for a cohort of 293 female patients with de novo breast cancer who were diagnosed and received treatment in Northern Ireland. Tissue from each patient was sampled in triplicate from different regions of the tumor, giving rise to a total of nine TMA blocks each containing 90–100 tissue cores. Full clinical and pathological follow-up was available for each patient, and full details regarding the cohort have been published previously.8 Of note, 169 cases were Grade 3 (58%), 119 cases Grade 2 (41%) and 5 cases Grade 1 (2%), while at time of diagnosis 175 tumors were ER +ve (60%), 115 ER -ve (39%) and 3 had unknown ER status (1%). All patients received anthracycline-based chemotherapy, with or without radiotherapy.8
Immunohistochemistry
All IHC was performed in a hybrid laboratory (Northern Ireland Molecular Pathology Laboratory) that has UK Clinical Pathology Accreditation, using established protocols described in detail elsewhere.8 The antibodies used were as follows: anti-ER (clone 6F11 mouse monoclonal antibody; Leica); anti-PR (clone PgR 636 mouse monoclonal antibody; Dako); anti-HER2 (clone CB11 mouse monoclonal antibody; Leica); anti-p53 (clone DO-7 mouse monoclonal antibody; Dako, Cambridgeshire, UK). In the case of Ki67, the original data set was stained with NCL-Ki67-MM1 antibody on a Leica staining platform with a polymer-based detection system. As a result of the recognized variation in Ki67 antibodies, and in particular their suitability for image analysis,9 additional tissue sections were also cut and stained with Confirm anti-Ki67 (clone 30-9) antibody on a Ventana platform with an Optiview detection kit for comparison.
Manual Scoring
All biomarkers in this data set were scored manually by a pathologist as described previously,8 and the pathologist also selected appropriate clinical cutoff thresholds that should be applied to these scores following standard guidelines in the literature.8 This allowed us to obtain a binary ‘clinical score’ variable distinguishing between positive and negative cases, which is not necessarily the modus operandi in clinical practice for some of these biomarkers but allowed a clear-cut comparison of the results. Briefly, ER and PR were evaluated using the Allred scoring method, giving scores in the range 0–8, where a score ≥3 was considered positive provided the proportion score was at least 2. Ki67 was evaluated using visual estimates of the percentage of positive staining tumor cells (range 0–100%), where ≥15% was considered positive. HER2 was scored in IHC according to current USA/UK guidelines10, 11 taking into consideration membranous staining only; cases scored as 0 and 1+ were considered negative, whereas 3+ was considered positive; equivocal cases (2+) were further subjected to DDISH analysis to identify amplification and categorized as positive or negative accordingly. p53 was assessed by calculating H-scores (range 0–300)12, 13 based upon nuclear staining. For p53 only, two cutoff thresholds (H-scores of 6 and 160) were required to separate extreme expression (both positive and negative) from the intermediate ‘wild-type’ phenotype. For the other biomarkers, a single cutoff was selected to separate strong/positive from weak/negative expression. Manual Ki67 scores were available for the MM1 antibody only.
Digital Image Analysis
All image analysis was performed using QuPath, which is a flexible software platform suitable for a range of digital pathology applications. In this study, the primary image analysis workflow comprised stain estimation, TMA dearraying, cell segmentation, feature computation and tumor cell identification with intensity classification (see Supplementary Figure 1). Application of this workflow resulted in both fine-grained cell-by-cell analysis and overall summary scores of biomarker expression for each TMA core. Dearraying consisted of an automated first step of core detection and grid assignment, followed by a manual quality assurance check during which the grid could be manually refined and cores removed if considered unsuitable for analysis (eg, because of insufficient tissue or dominant artifacts).
Cells are identified within QuPath using an custom, unsupervised algorithm based on stain separation using color deconvolution,14 followed by a sequence of image processing operations used to identify potential nucleus fragments either in the hematoxylin channel or from the sum of optical density values, depending upon stain characteristics. Morphological operations are then applied to merge and split fragments to identify individual nuclei. Each detected nucleus is expanded to give an approximation of the full cell area, constrained by distance and the proximity of neighboring nuclei, or by the presence of membrane staining in the case of HER2. A list of 103 (ER, PR, Ki67, p53) or 115 (HER2) features derived from cell morphometry and intensity measurements are then calculated for each cell (Supplementary Table 1). A random trees classifier15 is trained separately for each IHC biomarker using these features by having an experienced user annotate regions in a subset of the TMA cores across up to three slides, with interactive feedback on classification performance provided during training in the form of markup images. This limit of three sides was not imposed by QuPath, but was applied to ensure that the majority of the TMA cores used in the analysis were ‘unseen’ during training—as would normally be the case in high-throughput studies.
Cells were further subclassified according to staining intensity by applying manually selected thresholds to mean DAB optical density values determined by color deconvolution14 from the nuclear compartment (ER, PR, Ki67, p53) or membrane (HER2) of each cell. Cells close to the border of the core were removed by an iterative algorithm, which first calculated the convex hull for all cell centroids and then removed cells occurring on the boundary. This reduced the risk of artifacts impacting the final scores by excluding a thin layer of cells found at the outer edge of the tissue, whereas retaining the cells in the center.
Whole slide images were hosted remotely by PathXL (Belfast, UK), whereas analysis was performed locally on a desktop computer using a custom integration with PathXL’s API. After an initial interactive pre-analysis used to identify TMA cores and train up the tumor cell classification, scripts were generated for each biomarker to automate the analysis across the full data set.
Generation of Summary Scores
TMA cores identified as containing fewer than 100 tumor cells were excluded as unsuitable for scoring by DIA. After application of this selection criterion, the image analysis workflow resulted in 3911 tissue cores scored across all biomarkers, each containing an average of 3440 cells, of which 50.1% were classified as tumor cells. For each core, the tumor cell counts were combined into summary measurements incorporating information about DAB staining intensity, including the H-score (range 0–300), Allred score (range 0–8) and the percentage of cells staining positive (0–100%). For consistency, the H-score was used as the primary image analysis output for ER, PR and p53, whereas the percentage of positive tumor cells was used for Ki67—equivalent to the standard Ki67 labeling index.16 To more closely match clinical guidelines for HER2 scoring,17, 18 a modified H-score was calculated for HER2 as follows: cores having at least 10% tumor cells with strongly positive membranous staining were automatically assigned the maximum value (300) and cores exhibiting <10% moderate staining were assigned the minimum value (0), whereas all other cases were considered equivocal and the original H-score was retained. Where multiple summary measurements were available for a patient for each biomarker, the median was used as representative for that patient, so that each patient had at most one score per biomarker.
Statistical Analysis
Pearson’s and Spearman’s correlation coefficients were used to examine the relationship between the ‘raw’ image analysis results and the pathologist’s manual scores. Further comparisons were then performed after applying cutoff thresholds to both sets of scores and considering 5-year overall survival. In the case of the visual assessment, the pathologist who performed the scoring selected clinically relevant cutoff thresholds, as described above. For automated image analysis, a wider range of potential cutoff values was possible because of the more continuous nature of the scoring methodologies used. Therefore, several different methods were explored: (a) median (or tertiles, in the case of p53), (b) ‘minimized P-value’, based upon a log-rank test using overall survival and considering all possible thresholds, excluding the top and bottom 10% and (c) manual, in which a biologically plausible threshold was chosen that divided the patients into groups of roughly equal size to those previously identified by the pathologist to achieve comparability—while restricted to rounded values to reduce overfitting artifacts. It is important to note that method (b) inherently involved multiple testing, which leads to P-values, which are too small and cutoffs that potentially lack clinical meaning;19, 20, 21 consequently, we also report adjusted P-values to aid interpretation of the results.20 For p53, which required two cutoffs, method (b) was adapted to return one cutoff above and one cutoff below the median, and the log-rank test was performed comparing extreme positive and negative vs intermediate scores.
Analysis was performed and plots were created using QuPath’s built-in data exploration tools, and verified using R version 3.3.122 (with the ‘survival’ package23) or MATLAB (R2016a, The MathWorks, Inc., Natick, MA, USA) to confirm accuracy.
Results
Tumor Cell Classification
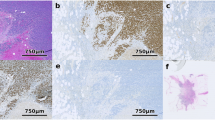
For all the biomarkers under investigation here, conventional scoring requires selective analysis only within the tumor cell population. For this reason, reliably and automatically identifying tumor cells is an essential component of any high-throughput image analysis of this type. To facilitate this, QuPath enables interactive training of cell classification, after which the classifier can be saved and run over multiple slides, and the results visualized via color-coded markup images. Representative examples of these markup images are shown in Figure 1 and in Supplementary Figures 2-4.

Example images showing QuPath’s trained tumor recognition applied to TMA cores stained for PR, Ki67 (Confirm) and HER2. Detected cells are color-coded according to their classification: red (tumor, strongly positive), orange (tumor, moderately positive), yellow (tumor, weakly positive), blue (tumor, negative) and green (non-tumor).
Concordance Between Pathologist and Image Analysis Scoring
The correlations between DIA and pathologist biomarker scores are summarized in Table 1. A high level of correlation between manual and DIA scores is demonstrated in all cases. The area under the receiver operating characteristic curve (AUC) provides a measure of how the DIA scores may be thresholded to align to the pathologist’s clinical (binary) score, where values above 0.949 indicate almost perfect agreement for the three clinical biomarkers ER, PR and HER2. Correlation remained high using both antibodies for Ki67, despite the pathologist’s scoring being based solely on the MM1-stained images. Correlation between DIA scores using each antibody against Ki67 was also high (PCC=0.794, rho=0.778).
Survival Analysis
When assessing the performance of any new approach to scoring IHC biomarkers, it is inadequate to use agreement with manual scoring as the only ‘gold standard’ because of the high inter- and intra-observer variability between pathologists.24, 25 Consequently, we incorporated survival analysis after stratifying patients using the cutoff methods described above to ascertain the extent to which DIA and manual scores are each able to independently identify a statistically significant stratification of patients for each biomarker. The results looking at 5-year overall survival are shown in Table 2, along with Kaplan–Meier curves using manual cutoffs in Figure 2 (comparable Kaplan–Meier curves based upon the pathologist’s scoring are given in Supplementary Figure 5). Although the analysis was applied to the same cohort, slightly lower numbers of patients are represented for the DIA scoring because of differences in core selection, for example, through the exclusion of small or folded tissue sections from DIA, or the automated rejection of cores containing fewer than 100 detected tumor cells. For all biomarkers considered, it was possible to achieve a stratification of patients with a similar or improved statistical significance (log-rank test, P-value<0.05) using DIA when compared with manual scoring using all of the cutoff determination methods that were applied, with the exception of the median cutoff for HER2—which did not provide a result, since more than half of the cases had the minimum score of 0. This is consistent with a prevalence of HER2 overexpression in breast cancers of 15–20%, which is not taken into consideration when using a 50% cutoff.17 However, in the case of Ki67 statistical significance was only seen with DIA when the Confirm antibody was used, but not MM1. This is consistent with reports suggesting that Ki67 is particularly challenging for DIA, with a dependency upon the specific antibody.9, 26

Kaplan–Meier curves showing 5-year overall survival based on patient stratification using DIA scoring with manually defined cutoff thresholds (see Table 2).
Robustness of Cutoff Thresholds
Table 2 demonstrates that a highly significant association with survival can be obtained using DIA in almost all cases, even when using quite different methods of cutoff selection that result in very different cutoff values. For example, the P-values obtained using thresholds of 5 and 126.5 were almost identical when applied to ER (both <0.002), despite the fact that changing the cutoff resulted in 43 patients (16%) being assigned to different groups.
To further explore the impact of cutoff selection upon the identified associations with survival, we generated plots showing the distribution of log-rank P-values for all possible thresholds for the different biomarkers (Figure 3). The plots for ER, PR and HER2 all show a wide range of values below the conventional threshold of 0.05 taken to denote statistical significance. For ER, this implies that any cutoff between 1 and 265 (a range encompassing the scores of 181 patients with 22 observed events) would yield a P-value below the 0.05 significance threshold. Similarly, any cutoff between 1 and 237 (140 patients and 16 observed events) would yield a significant difference for PR H-scores, and any cutoff between 1 and 150 (8 patients and 2 observed events) would be significant for HER2 (Figure 3, top 3 panels).

Distribution of P-values for all possible thresholds represented in the data for image analysis scoring of each biomarker. Horizontal (dotted) line indicates threshold for significance at 0.05 level.
Figure 3 also highlights the stark difference in Ki67 scores using each antibody. In the case of MM1, no single cutoff can be used to identify statistically significant differences (ie, the curve never dips below the 0.05 line), and indeed the curve fluctuates quite widely—implying limited prognostic value. However, the analysis using the Confirm antibody shows a pronounced dip precisely in the range expected based upon proposed Ki67 cutoffs of clinical relevance,24 so that a significant difference in overall survival would be seen by applying any cutoff between 7 and 26 (a range encompassing 98 patients and 13 observed events).
Alternative Scoring Methods
Having demonstrated the ability of DIA to robustly score all the biomarkers within this study, we next wanted to examine the importance of tumor recognition and the impact of different DIA scoring approaches and methodologies. To do this, we took advantage of the fact that QuPath generates data on every individual cell that it detects, and this information can easily be recombined to produce alternative scores. To assess the importance of tumor recognition, we created a set of H-scores where every cell—irrespective of tumor classification—contributed to the result. Furthermore, for ER, PR and p53 we calculated Allred scores based on tumor cells to provide an alternative to the H-score used otherwise.27 In all cases, clinically relevant cutoffs were determined automatically by selecting the integer threshold that maximized Youden’s J statistic, comparing DIA scores with the pathologist’s clinical score for each patient. This maximized the sum of the sensitivity and specificity, and ensured that all cutoffs were in a clinically meaningful range.
These results are summarized in Table 3. When comparing thresholded manual scores with H-scores generated by DIA using tumor identification, near perfect agreement is seen for ER, PR and HER2 (Cohen’s κ >0.86 in all cases), and good agreement for Ki67 and p53 (κ >0.6). Lower agreement is generally seen for all other methods of scoring all biomarkers, in addition to lower sensitivity, specificity and accuracy in most cases. However, lower agreement with the pathologist’s scoring does not always translate into a loss of statistical significance when associating the results with survival. Interestingly, a significant association with overall survival is seen when applying the H-score to all cells (ie, without tumor recognition) in most cases—indicating that, for at least some IHC biomarkers that are conventionally scored only within the tumor cell population, accurate tumor recognition is not a necessary prerequisite for identifying associations within a sufficiently large cohort for a TMA study, where a measure of tumor identification is already incorporated in the region selection applied when constructing the TMAs. Nevertheless, in most cases the inclusion of a digital tumor identification step has a substantial positive impact upon the quality of the results, and improved agreement with the pathologist’s scores. Furthermore, the range of cutoffs that may be applied to yield a significant stratification was always substantially larger whenever tumor recognition was applied—indicating its role in improving the robustness of the results, and increasing the likelihood that any cutoff used would generalize well to new data (Supplementary Table 2). Finally, it should be kept in mind that biomarkers exist for which expression within the stromal cells is of equal or greater importance to the expression within the tumor cells, and also that the automatic exclusion of cores containing fewer than 100 tumor cells is only possible whenever tumor identification is done; without this, a more time-consuming manual quality assurance would be required to remove cores lacking in sufficient tumor cells for reliable analysis.
In addition to the value of tumor recognition, Table 3 also shows the value of a DIA scoring mechanism with a wide dynamic range. Use of the H-score (range 0–300) consistently outperformed the Allred score (0–8), both in terms of agreement with the pathologist’s cutoffs and in associations with survival.
Application of Multiple Cutoffs to p53
Manual scoring of p53 within this data set made use of two cutoffs, as it was previously shown by our group that this was important to distinguish extreme positive and extreme negative from moderate staining patterns.8 Consequently, DIA scores were also generated here with two cutoffs. Nevertheless, the continuous nature of the H-score provided by DIA makes it amenable to alternative approaches to cutoff selection, which may be particularly valuable when biologically derived cutoffs are not available. One intuitive method is to consider ‘valleys’ present within P-value plots such as those shown in Figure 3 as being suggestive of where appropriate cutoff thresholds may lie. Applying this in the case of p53, the plot actually suggests two potential cutoffs to separate weak from moderate staining: at around H-scores of 15 and 50. Applying each of these independently leads to a significant stratification when comparing weak staining with moderate (P-values of 0.00038 and 0.0033, respectively; strong staining with an H-score >150 omitted). However, using all suggested cutoffs (15, 50 and 150) it is possible to identify four distinct patient subgroups according to different levels of staining: negative (<15; 49 patients), weak (≥ 15, <50; 63 patients), moderate (≥50, <150; 67 patients) and strong (≥150; 101 patients). The negative subgroup has the worst prognosis, with a statistically significant difference between this and the weak subgroup (P=0.0398). The separation between negative and moderate is highly significant (P=0.0002), whereas the separation between weak and moderate remains significant (P=0.049), as does the separation between moderate and strong (P=0.0057). To our knowledge, this additional separation between negative/weak sub-populations was not previously reported, and not identified using the manual scoring by a pathologist.8 This indicates another clear benefit in the continuous, reproducible scoring possible using DIA, where new expression phenotypes that may not be distinguished reproducibly by the naked eye can be identified using quantitative characterization of biomarkers (Figure 4). Example images are provided in Supplementary Figure 8.

Application of additional cutoffs to p53 data. (Left, center) Cutoffs to separate weak from moderate staining are set at H-scores of 15 and 50 respectively; cases exhibiting strong staining (H-score >150) are excluded (n=179). (Right) Three cutoffs (15, 50 and 150) are applied to stratify patients in the full data set.
Reevaluation of p53 and Ki67 According to ER Status
Although exploration of different cutoff values for categorizing scores produced by DIA, as described above, may provide additional insights regarding the staining of individual biomarkers in isolation, there is a risk of spurious findings by allowing survival information to inform cutoff choices because of the known problem of multiple testing20—particularly where a narrow range of cutoff values leads to a statistically significant result. Rather than applying numerous cutoffs to a full cohort, a clearer picture may emerge by considering patients separately according to different molecular subtypes.
To explore this, we repeated the survival analysis of p53 and Ki67 using DIA scores after separating patients according to ER status (Figures 5 and 6). For the ER-positive subgroup, the primary finding remained, in that applying lower cutoffs of either 15 or 50 combined with a higher cutoff of 150 could separate strong and weak from moderate expression, where moderate expression was associated with longer overall survival. However, in the ER-negative subgroup, it was not possible to identify a moderate subgroup for which prognosis was more favorable. Instead, any single cutoff between 12 and 270 led to a significant stratification for which high p53 expression correlated with better overall survival, relative to the other patients within the ER-negative subgroup. This is consistent with reports from the IBCSG Trials VIII and IX, where high p53 was also found to be associated with better survival among ER-negative patients, but worse survival among ER-positive patients.28

(Top) Kaplan–Meier curves showing 5-year overall survival when stratifying patients according to p53 IHC scores, after separating patients according to ER status. Cutoffs used are 15 and 150 (ER-positive; n=168, P-value compares extreme low and high vs moderate) and 50 only (ER-negative; n=112). (Bottom) Plots showing how P-values vary according to different cutoff thresholds that could be applied for patient stratification (log-rank test).

Kaplan–Meier curves showing 5-year overall survival when stratifying patients according to Ki67 labeling indices determined by DIA, after separating patients according to ER status.
For Ki67, the result was similar. For ER-positive cases, use of the same cutoff thresholds as applied to the whole data set identified an association between higher Ki67 labeling indices and worse overall survival, which was statistically significant for the Confirm antibody (P=0.0039, cutoff 25) and not quite significant for MM1 (P=0.0547, cutoff 15). However, within the ER-negative subgroup, the trend was reversed for both antibodies: higher Ki67 indices were associated with improved survival. This finding was statistically significant for both antibodies using similar cutoff thresholds (cutoff 35, P=0.0176 Confirm antibody; cutoff 30, P=0.0364 MM1). A significant association of higher Ki67 indices with improved prognosis was not evident within the pathologist’s manual scores.
Discussion
In this study, we have shown the ability of DIA to accurately score a range of important biomarkers in breast cancer TMAs. Using the QuPath digital pathology platform, we were able to identify associations between biomarker expression and overall survival with a similar or better level of statistical significance to that achieved through manual scoring by an experienced pathologist, while also uncovering additional insights that warrant further investigation and validation. Importantly, all of this has been achieved using automated tumor recognition algorithms that can be quickly and interactively trained to distinguish between different cell types.
Several commercial digital pathology solutions, such as Halo (Indica Labs, New Mexico, USA), Tissue Studio (Definiens AG, Munich, Germany) or Oncotopix (Visiopharm A/S, Hoersholm, Denmark), also provide TMA biomarker evaluation capabilities. Typically, the approach implemented in software applications such as these is to first identify regions of interest (eg, areas of tumor) based on an automated or semi-automated region identification using texture and color features, or alternatively by aligning a tumor mask derived from an adjacent tissue section stained with a cytokeratin marker. Cells within these regions are then segmented and scored according to staining intensity to produce the final result. Although QuPath offers some limited texture-based region identification, it currently does not provide image registration capabilities to align tissue sections automatically. Rather, the preferred approach within QuPath switches the order of operations, so that cell segmentation is placed first and then followed by a machine learning classification step to identified the sub-population of cells to be scored. In practice, this means that all cells may be segmented up-front by batch processing, after which a classifier to distinguish between cell types can be either loaded from disk and applied, or trained interactively based upon annotations provided by the user containing examples of different cell types (Supplementary Figure 1). In both cases, the final results are available immediately after classification.
The ability to perform algorithm training and verification of the results across a cohort in one sitting helps ensure an effective use of a pathologist’s time. During interactive training, the classification tends to stabilize quickly for common cell patterns, but an experienced pathologist browsing the slides may recognize examples of cells that differ markedly from those previously seen and have been misclassified; in such cases they may add new annotations to address the problem immediately and generate an improved set of results—without a need to repeat lengthy processing steps. This approach also avoids a reliance on cytokeratin staining, which substantially increases cost and laboratory effort, and limits the analysis to the intersection of tissue samples where both the biomarker of interest and cytokeratin staining has been successfully applied. On the other hand, the method of analysis described here is subject to the effectiveness of cell identification and classification, which may vary depending upon the stain quality and biomarker expression pattern. In particular, cell classification methods based on morphological or texture features may perform less robustly on lower grade tumors, where the features upon which the classification is based may vary less between tumor and benign cells. To this end, critical evaluation of the results and stringent quality control at all stages are essential to ensure the accuracy of the results, irrespective of the DIA software and approach used.
Related to the above, we also explored the issues involved when assessing the performance of DIA for biomarker scoring. We have focused on two separate measures of success: agreement with manual scoring by a trained pathologist, and the ability to explore statistically the relevance of marker stratification of patients by survival analysis. Neither measure alone is scientifically optimal. Considering manual scoring methods to be the gold standard fails to take into account that DIA may be capable of improved accuracy over that of a human. Also, demonstrating improved statistical significance in patient stratification may be a reflection on cohort size, patient event proportions and other factors indicating overfitting, and does not necessarily result in cutoff thresholds that are either generalizable or transferable to clinical practice. Furthermore, in the case of ER and PR, we have shown that the association with survival is sufficiently strong such that there is a considerable ‘margin for error’ in scoring when this is used to define successful analysis; so much so that including a large number of non-tumor cells in the evaluation, while somewhat diluting the robustness, can still lead to a highly significant result and excellent agreement with dichotomized manual scores. This ought to serve as a warning against using ER and PR alone as a demonstration that a new method of analysis is fit-for-purpose in a general setting for other biomarkers. For these reasons, we have presented our results across a range of different biomarkers, both from multiple viewpoints and with multiple different methods of cutoff selection, as a measure of the robustness of our analysis.
Our results also revealed two results of biological interest. The distinction between weak staining for p53 and no measurable expression was made by our group in a qualitative manner after careful validation of the p53 IHC,8 demonstrating the different mutation status associated to this pattern. For the first time to our knowledge, this is now reproduced using DIA from standard brightfield IHC staining. The importance of this finding requires a subsequent, independent cohort; however, this observation highlights the potential of DIA in generating results with a broader genomic relevance.
In addition, our initial results showed the strong dependence of Ki67 scoring on the specific antibody used, in keeping with previous reports.9 However, by separately analyzing ER-positive and ER-negative patients, we further demonstrated that substantial differences in the interpretation of Ki67 existed for this cohort. These results are perhaps surprising, given that it is well established that a high Ki67 labeling index is associated with worse prognosis.16 Nevertheless, it is also known that Ki67 score can be predictive of neoadjuvant chemotherapy response,29 and mean Ki67 labeling indices have been found to be higher in patients with a pathological complete response after neoadjuvant chemotherapy; this has led to the hypothesis that there may be a high cutoff above which prognosis is better than in patients with lower Ki67 values.30 As all patients within the cohort in this study received anthracycline-based chemotherapy,8 our findings that high Ki67 values within the ER-negative subgroup are significantly associated with better overall survival lends tentative support to this hypothesis, and warrants further investigation. In any case, the fact that the ‘correct’ interpretation of Ki67 may take on opposing roles depending upon the treatment and molecular subtype of the cancer may shed some further light on why it has proven so difficult to establish a standardized approach to Ki67 evaluation with a single accepted methodology and cutoff value,16, 24 as the best cutoff found within each study is highly dependent upon the makeup of the cohort. Our comparison of two antibodies against Ki67 has also reiterated the importance of antibody selection.
Based upon all our results, we are able to draw some conclusions regarding the application of DIA to biomarker evaluation. First, we have shown that it is possible to robustly score a range of biomarkers in TMAs without a need for a tumor identification marker. Second, we have demonstrated that tumor identification may not be essential in all cases whenever the goal is simply to ascertain an association between the biomarker and survival within TMAs; however, where it is applied, tumor recognition improves the robustness of the scoring by removing one confounding factor. Furthermore, for some biomarkers, it can be particularly important that the correct cells are being scored in order to identify subtle effects. Third, we have shown the benefits of using scoring metrics for DIA with a high dynamic range. This improves the ability to distinguish between more subtle differences, whereas also increasing the tolerance to a small number of artifacts or misclassifications. The H-score meets this criterion but the Allred score does not. Thus, it should be kept in mind when planning DIA studies that the preferred method of scoring may well differ from that which would be used by a pathologist scoring manually.
References
Salto-Tellez M . Principles of Molecular Diagnostics and Personalized Cancer Medicine. Tan D, Lynch HT (eds). Wolters Kluwer Health, 2013; 196–205.
Salto-Tellez M, James JA, Hamilton PW . Molecular pathology - the value of an integrative approach. Mol Oncol 2014;8:1163–1168.
Maxwell P, Salto-Tellez M . Validation of immunocytochemistry as a morphomolecular technique. Cancer Cytopathol 2016;124:540–545.
Elliott K, McQuaid S, Salto-Tellez M et al. Immunohistochemistry should undergo robust validation equivalent to that of molecular diagnostics. J Clin Pathol 2015;68:766–770.
Varga Z, Diebold J, Dommann-Scherrer C et al. How reliable is Ki-67 immunohistochemistry in grade 2 breast carcinomas? A QA study of the Swiss Working Group of Breast- and Gynecopathologists. PLoS ONE 2012;7:e37379.
Polley M-YC, Leung SC, McShane LM et al. An international Ki67 reproducibility study. J Natl Cancer Inst 2012;105:1897–1906.
Hamilton PW, Bankhead P, Wang Y et al. Digital pathology and image analysis in tissue biomarker research. Methods 2014;70:59–73.
Boyle DP, McArt DG, Irwin G et al. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology 2014;65:1–13.
Zabaglo L, Salter J, Anderson H et al. Comparative validation of the SP6 antibody to Ki67 in breast cancer. J Clin Pathol 2010;63:800–804.
Wolff AC, Hammond ME, Hicks DG et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med 2007;131:18–43.
Walker RA, Bartlett JM, Dowsett M et al. HER2 testing in the UK: further update to recommendations. J Clin Pathol 2008;61:818–824.
McCarty KS, Szabo E, Flowers JL et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res 1986;46:4244s–4248s.
Goulding H, Pinder S, Cannon P et al. A new immunohistochemical antibody for the assessment of estrogen receptor status on routine formalin-fixed tissue samples. Hum Pathol 1995;26:291–294.
Ruifrok AC, Johnston DA . Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 2001;23:291–299.
Breiman L . Random forests. Mach Learn 2001;45:5–32 2001.
Dowsett M, Nielsen TO, A'Hern R et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst 2011;103:1656–1664 (2011).
Wolff AC, Hammond ME, Hicks DG et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013.
Rakha EA, Pinder SE, Bartlett JM et al. Updated UK Recommendations for HER2 assessment in breast cancer. J Clin Pathol 2015;68:93–99.
Lausen B, Schumacher M . Evaluating the effect of optimized cutoff values in the assessment of prognostic factors. Comput Stat Data Anal 1996;21:307–326.
Altman DG, Sauerbrei W, Schumacher M . Dangers of using ‘optimal’ cutpoints in the evaluation of prognostic factors. J Natl Cancer Inst 1994;86:829–835.
Hilsenbeck SG, Clark GM . Practical p-value adjustment for optimally selected cutpoints. Stat Med 1996;15:103–112.
R Core Team R: A Language and Environment for Statistical Computing. R Core Team, R Foundation for Statistical Computing: Vienna, Austria, 2016, URL http://www.R-project.org/.
Therneau T A Package for Survival Analysis in S. version 2.38 2015 https://CRAN.R-project.org/package=survival.
Polley M-YC, Leung SC, McShane LM et al. An international Ki67 reproducibility study. J Natl Cancer Inst 2013;105:1897–1906.
Parker RL, Huntsman DG, Lesack DW et al. Assessment of interlaboratory variation in the immunohistochemical determination of estrogen receptor status using a breast cancer tissue microarray. Am J Clin Pathol 2002;117:723–728.
Stålhammar G, Fuentes-Martinez N, Lippert M et al. Digital image analysis outperforms manual biomarker assessment in breast cancer. Mod Pathol 2016;29:318–329.
Harvey JM, Clark GM, Osborne CK et al. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999;17:1474–1481.
Coates AS, Millar EK, O'Toole SA et al. Prognostic interaction between expression of p53 and estrogen receptor in patients with node-negative breast cancer: results from IBCSG Trials VIII and IX. Breast Cancer Res 2012;14:R143.
Brown JR, DiGiovanna MP, Killelea B et al. Quantitative assessment Ki-67 score for prediction of response to neoadjuvant chemotherapy in breast cancer. Lab Invest 2014;94:98–106.
Fasching PA, Heusinger K, Haeberle L et al. Ki67 chemotherapy response and prognosis in breast cancer patients receiving neoadjuvant treatment. BMC Cancer 2011;11:486.
Acknowledgements
The samples used in this research were received from the Northern Ireland Biobank, which is funded by HSC Research and Development Division of the Public Health Agency in Northern Ireland and Cancer Research UK through the Belfast CRUK Centre and the Northern Ireland Experimental Cancer Medicine Centre; additional support was received from the Friends of the Cancer Centre. The Northern Ireland Molecular Pathology Laboratory, which is responsible for creating resources for the NIB has received funding from Cancer Research UK, the Friends of the Cancer Centre and the Sean Crummey Foundation. The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007–2013/under REA grant agreement (285910 to PWH); Invest Northern Ireland (RDO0712612 to PWH); Cancer Research UK Accelerator (C11512/A20256 to PWH/MS-T). The Northern Ireland Molecular Pathology Laboratory is supported by Cancer Research UK, Experimental Cancer Medicine Centre Network, the NI Health and Social Care Research and Development Division, the Sean Crummey Memorial Fund, the Tom Simms Memorial Fund and the Friends of the Cancer Centre (to MS-T). The Northern Ireland Biobank is funded by the Health and Social Care Research and Development Division of the Public Health Agency in Northern Ireland and Cancer Research UK through the Belfast CRUK Centre and Northern Ireland Experimental Cancer Medicine Centre; additional support was received from the Friends of the Cancer Centre (to JAJ).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Peter W Hamilton is also Founder and Director in PathXL Ltd. Manuel Salto-Tellez is a senior advisor to PathXL. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Here the authors present the results of an automated digital image analysis using a new purpose-built software platform (QuPath) applied to breast cancer patients and five key biomarkers. Their findings demonstrate QuPath's suitability for fast, reproducible, high-throughput tissue microarray analysis, using tumor recognition algorithms for immunohistochemistry, trained automatically and interactively.
Supplementary information
Rights and permissions
About this article

Cite this article
Bankhead, P., Fernández, J., McArt, D. et al. Integrated tumor identification and automated scoring minimizes pathologist involvement and provides new insights to key biomarkers in breast cancer. Lab Invest 98, 15–26 (2018). https://doi.org/10.1038/labinvest.2017.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.131
This article is cited by
-
Histopathology images-based deep learning prediction of prognosis and therapeutic response in small cell lung cancer
npj Digital Medicine (2024)
-
Digital morphometry and cluster analysis identifies four types of melanocyte during uveal melanoma progression
Communications Medicine (2023)
-
Low TINAGL1 expression is a marker for poor prognosis in breast cancer
Journal of Cancer Research and Clinical Oncology (2023)
-
Prediction of Biomarker Expression on Primary Pancreatic Ductal Adenocarcinoma Tissues Using Fine-Needle Biopsies: Paving the Way for a Patient-Tailored Molecular Imaging Approach
Molecular Diagnosis & Therapy (2023)
-
Tissue microarrey: a potential cost-effective approach for mismatch repair testing in colorectal cancer
BMC Gastroenterology (2022)
